

Abstract
Studying autosomal dominant Alzheimer’s disease (ADAD), caused by gene mutations yielding nearly complete penetrance and a distinct age of symptom onset, allows investigation of presymptomatic pathological processes that can identify a therapeutic window for disease-modifying therapies. Astrocyte activation may occur in presymptomatic Alzheimer’s disease (AD) because reactive astrocytes surround β-amyloid (Aβ) plaques in autopsy brain tissue. Positron emission tomography was performed to investigate fibrillar Aβ, astrocytosis and cerebral glucose metabolism with the radiotracers 11C-Pittsburgh compound-B (PIB), 11C-deuterium-L-deprenyl (DED) and 18F-fluorodeoxyglucose (FDG) respectively in presymptomatic and symptomatic ADAD participants (n = 21), patients with mild cognitive impairment (n = 11) and sporadic AD (n = 7). Multivariate analysis using the combined data from all radiotracers clearly separated the different groups along the first and second principal components according to increased PIB retention/decreased FDG uptake (component 1) and increased DED binding (component 2). Presymptomatic ADAD mutation carriers showed significantly higher PIB retention than non-carriers in all brain regions except the hippocampus. DED binding was highest in presymptomatic ADAD mutation carriers. This suggests that non-fibrillar Aβ or early stage plaque depostion might interact with inflammatory responses indicating astrocytosis as an early contributory driving force in AD pathology. The novelty of this finding will be investigated in longitudinal follow-up studies.
Alzheimer’s disease (AD) is a progressive brain disorder with gradually occurring cognitive decline. The time course of the underlying pathological changes remains largely veiled. Increasing evidence argues that these changes start decades before the onset of clinical symptoms. The order and magnitude of these processes are hitherto not well understood. The typical histopathology of AD includes the presence of β-amyloid (Aβ) plaques, neurofibrillary tangles, neuronal cell dysfunction and loss, and the activation of glial cells. It has been hypothesized that the accumulation of Aβ plaques plays a causative role in the disease development1. However, increasing evidence suggests that neurodegeneration might be triggered by a combination of processes, including tau deposition and neuroinflammation in addition to plaque accumulation2,3. The rapid development of cerebrospinal fluid (CSF) and positron emission tomography (PET) biomarkers has allowed modelling of the hypothetical sequence of biomarker changes in the presymptomatic, prodromal, and symptomatic stages of AD4,5.
AD is sporadic (sAD) in the great majority of cases, but 1–5% of Alzheimer patients suffer from the autosomal dominant form of the disease (ADAD), which is caused by mutations in the presenilin 1 (PSEN1), presenilin 2 (PSEN2), or amyloid precursor protein (APP) genes6. Within families harbouring a specific mutation, the age of onset for ADAD or early-onset familial AD (eoFAD) is predictable, which provides an opportunity for determining the sequence and magnitude of pathological changes that culminate in symptomatic disease7. Given the importance of understanding very early pathological changes in AD, members of families harbouring these mutations can be studied long before they develop any symptoms, providing an invaluable tool for research of these processes.
There are strong indications that factors other than Aβ, such as the activation of astroglia and microglia, and subsequent neuroinflammation, contribute to AD genesis and progression8,9,10. Most of our current understanding of astrocytosis stems from immunohistochemical studies in postmortem brain tissue11,12. Reactive astrocytes undergo structural and functional changes regulated by specific signalling events that occur in a context-dependent manner13. It has been observed that Aβ plaques are surrounded by activated astrocytes, and that activated astrocytes produce reactive oxygen and nitrogen species, which may contribute to AD pathogenesis. Nevertheless, much is still unknown regarding the relationship between reactive astrocytes and Aβ pathology14.
The PET tracer 11C-deuterium-L-deprenyl (DED) binds to monoamine oxidase B (MAO-B) on the outer mitochondrial membrane in astrocytes; increased DED binding is thought to reflect reactive astrocytosis15,16. In a 11C-DED PET study of sAD patients, we found evidence for early astrocytosis in 11C-PIB positive patients with mild cognitive impairment (MCI PIB+)17. DED binding was increased in MCI PIB+ patients compared to sAD patients and controls, suggesting that increased astrocytosis occurs in the earlier prodromal stages of AD17.
This cross-sectional study reports the baseline results from a large, ongoing, longitudinal study aiming to examine the temporal and regional relationships between astrocytosis, Aβ deposition, and glucose metabolism as a measure of neurodegeneration in ADAD and sAD. Here we demonstrate for the first time the presence of significant astrocytosis decades before the occurrence of clinical symptoms.
Results
Subjects
The demographic data for the subjects are presented in Table 1. There were significant differences in age, education, and mini-mental state examination (MMSE) scores between the groups. The presymptomatic mutation carriers in particular were considerably younger than members of the other groups. Differences in MMSE were anticipated due to the different clinical stages of the groups. The patients with MCI were subdivided into PIB positive (PIB+) and PIB negative (PIB−) subjects according to their global-to-cerebellum gray matter PIB retention ratios using a cut-off point of 1.41 derived from a larger multicenter study of PIB PET18.
Table 1. Demographic information.
|
Clinical patients |
ADAD individuals |
|||||
|---|---|---|---|---|---|---|
| sAD | MCI PIB+ | MCI PIB− | Mutationnon-carriers | Presymptomaticmutationcarriers | Symptomaticmutationcarriers | |
| n | 7 | 8 | 3 | 12 | 6 | 3 |
| Age (y) | 64.1 ± 6.1C,D (55–73) | 61.9 ± 6.8H (53–75) | 64.3 ± 6.7K (60–72) | 54.3 ± 12.9M | 43.5 ± 9.3 | 58.7 ± 5.0 |
| Gender (m/f) | 5/2 | 4/4 | 2/1 | 8/4 | * | 2/1 |
| MMSE | 24.4 ± 5.7C,D,E (14–30) | 27.7 ± 1.9 (24–30) | 27.7 ± 2.3 (25–29) | 29.3 ± 1.1 (27–30) | 29.6 ± 0.6 (29–30) | 16.0 ± 8.7 (11–26) |
| Education (y) | 10.4 ± 1.8A,B (8–13) | 14.0 ± 2.5G (11–18) | 13.7 ± 2.1 (12–16) | 11.3 ± 2.3 | 13.2 ± 2.6 | 10.0 ± 1.0 |
| Mutations | PSEN1 H163Y; APPswe; APParc | PSEN1 H163Y; APPswe; APParc | ||||
| Time to expected age at symptom onset (y) | −11.8 ± 8.1 | 1.9 ± 2.6 | ||||
| APOE ε | 3/3: 2; 3/4: 2; 4/4: 3 | 3/3: 2; 3/4: 6 | 3/3: 2; 3/4: 1 | 2/3: 1; 3/3: 7; 3/4: 3; 4/4: 1 | 3/3: 3; 3/4: 3 | 3/3: 1; 2/4: 1; 3/4: 1 |
| FSIQ global cognition composite z-score | −2.3 ± 1.7 | −0.9 ± 1.8 | −0.2 ± 1.4 | −0.2 ± 1.5 | 0.3 ± 1.3 | −3.3 ± 1.9 |
| Episodic memory composite z-score# | −2.3 ± 0.8 | −1.3 ± 0.7 | −0.9 ± 0.9 | −0.1 ± 0.5 | 0.0 ± 0.5 | −2.2 ± 1.1 |
| ** | sAD | MCI PIB+ | MCI PIB− | Non-carriers | Presymptomatic carriers | Symptomatic carriers |
| sAD | A | B | C | D | E | |
| MCI PIB+ | A | F | G | H | I | |
| MCI PIB− | B | F | J | K | L | |
| Non-carriers | C | G | J | M | N | |
| Presymptomatic carriers | D | H | K | M | O | |
| Symptomatic carriers | E | I | L | N | O | |
All values are means ± SD (range), unless stated otherwise. Superscript letters indicate significant differences between groups (see legend ** for group comparison codes, Fisher’s LSD post-hoc test, p < 0.05). *Gender distribution of presymptomatic autosomal dominant AD mutation carriers is not revealed to preserve confidentiality; #sum of Rey auditory verbal learning test, total learning and delayed retention test, and Rey Osterrieth retention test scores. sAD = sporadic Alzheimer’s disease; ADAD = autosomal dominant Alzheimer’s disease; APOE = apolipoprotein E; FSIQ = full-scale intelligence quotient; MCI = mild cognitive impairment; MMSE = mini-mental state examination; PIB = Pittsburgh compound-B. Z-scores below −1.645 were considered outside normal range (italics).
