

Abstract
Cardiomyopathy is a common human disorder that is characterized by contractile dysfunction and cardiac remodeling. Genetic mutations and altered expression of genes encoding many signaling molecules and contractile proteins are associated with cardiomyopathy; however, how cardiomyocytes sense pathophysiological stresses in order to then modulate cardiac remodeling remains poorly understood. Here, we have described a regulator in the heart that harmonizes the progression of cardiac hypertrophy and dilation. We determined that expression of the myocyte-enriched protein cardiac ISL1-interacting protein (CIP, also known as MLIP) is reduced in patients with dilated cardiomyopathy. As CIP is highly conserved between human and mouse, we evaluated the effects of CIP deficiency on cardiac remodeling in mice. Deletion of the CIP-encoding gene accelerated progress from hypertrophy to heart failure in several cardiomyopathy models. Conversely, transgenic and AAV-mediated CIP overexpression prevented pathologic remodeling and preserved cardiac function. CIP deficiency combined with lamin A/C deletion resulted in severe dilated cardiomyopathy and cardiac dysfunction in the absence of stress. Transcriptome analyses of CIP-deficient hearts revealed that the p53- and FOXO1-mediated gene networks related to homeostasis are disturbed upon pressure overload stress. Moreover, FOXO1 overexpression suppressed stress-induced cardiomyocyte hypertrophy in CIP-deficient cardiomyocytes. Our studies identify CIP as a key regulator of cardiomyopathy that has potential as a therapeutic target to attenuate heart failure progression.
Introduction
Cardiovascular diseases continue to be leading causes of death and disability. Despite this alarming fact, there is a lack of effectual treatment, and the molecular mechanisms underlying these devastating diseases remain elusive (1, 2). Cardiomyopathy can be broken into several disease categories, including hypertrophic cardiomyopathy, in which the heart is enlarged and heart muscle cells (cardiomyocytes) increase their size, and dilated cardiomyopathy, in which the ventricle chambers are dilated (3–5). Many epigenetic regulators, transcription factors, signaling molecules, and genes that encode structure proteins have been implied in cardiomyopathy (2, 6, 7). Studies aiming at understanding the molecular mechanisms underlying the progression from cardiac hypertrophy to dilated cardiomyopathy will have important implications for human heart disease.
One of the major responses of the heart to biomechanical stress and pathological stimuli is to undergo cardiac hypertrophy, an increase in the thickness of the cardiac ventricular wall (8, 9). Initially, cardiac hypertrophy is an adaptive response that maintains cardiac output in the face of increased workload. However, chronic activation of hypertrophic pathways is associated with adverse consequences that may lead to heart failure and sudden death (10). However, the molecular mechanisms underlying the transition from cardiac hypertrophy to dilated cardiomyopathy and heart failure remain largely unknown. In this study, we describe what we believe to be a novel regulator in the heart that modulates cardiac remodeling in response to mechanical and pathological stress.
Results
Loss of CIP accelerates the progress from hypertrophy to heart failure during cardiac remodeling.
We and others have previously identified a cardiac ISL1-interacting protein (CIP, also identified and referred to as MLIP [muscle-enriched A-type lamin-interacting protein]) (11, 12). CIP is specifically expressed in embryonic and adult mouse myocytes and is highly conserved between mouse and human. We asked whether the expression of this gene is correlated with human cardiomyopathy. Indeed, we found that CIP transcripts were significantly reduced in the hearts of patients with dilated cardiomyopathy (Figure 1A and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI82423DS1). Similarly, we examined the expression of mouse CIP protein and found it reduced in mouse hearts with dilated cardiomyopathy (Figure 1B). However, CIP protein levels appeared unaltered in hypertrophic hearts (Supplemental Figure 1).
Figure 1. Loss of CIP accelerates the progress from hypertrophy to heart failure during cardiac remodeling.

