

Abstract
Variants in RNF213 lead to susceptibility to moyamoya disease, a rare cerebral angiopathy characterized by bilateral stenosis of the internal carotid arteries and development of a compensatory collateral network. We describe a 3-month-old female with seizures, arterial narrowing involving the internal carotid and intracranial arteries and inferior abdominal aorta, and persistently elevated transaminases. Whole exome sequencing demonstrated a novel de novo variant in RNF213, securing a molecular diagnosis and directing appropriate intervention. This report underscores the role of whole exome sequencing in cases for which a complex and atypical presentation may mask diagnosis. Furthermore, the early and severe presentation in our patient, in conjunction with a novel de novo RNF213 variant, suggests that specific variants in RNF213 may lead to a Mendelian form of disease rather than simply conferring susceptibility to multifactorial disease.
Keywords: Moyamoya disease, RNF213, whole exome sequencing
INTRODUCTION
Moyamoya disease (OMIM # 607151) is a progressive cerebral angiopathy characterized by stenosis of the bilateral internal carotid arteries with subsequent development of a fine network of abnormal compensatory collateral vessels [Suzuki and Takaku 1969; Currie et al., 2011; Fujimura et al., 2014]. This steno-occlusive change renders susceptibility to cerebral infarction or transient ischemic attacks, and rupture of the collateral vessels can cause intracranial hemorrhage [Kamada et al., 2011]. In fact, moyamoya disease accounts for approximately 6% of pediatric strokes [Smith and Scott, 2012], with a peak incidence rate at 5–9 years and a second higher peak in adulthood [Baba et al., 2008]. Strictly defined, moyamoya disease is bilateral arteriopathy in the absence of any other associated systemic disorder, whereas moyamoya syndrome includes intracranial arteriopathy in the setting of well-recognized systemic disorders such as sickle cell disease, neurofibromatosis type 1, Down syndrome, Alagille syndrome, congenital cardiac anomalies and renal artery stenosis [Smith and Scott, 2012]. The prevalence of moyamoya disease differs between populations and is ten times greater in East Asia as compared to Western countries, reaching a prevalence of approximately 3 cases per 100,000 children in Japan [Scott and Smith, 2009].
While the etiology of moyamoya disease is considered multifactorial, several lines of evidence support a genetic susceptibility to disease, including a positive family history in 10–15% of cases, a high concordance rate in monozygotic twins, and an apparent autosomal dominant mode of inheritance with incomplete penetrance in some families [Kamada et al., 2011]. The RNF213 gene has been identified as a susceptibility gene for Moyamoya, with a particular founder mutation p.R4810K showing strong association with disease in East Asian populations [Kamada et al., 2011; Liu et al., 2011]. Additional rare variants in the RNF213 gene have also been identified in moyamoya disease in ethnically diverse populations, including Europeans and Hispanics [Cecchi et al., 2014].
In the present report, we describe an atypical infantile presentation of moyamoya disease which was diagnosed molecularly by whole exome sequencing (WES) and only thereafter confirmed by diagnostic radiology. To the best of our knowledge, this is the first report of a de novo variant in RNF213 associated with infantile moyamoya, and adds to the spectrum of RNF213 private variants in the Hispanic population. We demonstrate the direct impact of early WES testing in pediatric stroke, affecting patient care and therapeutic intervention.
CASE PRESENTATION
The patient is a Hispanic (Mexican) female born to a non-consanguineous couple, a 47-year-old father and 38-year-old mother, at 33 6/7 weeks gestation due to cervical incompetency and premature contractions. Her birth weight was 2.27 kg (90th centile for gestational age) (Fig. 1). At three months of age, the patient developed acute onset of focal seizures with involvement of the right extremities and face. Further evaluation revealed age-appropriate development with no prior history of fever or trauma. Physical exam, including neurologic and ophthalmologic evaluation, demonstrated a well-appearing non-dysmorphic female with symmetric anthropometric measurements (weight, height and head circumference at percentiles 17, 17, and 10, respectively) without focal neurological deficits.