** Legend for pairwise comparisons using Fisher’s LSD post hoc test.
Neuropsychology
All raw test scores were transformed into z-scores (Table 1). Z-scores < −1.645 (fifth percentile) were considered outside the normal range. The sAD patients showed pathological z-scores in global cognition and episodic memory. Four of the eight MCI PIB+ patients had abnormal episodic memory compared to the population mean, and episodic memory had declined in the other four compared to previous assessments or they had abnormal values compared to their estimated premorbid function. One of the MCI PIB− patients showed abnormal episodic memory; the other two had abnormal performance in non-memory cognitive domains. The episodic memory z-scores were within the normal range for all non-carriers in ADAD families as well as for the six presymptomatic ADAD mutation carriers. Two of the three symptomatic ADAD carriers demonstrated results markedly outside the normal range in global cognition and episodic memory, while the third carrier had abnormal or close to abnormal results in two episodic memory tests. For detailed individual test scores, please refer to Table 2.
Table 2. Detailed neuropsychological test results.
|
Clinical patients |
ADAD individuals |
|||||
|---|---|---|---|---|---|---|
| sAD | MCI PIB+ | MCI PIB− | Mutationnon-carriers | Presymptomaticmutation carriers | Symptomaticmutation carriers | |
| WAIS-R Information | −2.00 (2.23) | −0.84 (0.60) | −0.89 (0.90) | −0.40 (1.03) | 0.18 (0.53) | −0.75 (1.85) |
| WAIS-R Similarities | −1.52 (1.54) | −0.64 (1.18) | 0.67 (0.79) | 0.08 (1.11) | −0.15 (0.79) | −0.94 (0.94) |
| WAIS-R Block Design | −2.23 (1.33) | −0.85 (1.17) | 0.23 (1.75) | 0.43 (1.14) | 0.73 (0.81) | −1.16 (1.42) |
| WAIS-R Digit Symbol | −1.17 (1.17) | −0.75 (1.07) | −0.19 (1.44) | 0.20 (0.51) | −0.03 (0.36) | −1.56 (1.49) |
| Rey-Osterrieth copy | −2.16 (3.79) | −0.40 (0.54) | −0.30 (0.88) | −0.15 (0.46) | −0.28 (0.35) | −3.55 (4.95) |
| Rey-Osterrieth recall | −2.43 (0.84) | −1.37 (0.71) | −0.74 (1.16) | −0.59 (0.85) | −0.22 (0.53) | −1.61 (2.39) |
| RAVLT trial 1–5 total | −1.89 (0.90) | −1.30 (0.90) | 0.62 (0.72) | −0.61 (0.67) | −0.50 (0.74) | −2.13 (0.90) |
| RAVLT delayed recall | −1.88 (1.54) | −1.74 (0.60) | −1.38 (0.66) | −0.38 (0.83) | −1.25 (1.07) | −1.99 (1.01) |
| Digit Span forward UD | −1.23 (0.73) | −0.75 (0.83) | −0.90 (1.29) | 0.20 (1.07) | −0.18 (1.66) | −1.02 (1.05) |
| Corsi Span UD | −2.20 (1.60) | −1.71 (1.70) | −0.49 (2.93) | 0.24 (1.20) | −0.93 (1.38) | −1.90 (2.70) |
| Trail Making A time (s) | −3.01 (6.37) | −1.14 (1.61) | −0.62 (0.65) | 0.85 (0.60) | 0.78 (0.47) | 0.27 (0.08) |
| Trail Making B time (s) | −1.69 (1.66) | −0.80 (1.36) | −0.44 (1.01) | 0.38 (0.42) | 0.09 (0.36) | −2.22 (2.92) |
All values are means (SD) of z-scores, z-scores below −1.645 were considered outside normal range (italics). WAIS-R: Wechsler Adult Intelligence Scale–Revised; RAVLT: Rey Auditory Verbal Learning Test.
Principal component analysis modeling and model quality
The principal component analysis PCA model accounted for 67% of the variance of the original data (R2(X)), and its cross-validated predictability, Q2(X), was 0.61 (considered a valid model)19. Figure 1a shows a scatter plot with the distribution of all participants’ data along two components. Figure 1b displays a simplified plot showing the means and standard deviations for each group. Supplementary Fig. S1 shows the influence of the 25 most important variables on each component. As demonstrated in Fig. 1a, the separation of the groups along component 1 shows a clear division between symptomatic ADAD mutation carriers, sAD patients, and MCI PIB+ patients on the one side and presymptomatic ADAD mutation carriers, MCI PIB− patients, and ADAD mutation non-carriers on the other. According to the loading plot, PIB retention and FDG uptake on the respective sides accounted for this separation (Fig. 1b; for detailed regional information, please see Supplementary Fig. S1). The order of rankings along the first component indicated highest PIB retention and lowest FDG metabolism for symptomatic ADAD mutation carriers, followed in order by sAD patients, MCI PIB+ patients, presymptomatic ADAD carriers, MCI PIB− patients, and ADAD non-carriers.
Figure 1.

(a) Scatter plot displaying results from all examined individuals according to Principal Component Analysis (PCA). Distribution along the first two components is shown. (b) The figure displays a simplified summary of the PCA data for each group; the central shapes represent the mean PCA score for each group, bars represent standard deviations for each group on each principal component.
DED slope values were most important for separation along component 2. Here, although the pattern was not as clear as for component 1, the order of rankings suggested highest slope values for the presymptomatic ADAD mutation carriers, followed by MCI PIB+ patients, symptomatic ADAD carriers, sAD patients, ADAD non-carriers, and MCI PIB− patients (for detailed regional information, please see Supplementary Fig. S1).