(A) qRT-PCR detection of the expression of human CIP transcripts in the hearts of patients with dilated cardiomyopathy (DCM). n = 5–10 for each group. (B) Western blot detection of the expression of mouse CIP proteins in dilated cardiomyopathy and control mouse hearts. GAPDH was used as a control. (C) Expression of CIP mRNA (n = 3 for each group) and protein in CIP-KO hearts detected by qRT-PCR and Western blot, respectively. Arrowheads mark multiple CIP protein products encoded by CIP mRNA splicing isoforms. (D) LV internal dimension at end-diastole (LVID;d) and FS of CIP-KO and control mice at indicated ages. (E) Gross heart morphology and H&E staining of CIP-KO and control hearts 4 weeks after TAC or sham operation. Scale bar: 2 mm. (F) LV posterior wall thickness at end-diastole (LVPW;d), LV internal dimension at end-diastole, and FS of TAC- or sham-operated CIP-KO and control mice. (G) Heart cross sections were stained with wheat germ agglutinin, and cardiomyocyte cross-sectional area was quantified. Scale bar: 50 μm. (H) Fast green and Sirius red staining of TAC- or sham-operated CIP-KO and control hearts. The fibrotic area was quantified. Scale bar: 1.5 mm. (I) qRT-PCR detection of expression of hypertrophy marker genes. n = 3–5 for each group. *P < 0.05, **P < 0.01, 1-way ANOVA with post-hoc Tukey’s test. The n number for each group is indicated.
Next, we investigated the function of CIP in mouse models of human cardiomyopathy (13). We generated CIP knockout mice by knocking a GFP-Cre cassette into the ATG-coding exon of the CIP locus (herein referred to as CIP-KO mice) (Supplemental Figure 2). Intercrossing of CIP heterozygous mice generated CIP-KO mice. We confirmed that CIP transcripts and proteins were abolished in mutant hearts (Figure 1C). Heterozygous intercrossing yielded progeny at the expected Mendelian ratio, indicating that CIP is dispensable for viability. The gross heart morphology of CIP-KO mice was indistinguishable from that of their littermate controls (Supplemental Figure 2). There were no detectable defects in cardiac morphology, histology, and cardiac function in young CIP-KO mice (Table 1). In addition, electrocardiography was performed, and no cardiac dysrhythmia was found in 3-month-old CIP-KO mice (Supplemental Table 2). However, cardiac function was depressed in aged CIP-KO mice, accompanied by increased left ventricular (LV) dimension, suggesting the development of dilated cardiomyopathy (Figure 1D and Table 1).
Table 1. Echocardiography examination of CIP-KO mice and their littermates at multiple time points under normal physiological conditions.

We hypothesized that loss of CIP sensitizes the heart to pathophysiological stresses. We applied transverse aortic constriction (TAC) to stimulate pressure overload–induced LV remodeling in both CIP-KO and control mice (14). Control mice developed significant cardiac hypertrophy 4 weeks after TAC. Histological echocardiographic measurements demonstrated an increase in LV wall thickness, without a significant increase in diastolic LV internal dimension in these mice. In contrast, TAC induced dramatic cardiac dilation in CIP-KO hearts. There was a significant increase in LV internal dimension and decrease in LV wall thickness in these mice (Figure 1, E and F, and Supplemental Table 3). While control mice exhibited a trend toward decreased cardiac contraction, the cardiac function of CIP-KO mice was dramatically impaired after TAC (Figure 1F and Supplemental Table 3). We measured the cross-sectional area of cardiomyocytes and found that TAC induced a greater increase in the size of CIP-KO cardiomyocytes compared with that of controls (Figure 1G). In addition, TAC induced more extensive fibrosis in CIP-KO hearts compared with that in controls (Figure 1H). Molecular markers for cardiac remodeling (9, 15), including Anp, Bnp, and Myh7, were markedly elevated in TAC-stressed CIP-KO hearts compared with controls (Figure 1I).
Genetic and functional interaction of CIP and calcineurin in the heart.