Figure 1.
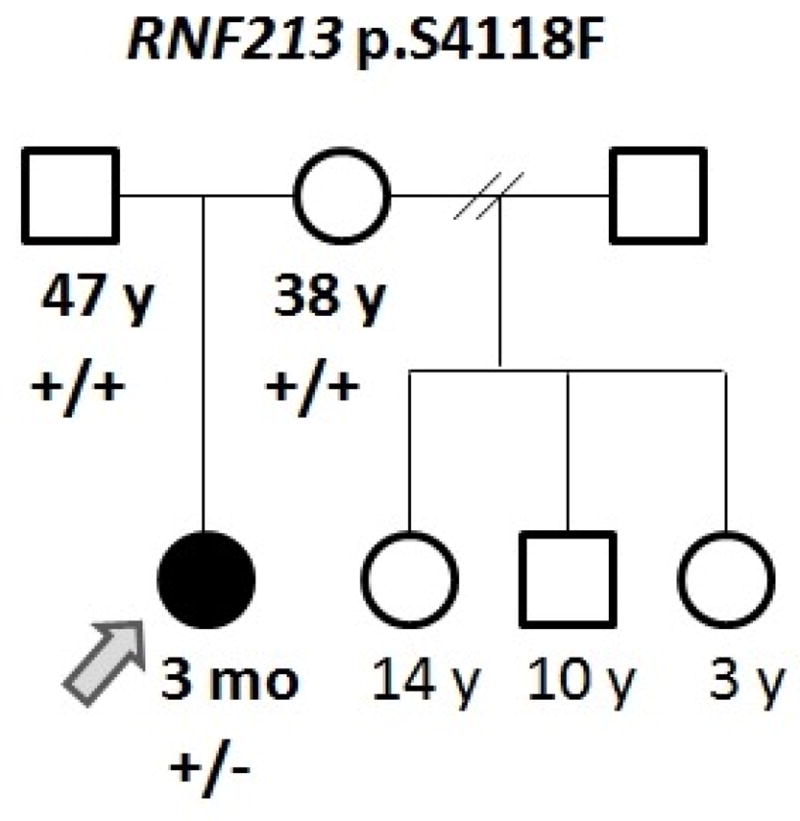
Pedigree of the reported family. (+) indicates a reference allele, (−) indicates a variant allele.
Initial labs were significant for an elevated AST (157 U/L; normal 20–60 U/L), ALT (143 U/L; normal 6–50 U/L), and GGT (190 U/L; normal 11–82 U/L); plasma homocysteine was also elevated (11.7 umol/L; normal 3.3–8.3 umol/L). Repeat total plasma homocysteine and methionine were normal. Lipid panels were within normal range, with LDL ranging from 48–94 (normal 0–109 mg/dL), VLDL 11 (normal 5–40 mg/dL), and maximal triglyceride level 124 (normal 20–150 mg/dL). HDL was 81 (normal >39 mg/dL). Infectious work-up including blood, urine, and cerebrospinal fluid cultures were negative. Electroencephalogram showed evidence of bilateral independent potentially epileptogenic foci in the posterior temporal regions; background activity appeared normal for age. Brain CT demonstrated hyperdensities, thought to represent calcifications within the bilateral distal branches of the middle and posterior cerebral arteries, and CT angiogram revealed narrowing of these vessels. Brain MRI/MRA confirmed normal parenchyma with marked narrowing at the bilateral internal carotid artery terminus and the supraclinoid internal carotid arteries, as well as narrowing of the vertebral arteries at the skull base with involvement of the posterior cerebral and superior cerebellar arteries (Fig. 2A). Imaging of extracranial vessels revealed moderate diffuse hypoplasia of the infrarenal abdominal aorta. The aortic root and thoracic aorta were normal in caliber. A diagnosis of generalized arterial calcification of infancy was considered, but molecular analysis of ENPP1 did not demonstrate pathologic mutations or copy number variants (Connective Tissue Gene Tests, Allentown, PA).
Figure 2.