11C-Pittsburgh compound-B PET region of interest analysis
PIB retention differed significantly between the groups (Fig. 2, Supplementary Table S1a); Kruskal-Wallis tests in each of 11 ROIs for the four groups being compared (presymptomatic ADAD carriers, MCI PIB+, sAD and ADAD non-carriers), were significant (p < 0.001) in all ROIs except for the hippocampus. Highest PIB retention values were seen in the symptomatic mutation carriers in all brain regions, with particularly increased retention in the putamen (z-score = 17.3) and in the hippocampus (z-score = 9.7) of one carrier.
Figure 2.
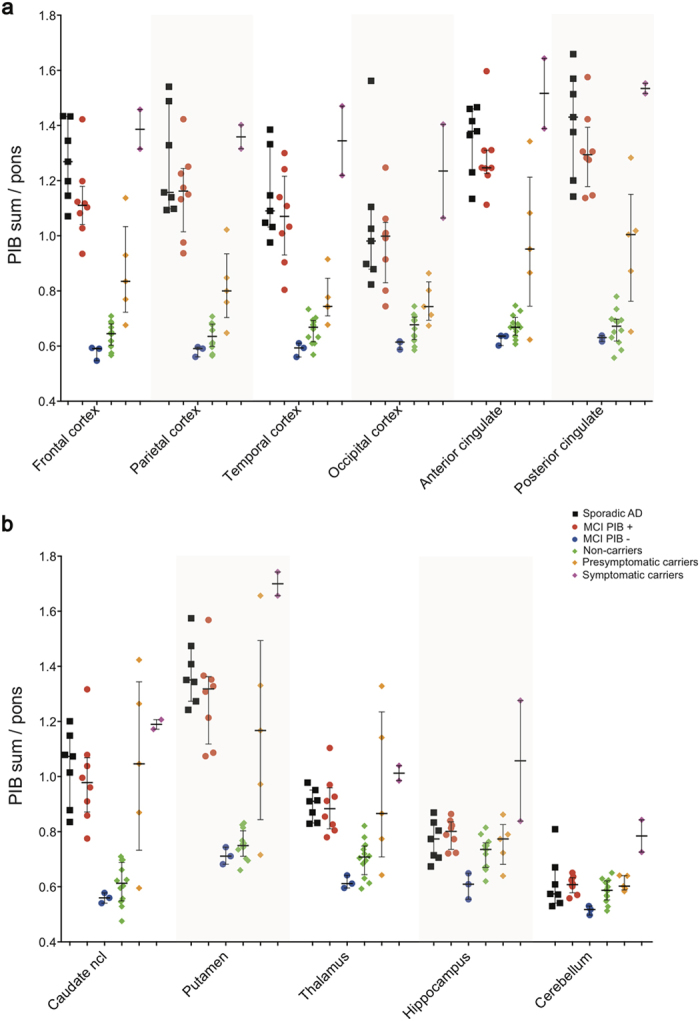
Scatter plots showing all individual PIB retention data in composite cortical (a) and subcortical (b) bilateral brain regions.
Following the highest PIB retention values observed in individual symptomatic carriers, the ranked order for PIB retention derived from Mann-Whitney pair-wise comparisons between groups and the z-scores for MCI PIB− patients was: sAD patients ≥ MCI PIB+ patients > presymptomatic ADAD mutation carriers > ADAD non-carriers ≥ MCI PIB−; this pattern is consistent with the results obtained from the PCA analysis. The ADAD non-carriers and the MCI PIB− subjects had persistently very low PIB retention scores in all the examined regions. Compared to ADAD non-carriers, the MCI PIB− patients had significantly lower PIB retention in the hippocampus (z-score = −2.1).
The sAD and MCI PIB+ patients had increased PIB retention compared to ADAD non-carriers with effect sizes r > 0.80 (p < 0.001) in all ROIs, except the hippocampus which did not show a significant difference. Both sAD and MCI PIB+ groups showed significantly increased PIB retention compared to presymptomatic ADAD mutation carriers in all cortical regions, most pronounced in parieto-temporal cortex (effect sizes r ~ 0.60–0.70, p < 0.05), but differences were not significant in subcortical regions. Presymptomatic mutation carriers had higher PIB than ADAD non-carriers in all ROIs (effect sizes r ~ 0.5–0.7, p < 0.05), except for the hippocampus.
11C-deuterium-L-deprenyl PET region of interest analysis
Comparisons of four groups (presymptomatic ADAD carriers, MCI PIB+, sAD and ADAD non-carriers) using Kruskal-Wallis tests in each of 11 ROIs (except for the cerebellum), showed a trend toward statistical significance for the anterior cingulate cortex, the thalamus and the hippocampus (p = 0.059–0.069) (Supplementary Table S1b). The highest DED slope values were seen in the presymptomatic ADAD mutation carriers on average. The largest differences among groups were found between presymptomatic ADAD-carriers and sAD, where Mann-Whitney comparisons were significant in temporal, anterior/posterior cingulate, thalamus and hippocampus (r ~ 0.6–0.7, p < 0.05) (Fig. 3). The ADAD non-carrier group showed high variance, whereof two individuals (31 and 43 in Fig. 1a) showed very high DED slope values in most brain regions, which accounts for the rather high mean values in the non-carriers. Subject 43 showed particularly high values, especially in the hippocampus and the parahippocampal gyrus (not shown). sAD had lower DED than MCI PIB+ in the hippocampus (r = 0.51, p < 0.05), consistent with the results from the PCA analysis. The Mann-Whitney comparisons between sAD and ADAD non-carriers revealed significantly lower DED binding in sAD in anterior cingulate, caudate and hippocampus (r =0.48-0.56, p < 0.05).
Figure 3.
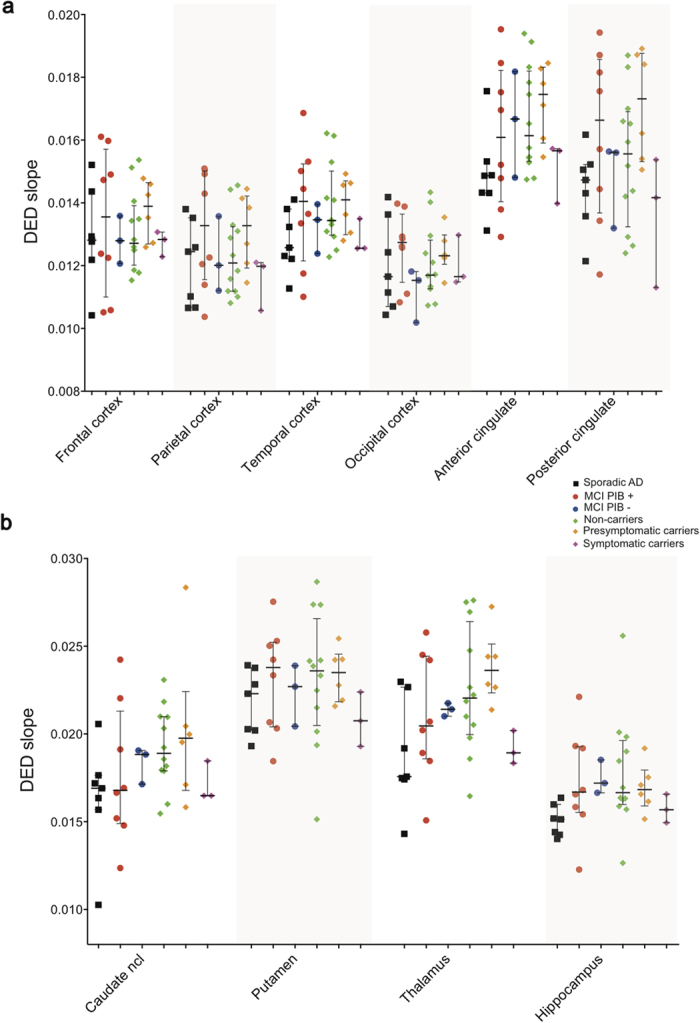
Scatter plots showing all individual DED slope data in composite cortical (a) and subcortical (b) bilateral brain regions. No data are shown for the cerebellum since this was used as the reference region in the modified Patlak reference tissue model.