The above observations prompted us to test the hypothesis that loss of CIP leads to premature heart failure during cardiac remodeling. We first determined the expression of CIP in the hearts of calcineurin transgenic mice [Tg(Myh6-Cna) mice, herein referred to as CnA-Tg mice], in which the active CnA transgene induces massive cardiac hypertrophy, which eventually progresses to heart failure (10), and we found that CIP protein levels were substantially reduced (Figure 2A). We verified this observation by crossing a CIP reporter mouse line, in which a lacZ reporter was knocked into the CIP locus (16), with the CnA-Tg mice. We detected a marked decrease in lacZ staining in CnA-Tg ventricular tissues (Figure 2B). Interestingly, more than 50% of the CIP-KO CnA-Tg mice died at less than 10 weeks of age, and all of these mice died prematurely by 18 weeks, whereas about 50% of CnA-Tg mice survived to 20 weeks (Figure 2C). CIP-KO CnA-Tg hearts had very poor cardiac performance, with only around 10% fractional shortening (FS), as well as increased LV dimension and decreased LV wall thickness, indicating progress to heart failure (Figure 2D and Supplemental Table 4). Histological examination confirmed the above observations (Figure 2E). Consistently, the expression of cardiac remodeling and heart failure markers, Anp, Bnp, and Myh7, was further elevated in CIP-KO CnA-Tg hearts when compared with CnA-Tg controls (Figure 2F). Together, these data indicated that loss of CIP induces premature death in animals and promotes the progression to heart failure under stress conditions.
Figure 2. Genetic and functional interaction of CIP and calcineurin in cardiac hypertrophy.

(A) Western blot detection of the expression of CIP proteins in CnA-Tg and control hearts. β-Tubulin was used as a control. (B) β-Gal staining of the CIP reporter gene in CnA-Tg and control hearts. Scale bar: 1 mm. (C) Survival curve of CIP-KO CnA-Tg mice and their control littermates. The n number for each group is indicated. P < 0.01, CIP-WT CnA-Tg vs. CIP-KO CnA-Tg; P < 0.01, CIP-Het CnA-Tg vs. CIP-KO CnA-Tg, log-rank (Mantel-Cox) test. (D) FS of 10-week-old CIP-KO CnA-Tg mice and their control littermates. (E) Gross heart morphology and H&E staining of 6-week-old CIP-KO CnA-Tg mice and their control littermates. Scale bar: 1.2 mm. (F) qRT-PCR detection of the expression of hypertrophy marker genes in 3-week-old CIP-KO CnA-Tg and control hearts. n = 3–7 for each group. *P < 0.05, **P < 0.01, 1-way ANOVA with post-hoc Tukey’s test.
CIP regulates cardiomyocyte hypertrophy in vitro.
To determine whether CIP modulates cardiomyocyte remodeling in vitro, we knocked down CIP in isolated neonatal cardiomyocytes using siRNA and tested cellular response to agonist-induced cardiomyocyte hypertrophy. siRNA significantly inhibited the expression level of endogenous CIP (Figure 3A). As shown in Figure 3, phenylephrine (PE) significantly promoted cardiomyocyte hypertrophy (17), which was further enhanced by CIP knockdown (Figure 3, A and B). Quantification of the size of cardiomyocytes confirmed that knockdown of CIP enhanced PE-induced cardiomyocyte hypertrophy (Figure 3C). The increased expression of Anp, Bnp, and Acta1 in CIP knockdown cardiomyocytes (Figure 3D) further supports the view that CIP is required for proper cardiomyocyte remodeling in response to stress. Collectively, these in vivo and in vitro data demonstrate that CIP is a key regulator for cardiomyocyte remodeling in such a manner that loss of CIP accelerates the progression from compensative remodeling to adverse remodeling in the heart.
Figure 3. CIP represses cardiomyocyte hypertrophy.

(A) Neonatal rat cardiomyocytes were transfected with siRNA-targeting CIP transcript (si-CIP) or control siRNA (si-Ctrl). Cells were treated (+) or not treated (–) with PE, and CIP expression in was examined by qRT-PCR. (B) Representative immunofluorescence images of α-actinin staining in neonatal rat cardiomyocytes. Scale bar: 30 μm. (C) Quantification of the size of cardiomyocytes. More than 100 cardiomyocytes were measure for each experimental group. (D) qRT-PCR detection of the expression of hypertrophy marker genes Anp, Bnp, and Acta1 in CIP knockdown and control cardiomyocytes with or without PE treatment. Data were obtained from 4 independent experiments. *P < 0.05, **P < 0.01, 1-way ANOVA with post-hoc Tukey’s test.