Brain imaging of patient showing (A) bilateral narrowing of internal carotid arteries at 3 months (black arrows); (B) carotid angiogram with typical ‘puff of smoke’ collateral vessels at 10 months; (C) right MCA distribution stroke, 10 months; and (D) left MCA distribution stroke, 10 months.
At 4 months of age, a liver biopsy was performed for persistently elevated transaminases and demonstrated lysosomal neutral fat droplets and cytoplasmic cholesterol crystals by electron microscopy, concerning for cholesteryl ester storage disease. Plasma lysosomal acid lipase activity (Seattle Children’s Hospital, Seattle, WA), as well as liver tissue acid lipase content and LIPA gene sequencing (Medical Genetics Laboratories, Baylor College of Medicine, Houston, TX) were normal. Due to lack of an underlying diagnosis in the setting of a complex phenotype including intracranial vasculopathy, narrowing of the distal abdominal aorta, persistently elevated liver enzymes, and negative targeted gene testing, chromosomal microarray and clinical whole exome sequencing (WES) were sent at 7 months of age (Medical Genetic Laboratories, Baylor College of Medicine).
At ten months of age, the patient presented with bilateral acute MCA strokes, occurring seven days apart. Review of her molecular testing demonstrated a normal chromosome microarray (v9.1.1). Exome sequencing of the proband, followed by Sanger sequencing of proband and parental samples for validation and segregation, revealed a de novo heterozygous c.12353C>T (p.S4118F) novel variant in RNF213 (Table I and Fig. 3). This finding prompted further investigation with a carotid angiogram that confirmed moyamoya changes within the distal internal carotid arteries with associated arterial-arterial collaterals and slow flow distally (Fig. 2). The patient underwent bilateral dural inversions at ten months of age. She was seen at a follow-up clinic visit at 16 months of age, and was cruising but not yet walking and had no specific words, concerning for mild motor and language delay.
Table I.
Whole exome sequence results: variants in disease genes possibly related to patient’s phenotype.
| Disease | Inheritance | Gene | Nucleotide/Amino Acid | Zygosity | ESP5400 AA/EA | SIFT/PolyPhen-2 |
|---|---|---|---|---|---|---|
| {Moyamoya disease 2, susceptibility to} [MIM:607151] | Complex | RNF213 | c.12353C>T p.S4118F |
Het | N/R | Damaging/Probably damaging |
| Homocystinuria, cblD type, variant 1 [MIM:277410] | AR | MMADHC | c.41A>G p.Y14C |
Het | N/R | Damaging/Probably damaging |
| Homocystinuria- megaloblastic anemia, cblG complementation type [MIM:250940] | AR | MTR | c.1862A>G p.D621G |
Het | 1/3737 10/7010 |
Damaging/Benign |
AR - autosomal recessive; Het - heterozygous; N/R - not reported; AA/EA - African Americans/European Americans
Figure 3.
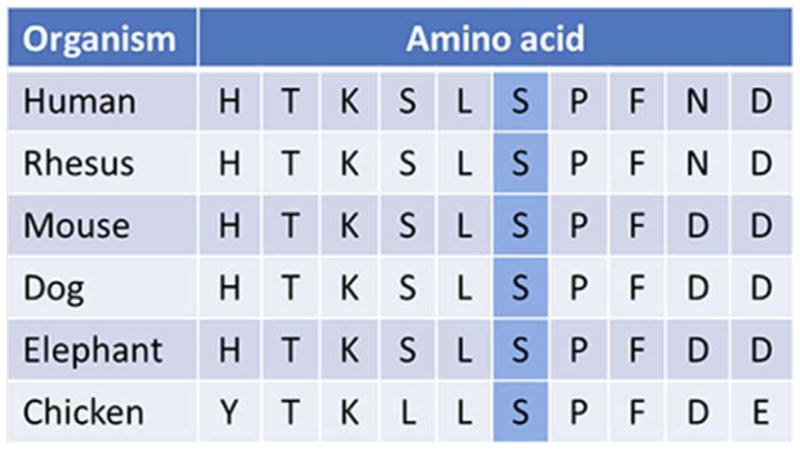
Evolutionary conservation of the altered amino acid (p.S4118F) in the RNF213 gene (adapted from http://genome.ucsc.edu)
DISCUSSION
The discovery of the p.R4810K East Asian founder mutation established RNF213 as a key susceptibility gene for moyamoya disease. Moyamoya disease can be inherited in an autosomal dominant fashion with incomplete penetrance, and case-control studies have demonstrated that p.R4810K confers at least a 100-fold risk of disease [Kamada et al., 2011; Liu et al., 2011; Miyatake et al., 2012].