18F-fluorodeoxyglucose PET region of interest analysis
The pattern of cerebral glucose metabolism as measured by FDG uptake was significantly different between the groups (Fig. 4, Supplementary Table S1c); the Kruskal-Wallis tests between the four groups being compared (presymptomatic ADAD carriers, MCI PIB+, sAD and ADAD non-carriers) showed significant results in all ROIs, most pronounced in parieto-temporal, posterior cingulate, and thalamus (p < 0.001). FDG uptake was lowest in individual symptomatic mutation carriers (most pronounced in parieto-temporal and posterior cingulate with z-scores between −3.2 and −4.2), followed by sAD and MCI PIB+ patients in almost all examined brain regions (Fig. 4). There were no significant differences in FDG uptake between sAD and MCI PIB+ patients. As expected, the highest FDG uptake occurred in the non-carriers, which was significantly higher (p < 0.05) than in sAD and in MCI PIB+ groups in all ROIs. The group of presymptomatic carriers showed to be statistically comparable to MCI PIB+ patients in anterior and posterior cingulate cortices as well as in the occipital region, indicating incipient hypometabolism in presymptomatic carriers. The pattern of glucose metabolism across groups was consistent with results from the PCA analysis.
Figure 4.
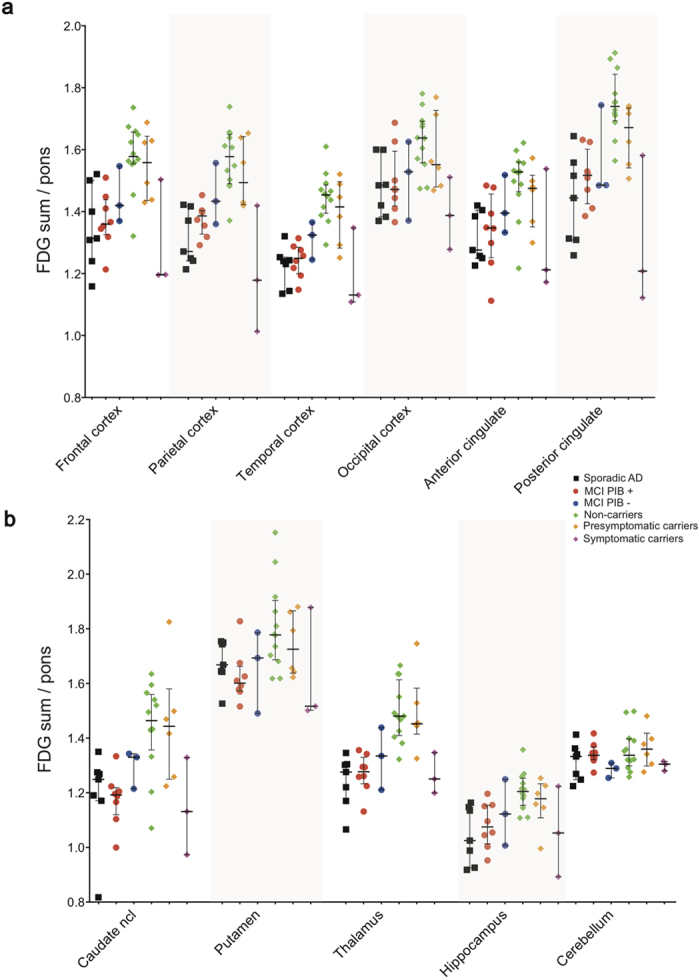
Scatter plots showing all individual FDG uptake data in composite cortical (a) and subcortical (b) bilateral brain regions.
Statistical parametric mapping analysis
The statistical parametric mapping (SPM) results generally demonstrated ongoing progressive pathology in ADAD mutation carriers as the expected age at symptom onset approached (Fig. 5), consistent with the observed regional PET retention values in individual presymptomatic mutation carriers (Supplementary Table S2). The data demonstrated that PIB retention increases, DED binding decreases, and FDG uptake decreases close to and after the expected age of symptom onset. Increased DED binding was detectable at the earliest measured time point, nearly three decades before expected symptom onset. The pattern of changes for each PET tracer was not homogeneous for each individual.
Figure 5. Progression of PET biomarkers in ADAD mutation carriers.

Statistical parametric mapping (SPM) results are displayed for ADAD mutation carriers (n = 7). Each pair of columns represents an individual mutation carrier compared to five age-matched non-carriers. Each pair of rows represents a different PET biomarker. The scale from left to right represents the approximate time (in years) to the expected onset of clinical symptoms. All SPM clusters shown are significant at p < 0.001 (uncorrected). The figure demonstrates the progressive and heterogeneous nature of Alzheimer’s disease pathology as the expected onset of clinical symptoms approaches; neocortical fibrillar Aβ increases (PIB retention, top two rows), astrocytosis decreases (DED binding, middle two rows), glucose metabolism decreases (FDG hypometabolism shown, bottom two rows).
Discussion
The rapid development of molecular imaging has provided powerful tools for the detection of AD pathology at an early stage of the disease. These tools, in combination with the currently available biomarkers, provide an unprecedented opportunity to define a therapeutic window for use in the development of potential preventive/disease-modifying therapies. For example, high retention of specific Aβ PET tracers such as Pittsburgh compound-B in patients with mild cognitive impairment seems to predict a high risk of developing Alzheimer’s disease18,20, at a stage of the disease when cerebral glucose metabolism is less impaired. The new research diagnostic criteria for Alzheimer’s disease suggested by the International Working Group and the US National Institute on Aging–Alzheimer’s Association take into account the recent improved accessibility of CSF and imaging biomarkers21,22,23,24. Amyloid PET has been defined as a diagnostic marker that reflects in vivo pathology, while cerebral glucose hypometabolism is a downstream marker that monitors the course of neurodegeneration23. Studies in presymptomatic AD patients are important for further insight into the time course of these pathophysiological processes.