Genetic and functional interaction of CIP and lamin A/C in dilated cardiomyopathy.
Pathological stress often induces dilated cardiomyopathy (3, 18). CIP was previously shown to interact with ISL1 (11) and lamin A/C (LMNA) (12). Intriguingly, mutations in LMNA are associated with cardiomyopathy, and genetic deletion of the Lmna gene results in dilated cardiomyopathy and premature death in mice (19). We asked whether the genetic interaction between CIP and LMNA contributes to dilated remodeling. We found that Lmna+/– CIP-KO mice exhibited dilated cardiomyopathy and decreased cardiac function, whereas either Lmna+/– or CIP-KO mice alone had normal cardiac function (Figure 4, A and B, and Supplemental Table 5). Interestingly, both Lmna–/– CIP-Het and Lmna–/– CIP-KO mice died sooner than Lmna–/– mice (Figure 4C). Furthermore, Lmna–/– CIP-KO double-knockout mice developed much more severe ventricular dilation and cardiac dysfunction when compared with Lmna-null mice alone (Figure 4D). These observations indicate that both CIP and LMNA contribute to cardiac function in an allele dosage-dependent manner.
Figure 4. Genetic interaction of CIP and LMNA in dilated cardiomyopathy.

(A) H&E staining of 12-month-old Lmna+/– CIP-KO compound mice and their control littermates. Scale bar: 1.5 mm. (B) Progressive increase of LV internal dimension at end-diastole and decrease of FS in Lmna+/– CIP-KO mice from 6 to 12 months. (C) Survival curve of Lmna–/– CIP-KO mice and their control littermates. P < 0.05, for Lmna–/– vs. Lmna–/– CIP-Het; P < 0.05, for Lmna–/– vs. Lmna–/– CIP-KO, log-rank (Mantel-Cox) test. (D) Ratio between LV internal dimension at end-systole and tibial length (LVID;s/TL) and FS of Lmna–/– CIP-KO double mutant mice and their control littermates at the age of 5 weeks. (E) qRT-PCR detection of the expression of CIP and Lmna in CIP-KO and control hearts. (F) qRT-PCR detection of the expression of CIP in Lmna–/– and control (Lmna+/+) hearts. (G) Immunofluorescence images of CIP (CIP-GFP, GFP-tagged CIP), LMNA, and α-actinin in cardiomyocytes isolated from LMNA-KO and control (LMNA-WT) hearts. DAPI marked nuclei. Scale bar: 20 μm. (H) Immunoprecipitation of CIP and LMNA interaction. The n number for each group is indicated. *P < 0.05, **P < 0.01, 1-way ANOVA with post-hoc Tukey’s test.
We asked whether loss of CIP would affect the LMNA expression level. qRT-PCR analyses indicate that Lmna transcript levels were not altered in the hearts of CIP-KO mice (Figure 4E). Conversely, we asked whether LMNA could regulate the expression of CIP. We performed qRT-PCR and found that the CIP mRNA level was unchanged in Lmna mutant hearts (Figure 4F). Next, we investigated whether loss of Lmna might change the cellular location of CIP proteins in cardiomyocytes. We transfected GFP-tagged CIP into isolated neonatal mouse cardiomyocytes from either Lmna–/– or control mice and examined GFP-CIP protein expression in both the nucleus and cytoplasm. Deletion of Lmna did not appear to alter the cellular distribution of CIP proteins (Figure 4G). We demonstrated, using coimmunoprecipitation assays, that CIP and LMNA proteins interact directly (Figure 4H), consistent with a prior report (12). Together, our results show that CIP and LMNA interact genetically and physically to modulate cardiac remodeling.
Overexpression of CIP suppresses stress-induced cardiac mal-remodeling.