This report describes a Hispanic female who presented at 3 months of age with acute onset of focal seizures, elevated transaminases, and abnormal vascular imaging including marked narrowing of the bilateral internal carotid arteries in addition to involvement of the posterior cerebral and superior cerebellar arteries and mild stenosis of the infrarenal abdominal aorta. After initial targeted gene testing (LIPA, ENPP1) resulted negative, the unusual combination of arterial narrowing and progressive hepatitis prompted a thorough genetic evaluation including exome sequencing which identified a heterozygous de novo variant in the RNF213 gene that is predicted to be damaging/probably damaging by SIFT/PolyPhen-2, respectively. These results prompted a carotid angiogram that confirmed moyamoya disease and subsequent revascularization surgery using the dural inversion procedure [Dauser et al., 1997]. Whole exome sequencing did not provide a molecular explanation for the elevated transaminases and abnormal liver biopsy seen in our patient; however, over time, her liver enzymes almost normalized (AST 57 U/L; normal for age 3–69 U/L), ALT (76 U/L; normal for age 5–30 U/L).
Early onset of moyamoya disease has been described in patients homozygous for the RNF213 p.R4810K allele, with 60% of homozygotes diagnosed before age 4 [Miyatake et al., 2012]. A study of 19 moyamoya patients diagnosed before the age of 2 yrs with no molecular diagnosis revealed that the majority (58%) had an associated disease or genetic condition [Jackson et al., 2014], better defining these as moyamoya syndrome. As our patient presented at 3 months of age but was non-dysmorphic and had no obvious associated genetic disease, we screened the exome data for additional risk factors that may have contributed to her early morbidity. Two variants of unknown significance, both predicted damaging by SIFT, were found in the MMADHC and MTR genes, associated with autosomal recessive homocystinuria (Table I). No second hit in these genes was identified. This is of special interest in light of the initial elevation of homocysteine in our patient, albeit follow-up studies including plasma homocysteine and methionine were normal.
The de novo p.S4118F variant identified in our patient has not been reported previously. It affects an amino acid in the C-terminus of the RNF213 gene, consistent with previous observations that pathogenic mutations affecting the RNF213 protein are clustered in this region [Kamada et al., 2011; Liu et al., 2011; Cecchi et al., 2014]. Ours is only the second report of a moyamoya patient with a documented de novo RNF213 variant; the first patient presented at 10 months of age and was found to have a nonframeshift 3 bp deletion (p.K4115del) [Cecchi et al., 2014] positioned three residues from the amino acid altered in our patient. Although further subjects are needed for genotype-phenotype correlations, this may be indicative of the importance of this specific highly conserved region of the protein. The extracranial vascular involvement in our patient in conjunction with the previously reported premature coronary artery disease [Cecchi et al., 2014] suggest that RNF213 may be associated with a more generalized vasculopathy than other moyamoya genes, and would advocate for possible inclusion of RNF213 in vascular disease gene panels as well as interrogation of the extracranial vasculature in patients with RNF213 mutations. Early identification of at-risk individuals is instrumental in prevention of stroke and co-morbidities, as various treatment options are available [Dauser et al, 2007, Jackson et al., 2014].