The amyloid cascade hypothesis, the focus of AD research for decades1, has received increasing scrutiny due to the recent unsuccessful treatment trials based on this line of thought25, and several other mechanisms including neuroinflammation have been suggested as promising alternative therapeutic targets26. Astrocyte activation has received increased attention due to its role in neuroinflammation8,27,28. However, astrocytosis is not completely understood; reactive astrocytes appear to have either neuroprotective or neurodegenerative effects at different stages of the evolution of the disease8,29.
The chronological order and relative causality of astrocyte activation, fibrillar Aβ deposition and glucose hypometabolism, have not been established, yet. Interestingly, it has been shown in cultured rat astrocytes that Aβ induces MAO-B expression30, which indicates a link between astrocyte activation and amyloid pathology. Astrocyte activation is a complex, regionally and temporally dependent phenomenon31, and MAO-B expression in reactive astrocytes differs across brain regions16. In addition, different markers of astrocytosis could detect different subpopulations of astrocytes and/or different stages in the disease. Reactive astrocytes have been measured in postmortem AD brain tissue using 3H-DED autoradiography and glial fibrillary acidic protein (GFAP) immunohistochemistry31,32,33. A strong regional correlation was observed between postmortem GFAP reactive astrocytes and both in vivo11C-PIB and in vitro3H-PIB binding. However, no correlation was found between postmortem3H-DED and in vivo11C-PIB33. There was no correlation either between 3H-DED reactive astrocytes and fibrillar Aβ in AD autopsy brains, using regional and laminar distribution analyses31. Evidence that neurodegenerative processes might not be solely dependent on Aβ plaque pathology is also based on in vivo PET imaging findings such as a lack of correlation between plaque deposition, metabolic dysfunction, atrophy, and clinical outcome (e.g. cognitive dysfunction) in AD patients34.
While there are several in vitro studies on reactive glia, less is known about their function in vivo. PET imaging of astrocytosis in mild cognitive impairment and sAD patients using 11C-DED has demonstrated elevated astrocytosis in MCI PIB+ patients (prodromal AD), suggesting that astrocytosis is an early event in sAD patients17. We have also recently reported that increased 11C-DED binding correlated with decreased gray matter density in the parahippocampus of MCI PIB+ patients35. However, no in vivo studies using 11C-DED in ADAD have been reported to date. Moreover, in vivo studies on astrocytosis are required to investigate the different roles of the reactive astrocytes across different disease stages, especially with regard to neuroprotection vs. neurotoxicity. Furthermore, microglia and astrocytes show differential relationships to Aβ pathology and seem to play different roles in inflammation in AD14,36. In vivo PET studies using tracers for the translocator protein (TSPO) as a marker for microglial activation, showed discordant results with both increased tracer uptake in AD patients as compared to healthy controls and no difference between patients and controls37,38,39,40. This discrepancy can partly be explained by a polymorphism of TSPO41. The time course and interrelationship of microglial and astrocytic involvement in AD-related inflammation are yet to be elucidated.
In this study, the finding of early astrocytosis roughly coinciding temporally but not necessarily spatially with fibrillar Aβ deposition is consistent with evidence from postmortem and in vivo studies suggesting a general lack of correlation between reactive astrocytes and fibrillar Aβ. The observed early astrocytosis might have been caused by Aβ oligomers or other pathological features such as intraneuronal hyperphosphorylated tau protein and adds support to the idea of glial activation as independent of other pathological substrates in AD.
The finding of early astrocytosis in the AD continuum is strengthened by recent findings of YKL-40, a potential astrocyte-derived biomarker measured in CSF, being strongly related to Aβ in AD patients and with tau pathology as a marker for neurodegeneration, most pronounced at early pre-dementia stages of AD42,43,44,45. Increased levels of CSF YKL-40 were furthermore related to cognitive decline and cortical thinning43,46.
We also compared cases of ADAD and sAD in this study, since a common neuropathological pathway has been suggested47, although the onset of symptoms occurs earlier and disease severity is greater in familial cases48. In recent cross-sectional49 and longitudinal50 ADAD studies, PIB, FDG, and atrophy were evaluated and compared in order to investigate regional and temporal differences in biomarker levels, in relation to the estimated years to symptom onset (EYO). PIB retention was increased in almost every cortical region, starting earlier than 15 EYO, in ADAD patients. Reduced FDG and cortical thinning were detected especially in the parietal and posterior cingulate cortex (PCC)/precuneus regions from 10 to 5 EYO. In our study in ADAD, MCI patients and sAD patients, we included assessment of 11C-DED binding in addition to PIB and FDG examinations. The PCA model showed a clear separation pattern between the different groups using the three different PET tracers in 24 brain regions allowed. Interestingly, the direction of the measured signal for the PET biomarkers PIB and FDG (which indicated worsening of amyloid deposition and glucose hypometabolism with time) differed from that for DED (which indicated decreased astrocytosis). This observation clearly reveals an opposite direction in time course between PIB and DED although these PET tracers show similar early presymptomatic presence but change differently in disease progression.
The highest level of DED binding was observed in presymptomatic ADAD mutation carriers, while DED binding was low in symptomatic ADAD mutation carriers. Interestingly, earlier studies in healthy subjects have reported an age-related increase in MAO-B activity in healthy human brains, as investigated in vitro at autopsy16 and in vivo using PET DED imaging51.
The increase in PIB retention occurred early in presymptomatic ADAD carriers, predominantly in the anterior and posterior cinguli and the basal ganglia. This pattern of early changes in the subcortical brain regions is in agreement with recent ADAD studies50, where high PIB retention was detected in regions such as the caudate nucleus and the pallidum, in the absence of atrophy. An early hypermetabolic phase 25 years before estimated symptom onset was detected in the precuneus and PCC, based on a linear model of FDG vs. EYO50. Furthermore, previous studies in ADAD patients have shown thalamic hypometabolism 20 years before estimated symptom onset7 and atrophy on average 6 years before symptom onset52 in asymptomatic PSEN1 mutation carriers, suggesting that the thalamus may also be involved in early disease mechanisms in ADAD patients. Subtle cognitive dysfunction in ADAD patients has been detected relatively close to the onset of symptoms (from about five years EYO)53. However, our study observed that performance can be quite variable in subjects from five years before symptom onset, with some subjects showing greater impairment and others remaining cognitively normal.
To conclude, we have demonstrated that measures of astrocytosis in ADAD mutation carriers can be observed decades before symptom onset, possibly coinciding with early fibrillar Aβ plaque deposition, both of which are followed later on by impaired glucose metabolism. Multivariate analysis of the PET results clearly separated the subject groups in a different order for the PIB/FDG tracers compared to the DED tracer. The analysis also suggested that DED might not yield equally high levels of sensitivity or specificity as PIB/FDG. It appears, however, that astrocytosis is initiated very early on, possibly before or at a similarly early stage as fibrillar Aβ deposition; this supports the notion that non-fibrillar forms of amyloid might cause inflammatory responses but also supports the possibility that astrocytosis is an important early contributory driving force in AD pathology. The novelty and importance of these findings will be confirmed in ongoing longitudinal studies.