Next, we generated CIP conditional knockin mice, in which a conditional Flag-tagged CIP transgene was knocked into the Rosa26 locus (20) (referred to herein as CIPKI mice). Cardiac-specific overexpression of CIP was achieved in CIPKI cTNT-Cre mice (referred to herein as CIP-OE mice hereafter). These mice were grossly normal and did not exhibit a detectable cardiac phenotype (Supplemental Figure 3). To test whether overexpression of CIP protects the heart from developing hypertrophy in response to stress, we subjected the CIP-OE mice to TAC surgery. Control mice developed significant cardiac hypertrophy accompanied by a decrease in cardiac function 4 weeks after TAC. Remarkably, cardiac hypertrophy was suppressed and cardiac function was preserved in CIP-OE mice (Figure 5A and Table 2). Overexpression of CIP prevented the TAC-induced increases in ventricle weight/body weight ratio (Figure 5B), cardiomyocyte size (Figure 5C), and cardiac fibrosis (Figure 5D). TAC-induced elevation of hypertrophic marker gene expression was significantly inhibited in CIP-OE hearts (Figure 5E). Of note, no difference was observed between CIP-OE and control hearts in sham-operated groups, suggesting that CIP overexpression does not have deleterious effect.
Figure 5. Gain of function of CIP protects the heart from mal-remodeling.

(A) LV posterior wall thickness at end-diastole, LV internal dimension at end-diastole, and FS of TAC- or sham-operated (4 weeks) CIP-OE and control mice. (B) The ventricle weight/body weight (Vw/Bw) ratio of CIP-OE and control hearts 4 weeks after TAC or sham operation. (C) Wheat germ agglutinin staining and cardiomyocyte cross-sectional area quantification of indicated hearts. Scale bar: 50 μm. (D) Fast green and Sirius red staining of CIP-OE and control hearts 4 weeks after TAC or sham operation. Scale bar: 1.5 mm. (E) qRT-PCR detection of the expression of hypertrophy marker genes in TAC- or sham-operated (4 weeks) CIP-OE and control hearts. n = 4–5 for each group. (F) Gross heart morphology and H&E staining of CIP-OE and control hearts 10 weeks after TAC or sham operation. Scale bar: 1.6 mm. (G) Dynamics of LV dimension and cardiac function 4 and 10 weeks after TAC or sham operation in CIP-OE and control mice. n = 8–15 for each group. (H) H&E staining of 4-week-old CnA-Tg and control mice injected with AAV9-CIP or control virus (AAV9-GFP). Scale bar: 1.5 mm. (I) Ventricle weight/body weight ratio of CnA-Tg and control mice injected with AAV9-CIP or control virus. (J) qRT-PCR detection of the expression of hypertrophy marker genes and CIP in CnA-Tg and control hearts with AAV9-CIP virus or control virus. n = 5–8 for each group. *P < 0.05, **P < 0.01, 1-way ANOVA with post-hoc Tukey’s test.