RNF213 encodes a 526-amino acid protein harboring a RING (really interesting new gene) finger motif and an AAA ATPase domain. It is presumed to function as an E3 ubiquitin ligase and is highly expressed in spleen and leukocytes, with low but significant expression in endothelial cells [Kamada et al., 2011]. It is still undetermined whether the p.R4810K allele and additional variants in RNF213 are loss of function mutations, as initially suggested by zebrafish morfants which exhibited aberrant angiogenesis in the head region [Liu et al., 2011]; or gain of function mutations. Rnf213 knockout mice did not develop an abnormal vascular network at the base of the brain, but did exhibit abnormal vascular remodeling after carotid artery ligation [Sonobe et al., 2014]. Several hypotheses have been generated from the animal studies, including that RNF213 deficiency could lead to vascular fragility with vulnerability of vessels to hemodynamic stress and secondary insults; proposal of a two-hit model where an additional insult such as an autoimmune response or infection/inflammation may be necessary for the development of moyamoya disease; or alternatively, that the mechanism involves a gain of function not yet studied in a model system [Fujimura et al., 2014]. The latter is supported by the observation that only missense variants and not frame-shift or nonsense mutations have been associated with moyamoya disease [Cecchi et al., 2014].
There is ongoing debate over whether Mendelian disorders and multifactorial traits present opposite ends of the disease spectrum in humans or a continuum, and new genomic technologies are allowing for renewed assessment of this question [Antonarakis et al., 2010]. Much of the speculation about missing heritability from genome-wide association studies (GWAS) has focused on the possible contribution of variants of low minor allele frequency (MAF) which may not be sufficiently frequent to be captured by GWAS nor carry sufficiently large effect sizes to be detected by classical linkage analysis in family studies [Manolio et al., 2009]. The RNF213 p.R4810K allele was first identified in a genome wide association study with a frequency of 1/72 (0.014) and an odds ratio of 190.8 in cases versus controls [Kamada et al., 2011], presenting a prime example of a variant of low minor allele frequency with a high effect on a common disease. This mirrors a recent publication on the TM4SF20 ancestral deletion and susceptibility to early language delay and cerebral white matter hyperintensities, where a population specific allele (MAF~1%) in the Vietnamese Kinh population was found to underlie an apparent common disease trait inherent to a distinct world population [Wiszniewski et al., 2013].
The wide phenotypic variability in terms of severity and age at presentation in moyamoya patients with the p.R4810K allele [Cecchi et al., 2014] further highlights the question of penetrance and the contribution of modifier genes or environmental factors. Another yet unanswered question is whether certain alleles of RNF213 lead to a Mendelian disease of early onset, whereas other alleles confer susceptibility to moyamoya disease. Such a paradigm is apparent for many neurodegenerative diseases (e.g., MAPT in autosomal dominant frontotemporal dementia, sporadic progressive supranuclear palsy, and late-onset Parkinson disease [Antonarakis et al., 2010; Martin et al., 2001]) and for disorders of lipid metabolism (e.g., LDLR variants in familial hypercholesterolemia and sporadic disease [Kathiresan et al., 2009; Varret et al., 2008]). This perhaps can be seen to complement another situation wherein a homozygous mutation gives rise to a monogenic Mendelian disorder while a heterozygous carrier state leads to susceptibility to a multifactorial disease, as evident for Gaucher disease and susceptibility to Parkinson disease (GBA [Sidransky et al., 2009]), cystic fibrosis and susceptibility to pancreatic insufficiency (CFTR [Cohn et al., 1998; Sharer et al., 1998], Stargardt disease and susceptibility to age-related macular degeneration (ABCA4 [Shroyer et al., 1999; Allikmets et al., 1997]), and others.
Genotype-phenotype correlation studies of RNF213 and moyamoya disease in the future will likely concentrate on modifiers of the p.R4810K allele at other loci as well as on additional variants identified in different ethnicities. Publication of novel alleles that lead to early and severe onset disease as reported here, in conjunction with functional studies, may be especially helpful in ultimately understanding the possible continuum of Mendelian and multifactorial inheritance in moyamoya disease.
Acknowledgments
T.H. and J.E.P. were supported by the Medical Genetics Research Fellowship Program NIH/NIGMS NIH T32 GM07526.