Methods
Participants
Forty-four participants were recruited from the Department of Geriatric Medicine, Karolinska University Hospital Huddinge, Stockholm, Sweden; four were excluded for missing data and one PSEN1 carrier was excluded for incomplete penetrance of the mutation. Eleven MCI patients, seven sAD patients, and 21 ADAD family members were eligible for inclusion (demographic details are provided in Table 1).
The ADAD families are part of an ongoing longitudinal clinical study at the Karolinska Institute and were recruited without reference to their mutation status. The family members are regularly followed up clinically and examined using neuropsychological assessment, MRI, collection of CSF, and blood tests54. In this study, we included mutation carriers and non-carriers from families harbouring the Arctic APP (APParc), Swedish APP (APPswe), and presenilin 1 (PSEN1) (p.H163Y) mutations. Among the 21 family members, nine were carrying an ADAD mutation: two carried the APParc, two the APPswe, and five the PSEN1 mutations. The presence of the mutations in the subjects was confirmed by sequencing.
All participants underwent complete clinical examination, DED-, PIB-, and FDG-PET, MRI, and neuropsychological testing. The PIB PET data from the two APParc mutation carriers were not included in the analyses because, according to our previously published results, symptomatic carriers of this mutation do not show PIB PET retention, while exhibiting other clinical and biomarker features comparable with other mutation carriers55. The sAD subjects and one mutation carrier fulfilled the criteria for AD as outlined by the NINCDS-ADRDA56. The MCI patients and one symptomatic mutation carrier fulfilled the criteria for MCI as outlined by Petersen57. All subjects provided written informed consent to participate in the study, which was conducted according to the Declaration of Helsinki and subsequent revisions and was approved by the Regional Human Ethics Committee of Stockholm and the Isotope Committee of Uppsala University, Sweden.
Neuropsychological evaluation
All participants underwent routine clinical neuropsychological testing, which involved tests for global cognitive function, language, visuospatial function, episodic memory, attention, and executive ability, typically within six months of the PET examinations. Test scores were converted into z-scores in comparison with a reference group of healthy elderly people from the Karolinska University Hospital Huddinge58, while controlling for demographic conditions. Table 1 presents the composite z-scores for global cognition (full-scale intelligence quotient; FSIQ) and episodic memory performance (average of three scores: Rey auditory verbal learning test, total learning and delayed retention test, and Rey Osterrieth retention test), while Table 2 provides detailed individual test results.
PET image acquisition and processing
The PET investigations were performed at Uppsala PET center on ECAT EXACT HR+ (Siemens/CTI) and GE discovery ST PET/CT (GE Healthcare) scanners. The PET scans for all three radiotracers were commonly performed in the order PIB, DED, and FDG in each subject on the same day with 2–3 hours between tracer injections. For a few subjects, and due to tracer synthesis failures, two scans were within four weeks of the others. The orbito-meatal line was used to center the heads of the participants. The emission scans for the DED investigation consisted of 19 time frames (4 × 30 s, 8 × 60 s, 4 × 300 s and 3 × 600 s) with a total duration of 60 min; the emission scans for the PIB investigations consisted of 24 frames (4 × 30, 9 × 60, 3 × 180 and 8 × 300 s) over 60 min. A late 40–60 min PIB sum image was created and used for subsequent image analysis. For each FDG emission scan, seven frames (1 × 60 s, 1 × 1140 s, 5 × 300 s) were acquired over 45 min. A late 30–45 min FDG sum image was created and used for subsequent analysis. Patients were required to fast for 4 h preceding the FDG scan, which was performed in a quiet room with dimmed light and eyes closed. The mean injected doses for each tracer were DED: 209 ± 57 MBq, PIB: 217 ± 74 MBq, and FDG: 232 ± 45 MBq. All emission data were acquired in 3D mode and reconstructed with filtered back-projection using a 4 mm Hanning filter, resulting in a transaxial spatial resolution of 5 mm in the field of view. The matrix included 128 × 128 pixels, and a 2.5 zoom factor was used. The reconstructed frames were re-aligned to correct for patient motion during each PET scan. The PET protocol followed the protocol established in our previous publication17.
MRI image acquisition
All patients and ADAD family members underwent structural T1 MPRAGE MRI scanning using a 3T (Siemens Trio) scanner at the Karolinska University Hospital Huddinge, Stockholm, on average within five months of the PET examinations.
Region of interest PET image analysis
All images were processed and analysed using a probabilistic atlas approach, as described previously17. In short, all image analyses were performed in the space of the DED PET images to preserve the fidelity of these PET data. First, all DED data from 10–60 min for each participant was summed to create a DED PET sum image in native space. The individual T1 MR images were co-registered and re-sliced to their corresponding DED sum image (using SPM8; Functional Imaging Laboratory, Wellcome Department of Imaging Neuroscience, University College London). This step created a T1-weighted MR reference image for each participant in DED PET space. Subsequently, each patient’s PIB and FDG images were co-registered and re-sliced to their individual T1 MR reference image.
All T1 MR reference images were then segmented into gray and white matter tissue classes using SPM8 59. The resultant probabilistic gray matter map was thresholded at 0.5 to create a binary gray matter mask. An inverse non-linear transform parameter file was generated as part of the segmentation algorithm. The inverse parameter file allowed data in the MNI (Montreal Neurological Institute) space to be transformed back into a native DED PET image space. The inverse parameter file from each participant was used to transform a simplified digital probabilistic atlas60, consisting of 24 cortical and subcortical regions, into native DED PET space. These atlases were multiplied by the corresponding binary gray matter mask, which generated a specific gray matter digital atlas for each participant. This step resulted in a single digital atlas for each participant that could be easily applied to analyse all three sets of PET data without additional manipulation.
Raw co-registered and re-sliced FDG (Bq/ml) and PIB (Bq/ml) PET data for each patient were sampled using the same individual digital atlases. Using this method, mean FDG uptake and PIB retention values were measured for each atlas region, as described previously17. Regional gray matter ratio values were created for FDG and PIB by dividing by the respective mean uptake in the pons.
11C-deuterium-L-deprenyl PET data modelling
The PET data for each participant were analysed using the individual brain atlases generated in the steps described above. Regional parametric data were generated from dynamic DED data from 20 to 60 minutes. No arterial blood samples were available as an input function for the DED modelling because of the clinical character of the study. Instead, a modified Patlak reference tissue model was used for kinetic analysis, according to earlier study methods17,61. Cerebellar gray matter from the individual atlases was used as the reference region in this model. Because net tracer accumulation also occurred in the cerebellum, the model was modified by correcting k3 in the reference region for irreversible binding with a fixed correction factor of 0.01, which was the minimum value for correction still leading to linearization in the model. This graphical reference Patlak model resulted in two measurements: the intercept (initial tracer distribution volume) and the slope (kI = net DED binding to MAO-B). Since our main interest was to evaluate DED binding we used the slope value in subsequent analyses.
Principal component analysis
Principal component analysis (PCA)62 is a multivariate method implemented in the software package SIMCA-P+; Umetrics AB, Umea, Sweden19. It is an unsupervised method meaning it does not use a priori information about groups for the analysis. Statistically, PCA reduces the dimensionality and complexity of the data by finding lines and planes in the n-dimensional space (n = number of variables in the model) that approximates the data in the best way possible in the least squares sense. This gives us the opportunity to get an overview of the data to observe group belonging, trends and outliers. It is also possible to view relationships between the observations and the variables. One of the advantages of multivariate methods like PCA is that it can handle many more variables than observations19.
The PCA model was created to include all individuals: sAD, MCI PIB+ patients, MCI PIB− patients, ADAD mutation non-carriers, presymptomatic ADAD mutation carriers, and symptomatic ADAD mutation carriers. In total 72 variables (24 regions for PIB retention, FDG uptake, and DED binding slope) were included for each subject in the PCA analysis. The pre-processing steps mean-centering and unit variance scaling were performed. Mean-centering improves the interpretability of the data by subtracting the variable average, which repositions the data set around the origin. Large variance variables are more likely to be expressed in modelling than low variance variables. Consequently, unit variance scaling was selected to scale the data appropriately. This scaling method calculates the standard deviation of each variable. The inverse standard deviation is used as a scaling weight for each PET measurement. The results from the PCA were visualized by plotting the first two components in a scatter plot. Each point in the scatter plot represents one individual subject. Loading plots were also created to illustrate how the PET variables included in the model influenced the observed pattern in the scatter plot along each component (only the 25 most important variables are shown).
Region of interest statistical analyses
For each PET tracer, regional mean uptake values were compared between groups using two-tailed non-parametric Kruskal-Wallis tests, followed by post-hoc Mann-Whitney U tests in SPSS software. Non-parametric tests were applied due to the small sample sizes. The groups with n ≥ 5 being compared were: presymptomatic ADAD mutation carriers, MCI PIB+, sporadic AD (sAD) and ADAD mutation non-carriers. The comparisons were performed for 11 bilateral ROIs: frontal, parietal, temporal, occipital, anterior and posterior cingulate cortices, caudate nucleus, putamen, thalamus, hippocampus and cerebellum. Significance level was set at p < 0.05. Size effects (r) of Mann-Whitney comparisons were calculated using r = z/(√N), where z is the Mann-Whitney z and N the sum of individuals from the two groups being compared. Due to the small sample sizes of the MCI PIB− and symptomatic ADAD mutation carrier groups, individual z-score values were obtained instead with reference to the ADAD non-carrier group; z-score values were considered abnormal at |z| > 1.645.
Statistical parametric modeling analysis
The PET data (PIB, FDG, DED) from each participant, which had been realigned to the DED image in native space, were spatially normalized using non-linear transformation from the segmentation of the T1 MRI data. This resulted in PIB and FDG ratio (/pons) images and DED slope (binding; min−1) images in standard MNI space for each participant. After spatial normalization, each individual PET image from each ADAD mutation carrier was compared separately, using a two-sample t-test (SPM), with images from a group of five non-carriers (controls) who were the most proximal in age to the mutation carrier investigated. An explicit binary gray matter mask was used so that only voxels within the mask were compared in the SPM analysis. All SPM results for each tracer were analysed at a p value of <0.001 (uncorrected for multiple comparisons).
Additional Information
How to cite this article: Schöll, M. et al. Early astrocytosis in autosomal dominant Alzheimer's disease measured in vivo by multi-tracer positron emission tomography. Sci. Rep. 5, 16404; doi: 10.1038/srep16404 (2015).
Supplementary Material
Acknowledgments
We would like to express our gratitude to all participants who have made this study possible. Dr. Anne Kinhult Ståhlbom is acknowledged for professional help and Mr. Johan Lilja is acknowledged for support related to the imaging software VOIager. Funding: This work was supported by grants from the Knut and Alice Wallenberg foundation, GE Healthcare (unrestricted grant), the Swedish Research Council (projects 05817, 521-2010-3134), the Regional Agreement on Medical Training and Clinical Research (ALF) between Stockholm County Council and Karolinska Institutet, the Strategic Research Program in Neuroscience at Karolinska Institutet, Karolinska Institutet’s Doctoral Funding, the Swedish Foundation for Strategic Research (SFF), the Swedish Brain Power network, the Old Servants foundation, Gun and Bertil Stohne’s foundation, the Alzheimer Foundation in Sweden, the King Gustaf V and Queen Victoria’s Foundation of Freemasons, and The Brain Foundation.
Footnotes
Author Contributions A.N., M.S., B.L. and C.G. designed and planned, and A.N. coordinated the study. S.T., C.G. and A.N. conducted clinical investigations. A.W. performed the PET scans. O.A. distributed neuropsychological testing. M.S., S.C., E.W., A.W. and E.R.-V. collected and analysed all data. First manuscript was drafted by M.S. and A.N., M.S., S.C., E.W., E.R.-V., O.A., S.T., A.W., C.G., B.L. and A.N. revised the manuscript.