References
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, Dean M, Lupski JR, Leppert M. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- Antonarakis SE, Chakravarti A, Cohen JC, Hardy J. Mendelian disorders and multifactorial traits: the big divide or one for all? Nat Rev Genet. 2010;11:380–384. doi: 10.1038/nrg2793. [DOI] [PubMed] [Google Scholar]
- Baba T, Houkin K, Kuroda S. Novel epidemiological features of moyamoya disease. J Neurol Neurosurg Psychiatry. 2008;79:900–904. doi: 10.1136/jnnp.2007.130666. [DOI] [PubMed] [Google Scholar]
- Dauser RC, Tuite GF, McCluggage CW. Dural inversion procedure for moyamoya disease. Technical note. J Neurosurg. 1997;86:719–723. doi: 10.3171/jns.1997.86.4.0719. [DOI] [PubMed] [Google Scholar]
- Cecchi AC, Guo D, Ren Z, Flynn K, Santos-Cortez RL, Leal SM, Wang GT, Regalado ES, Steinberg GK, Shendure J, Bamshad MJ, Grotta JC, Nickerson DA, Pannu H, Milewicz DM University of Washington Center for Mendelian Genomics. RNF213 Rare Variants in an Ethnically Diverse Population With Moyamoya Disease. Stroke. 2014;45:3200–3207. doi: 10.1161/STROKEAHA.114.006244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–658. doi: 10.1056/NEJM199809033391002. [DOI] [PubMed] [Google Scholar]
- Currie S, Raghavan A, Batty R, Connolly DJ, Griffiths PD. Childhood moyamoya disease and moyamoya syndrome: a pictorial review. Pediatr Neurol. 2011;44:401–413. doi: 10.1016/j.pediatrneurol.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Sonobe S, Nishijima Y, Niizuma K, Sakata H, Kure S, Tominaga T. Genetics and Biomarkers of Moyamoya Disease: Significance of RNF213 as a Susceptibility Gene. J Stroke. 16:65–72. doi: 10.5853/jos.2014.16.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EM, Lin N, Manjila S, Scott RM, Smith ER. Pial synangiosis in patients with moyamoya younger than 2 years of age. J Neurosurg Pediatr. 2014;13:420–425. doi: 10.3171/2014.1.PEDS13251. [DOI] [PubMed] [Google Scholar]
- Kamada F, Aoki Y, Narisawa A, Abe Y, Komatsuzaki S, Kikuchi A, Kanno J, Niihori T, Ono M, Ishii N, Owada Y, Fujimara M, Mashimo Y, Suzuki Y, Hata A, Tsuchiya S, Tominaga T, Matsubara Y, Kure S. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56:34–40. doi: 10.1038/jhg.2010.132. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, Voight BF, Bonnycastle LL, Jackson AU, Crawford G, Surti A, Guiducci C, Burtt NP, Parish S, Clarke R, Zelenika D, Kubalanza KA, Morken MA, Scott LJ, Stringham HM, Galan P, Swift AJ, Kuusisto J, Bergman RN, Sundvall J, Laakso M, Ferrucci L, Scheet P, Sanna S, Uda M, Yang Q, Lunetta KL, Dupuis J, de Bakker PI, O’Donnell CJ, Chambers JC, Kooner JS, Hercberg S, Meneton P, Lakatta EG, Scuteri A, Schlessinger D, Tuomilehto J, Collins FS, Groop L, Altshuler D, Collins R, Lathrop GM, Melander O, Salomaa V, Peltonen L, Orho-Melander M, Ordovas JM, Boehnke M, Abecasis GR, Mohlke KL, Cupples LA. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Morito D, Takashima S, Mineharu Y, Kobayashi H, Hitomi T, Hashikata H, Matsuura N, Yamazaki S, Toyoda A, Kikuta K, Takagi Y, Harada KH, Fujiyama A, Herzig R, Krischek B, Zou L, Kim JE, Kitakaze M, Miyamoto S, Nagata K, Hashimoto N, Koizumi A. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One. 2011;6:e22542. doi: 10.1371/journal.pone.0022542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ER, Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons K, Pahwa R, Stern MB, Colcher A, Hiner BC, Jankovic J, Ondo WG, Allen FH, Jr, Goetz CG, Small GW, Masterman D, Mastaglia F, Laing NG, Stajich JM, Ribble RC, Booze MW, Rogala A, Hauser MA, Zhang F, Gibson RA, Middleton LT, Roses AD, Haines JL, Scott BL, Pericak-Vance MA, Vance JM. Association of single-nucleotide polymorphisms of the tau gene with late-onset Parkinson disease. JAMA. 2001;286:2245–2250. doi: 10.1001/jama.286.18.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake S, Miyake N, Touho H, Nishimura-Tadaki A, Kondo Y, Okada I, Tsurusaki Y, Doi H, Sakai H, Saitsu H, Shimojima K, Yamamoto T, Higurashi M, Kawahara N, Kawauchi H, Nagasaka K, Okamoto N, Mori T, Koyano S, Kuroiwa Y, Taguri M, Morita S, Matsubara Y, Kure S, Matsumoto N. Homozygous c.14576G>A variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology. 2012;78:803–810. doi: 10.1212/WNL.0b013e318249f71f. [DOI] [PubMed] [Google Scholar]
- Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360:1226–1237. doi: 10.1056/NEJMra0804622. [DOI] [PubMed] [Google Scholar]
- Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- Shroyer NF, Lewis RA, Allikmets R, Singh N, Dean M, Leppert M, Lupski JR. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: from monogenic to multifactorial. Vision Res. 1999;39:2537–2544. doi: 10.1016/s0042-6989(99)00037-1. [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Scott RM. Spontaneous occlusion of the circle of Willis in children: pediatric moyamoya summary with proposed evidence-based practice guidelines. A review. J Neurosurg Pediatr. 2012;9:353–360. doi: 10.3171/2011.12.PEDS1172. [DOI] [PubMed] [Google Scholar]
- Sonobe S, Fujimura M, Niizuma K, Nishijima Y, Ito A, Shimizu H, Kikuchi A, Arai-Ichinoi N, Kure S, Tominaga T. Temporal profile of the vascular anatomy evaluated by 9.4-T magnetic resonance angiography and histopathological analysis in mice lacking RNF213: a susceptibility gene for moyamoya disease. Brain Res 2014. 2014;1552:64–71. doi: 10.1016/j.brainres.2014.01.011. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Takaku A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20:288–299. doi: 10.1001/archneur.1969.00480090076012. [DOI] [PubMed] [Google Scholar]
- Varret M, Abifadel M, Rabes JP, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet. 2008;73:1–13. doi: 10.1111/j.1399-0004.2007.00915.x. [DOI] [PubMed] [Google Scholar]
- Wiszniewski W, Hunter JV, Hanchard NA, Willer JR, Shaw C, Tian Q, Illner A, Wang X, Cheung SW, Patel A, Campbell IM, Gelowani V, Hixson P, Ester AR, Azamian MS, Potocki L, Zapata G, Hernandez PP, Ramocki MB, Santos-Cortez RL, Wang G, York MK, Justice MJ, Chu ZD, Bader PI, Omo-Griffith L, Madduri NS, Scharer G, Crawford HP, Yanatatsaneejit P, Eifert A, Kerr J, Bacino CA, Franklin AI, Goin-Kochel RP, Simpson G, Immken L, Haque ME, Stosic M, Williams MD, Morgan TM, Pruthi S, Omary R, Boyadjiev SA, Win KK, Thida A, Hurles M, Hibberd ML, Khor CC, Van Vinh Chau N, Gallagher TE, Mutirangura A, Stankiewicz P, Beaudet AL, Maletic-Savatic M, Rosenfeld JA, Shaffer LG, Davis EE, Belmont JW, Dunstan S, Simmons CP, Bonnen PE, Leal SM, Katsanis N, Lupski JR, Lalani SR. TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am J Hum Genet. 2013;93:197–210. doi: 10.1016/j.ajhg.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]