References
- Hardy J. A. & Higgins G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992). [DOI] [PubMed] [Google Scholar]
- Nordberg A. Molecular imaging in Alzheimer’s disease: new perspectives on biomarkers for early diagnosis and drug development. Alzheimers Res Ther 3, 34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G. Alzheimer disease: Abeta-independent processes-rethinking preclinical AD. Nat Rev Neurol 9, 123–124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R. Jr. et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R. Jr. et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekris L. M., Yu C. E., Bird T. D. & Tsuang D. W. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23, 213–227 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöll M. et al. Glucose metabolism and PIB binding in carriers of a His163Tyr presenilin 1 mutation. Neurobiol Aging 32, 1388–1399 (2011). [DOI] [PubMed] [Google Scholar]
- Verkhratsky A., Olabarria M., Noristani H. N., Yeh C. Y. & Rodriguez J. J. Astrocytes in Alzheimer’s disease. Neurotherapeutics 7, 399–412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner F. L., Ransohoff R. M. & Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16, 358–372 (2015). [DOI] [PubMed] [Google Scholar]
- Heneka M. T., Golenbock D. T. & Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol 16, 229–236 (2015). [DOI] [PubMed] [Google Scholar]
- McGeer P. L. & McGeer E. G. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 21, 195–218 (1995). [DOI] [PubMed] [Google Scholar]
- McGeer E. G. & McGeer P. L. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis 19, 355–361 (2010). [DOI] [PubMed] [Google Scholar]
- Burda J. E. & Sofroniew M. V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A. et al. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropath Exp Neur 72, 462–471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler J. S., Logan J., Volkow N. D. & Wang G. J. Translational neuroimaging: positron emission tomography studies of monoamine oxidase. Mol Imaging Biol 7, 377–387 (2005). [DOI] [PubMed] [Google Scholar]
- Tong J. et al. Distribution of monoamine oxidase proteins in human brain: implications for brain imaging studies. J Cereb Blood Flow Metab 33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S. F. et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med 53, 37–46 (2012). [DOI] [PubMed] [Google Scholar]
- Nordberg A. et al. A European multicentre PET study of fibrillar amyloid in Alzheimer’s disease. Eur J Nucl Med Mol I 40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson L. et al. Multi- and Megavariate Data Analysis (Part I -Basics and Principals and Applications). 2nd edn, 95–98 (Umetrics AB, 2006). [Google Scholar]
- Forsberg A. et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging 29, 1456–1465 (2008). [DOI] [PubMed] [Google Scholar]
- Dubois B. et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 9, 1118–1127 (2010). [DOI] [PubMed] [Google Scholar]
- Dubois B. et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 6, 734–746 (2007). [DOI] [PubMed] [Google Scholar]
- Dubois B. et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol 13, 614–629 (2014). [DOI] [PubMed] [Google Scholar]
- McKhann G. M. et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi V. T. Alzheimer’s disease 2012: the great amyloid gamble. Am J Pathol 180, 1762–1767 (2012). [DOI] [PubMed] [Google Scholar]
- Pimplikar S. W. Neuroinflammation in Alzheimer’s disease: from pathogenesis to a therapeutic target. J Clin Immunol 34 Suppl 1, S64–69 (2014). [DOI] [PubMed] [Google Scholar]
- Eikelenboom P. et al. Neuroinflammation - an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener Dis 7, 38–41 (2010). [DOI] [PubMed] [Google Scholar]
- Steele M. L. & Robinson S. R. Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol Aging 33, 423 e421-413 (2012). [DOI] [PubMed] [Google Scholar]
- Verkhratsky A., Marutle A., Rodriguez-Arellano J. J. & Nordberg A. Glial Asthenia and Functional Paralysis: A New Perspective on Neurodegeneration and Alzheimer’s Disease. Neuroscientist (2014). [DOI] [PubMed] [Google Scholar]
- Song W., Zhou L. J., Zheng S. X. & Zhu X. Z. Amyloid-beta 25-35 peptide induces expression of monoamine oxidase B in cultured rat astrocytes. Acta Pharmacol Sin 21, 557–563 (2000). [PubMed] [Google Scholar]
- Marutle A. et al. 3H-Deprenyl and 3H-PIB autoradiography show different laminar distributions of astroglia and fibrillar beta-amyloid in Alzheimer brain. J Neuroinflammation 10, 90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossan S. S. et al. Quantitative localization of human brain monoamine oxidase B by large section autoradiography using L-[3H]deprenyl. Brain Res 547, 69–76 (1991). [DOI] [PubMed] [Google Scholar]
- Kadir A. et al. Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh Compound B positron emission tomography patient with Alzheimer’s disease. Brain 134, 301–317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Joie R. et al. Region-specific hierarchy between atrophy, hypometabolism, and beta-amyloid (Abeta) load in Alzheimer’s disease dementia. J Neurosci 32, 16265–16273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo I. H., Carter S. F., Schöll M. L. & Nordberg A. Astrocytosis measured by (11)C-deprenyl PET correlates with decrease in gray matter density in the parahippocampus of prodromal Alzheimer’s patients. Eur J Nucl Med Mol I 41, 2120–2126 (2014). [DOI] [PubMed] [Google Scholar]
- von Bernhardi R., Eugenin-von Bernhardi L. & Eugenin J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci 7, 124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl W. C. et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 136, 2228–2238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrone A. et al. In vivo imaging of the 18-kDa translocator protein (TSPO) with [18F]FEDAA1106 and PET does not show increased binding in Alzheimer’s disease patients. Eur J Nucl Med Mol I 40, 921–931 (2013). [DOI] [PubMed] [Google Scholar]
- Varley J., Brooks D. J. & Edison P. Imaging neuroinflammation in Alzheimer’s and other dementias: Recent advances and future directions. Alzheimers Dement 11, 1110–20 (2014). [DOI] [PubMed] [Google Scholar]
- Golla S. S. et al. Quantification of [18F]DPA-714 binding in the human brain: initial studies in healthy controls and Alzheimer’s disease patients. J Cereb Blood Flow Metab 35, 766–772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R. et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab 32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau A. et al. Expression of oligodendroglial and astrocytic lineage markers in diffuse gliomas: use of YKL-40, ApoE, ASCL1, and NKX2-2. J Neuropath Exp Neur 65, 1149–1156 (2006). [DOI] [PubMed] [Google Scholar]
- Craig-Schapiro R. et al. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry 68, 903–912 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonell A. et al. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J Alzheimers Dis 42, 901–908 (2014). [DOI] [PubMed] [Google Scholar]
- Rosen C. et al. Increased Levels of Chitotriosidase and YKL-40 in Cerebrospinal Fluid from Patients with Alzheimer’s Disease. Dement Geriatr Cogn Dis Extra 4, 297–304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcolea D. et al. Relationship between cortical thickness and cerebrospinal fluid YKL-40 in predementia stages of Alzheimer’s disease. Neurobiol Aging 36, 2018–2023 (2015). [DOI] [PubMed] [Google Scholar]
- Bateman R. J. et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther 3, 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa C. F. et al. Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology 46, 406–412 (1996). [DOI] [PubMed] [Google Scholar]
- Bateman R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367, 795–804 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger T. L. et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. P Natl Acad Sci USA 110, E4502–4509 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler J. S. et al. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging 18, 431–435 (1997). [DOI] [PubMed] [Google Scholar]
- Ryan N. S. & Fox N. C. Reply: Implications of presymptomatic change in thalamus and caudate in Alzheimer’s disease. Brain 136, e259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storandt M., Balota D. A., Aschenbrenner A. J. & Morris J. C. Clinical and psychological characteristics of the initial cohort of the Dominantly Inherited Alzheimer Network (DIAN). Neuropsychology 28, 19–29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thordardottir S. et al. Preclinical Cerebrospinal Fluid and Volumetric Magnetic Resonance Imaging Biomarkers in Swedish Familial Alzheimer’s Disease. J Alzheimers Dis 43, 1393–402 (2014). [DOI] [PubMed] [Google Scholar]
- Schöll M. et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology 79, 229–236 (2012). [DOI] [PubMed] [Google Scholar]
- McKhann G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944 (1984). [DOI] [PubMed] [Google Scholar]
- Petersen R. C. Mild cognitive impairment as a diagnostic entity. J Intern Med 256, 183–194 (2004). [DOI] [PubMed] [Google Scholar]
- Bergman I., Blomberg M. & Almkvist O. The importance of impaired physical health and age in normal cognitive aging. Scand J Psychol 48, 115–125 (2007). [DOI] [PubMed] [Google Scholar]
- Ashburner J. & Friston K. J. Unified segmentation. Neuroimage 26, 839–851 (2005). [DOI] [PubMed] [Google Scholar]
- Hammers A. et al. Three-dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp 19, 224–247 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson A. et al. Evidence for astrocytosis in ALS demonstrated by [11C](L)-deprenyl-D2 PET. J Neurol Sci 255, 17–22 (2007). [DOI] [PubMed] [Google Scholar]
- Pearson K. On Lines and Planes of Closest Fit to Systems of Points in Space. Philos Mag A 2, 559–572 (1901). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.