Table 2. Echocardiography examination of CIP-OE mice and their control littermates 4 weeks after TAC or sham operation.
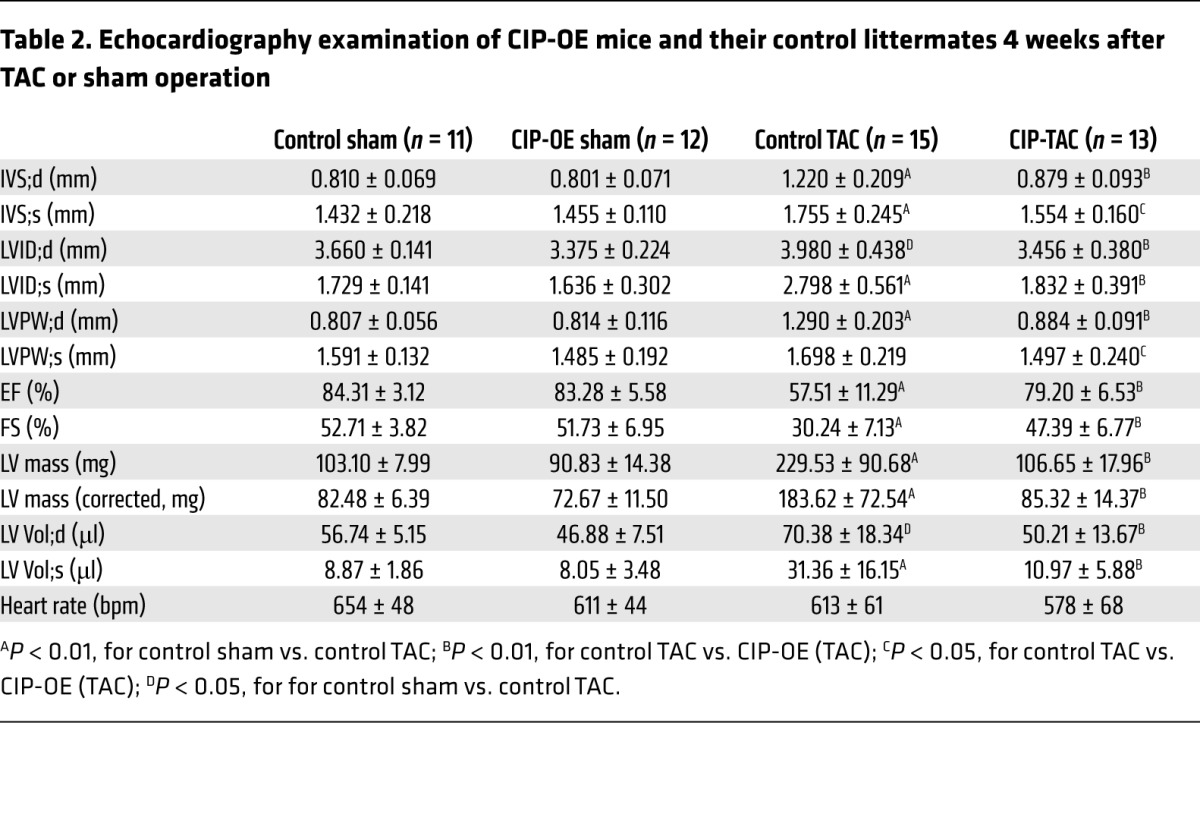
Prolonged cardiac stress often induces the transition from compensative remodeling to adverse remodeling (21). In order to determine whether overexpression of CIP is able to delay this transition and prevent adverse remodeling in the heart, we prolonged the duration of TAC-induced pressure overload to 10 weeks. In control mice, LV dimension was markedly increased, while LV wall thickness was decreased, indicating progression to the adverse remodeling and heart failure (Figure 5, F and G). In contrast, cardiac function was well preserved, with no sign of LV chamber dilation in CIP-OE mice (Figure 5, F and G, and Supplemental Table 6). Morphologically and histologically, CIP-OE hearts were indistinguishable from those of sham-operated controls (Figure 5F). Therefore, cardiac overexpression of CIP clearly has a beneficial effect on the heart under stress conditions.
To further test the therapeutic potential of CIP in suppressing pathological hypertrophy induced by CnA, we engineered an adeno-associated virus serotype 9 (AAV9) vector directed by the cardiac-specific cTNT promoter to drive the expression of the CIP gene in the heart (AAV9-CIP) (22, 23). Neonatal CnA-Tg and control littermate mice were transduced with the AAV vector, and phenotypes were analyzed 4 weeks later. We first confirmed the specificity and efficacy of the AAV system, showing that control AAV9-cTNT-GFP was only expressed in the heart and not in any other organs tested (Supplemental Figure 4). AAV9-CIP did not cause any overt phenotype in wild-type mice, consistent with our observations for CIP-OE mice. As expected, the control AAV9-GFP–treated CnA-Tg hearts developed massive cardiac hypertrophy comparable to that of untreated CnA-Tg hearts. However, AAV9-CIP–mediated cardiac overexpression of CIP attenuated the cardiac hypertrophy in CnA-Tg hearts, as evidenced by smaller heart size and reduced ventricle weight/body weight ratio (Figure 5, H and I). Additionally, the expression of cardiac remodeling markers (Anp, Bnp, and Myh7), which was dramatically induced in CnA-Tg hearts, was substantially repressed in CIP-overexpressed CnA-Tg hearts (Figure 5J). Together, these data demonstrated that cardiac overexpression of CIP benefits the heart by repressing adverse remodeling, including the progression to dilation and heart failure.
CIP mediated stress-responsive signaling pathways in the heart.
We carried out unbiased transcriptome profiling studies in the hearts of CIP-null mice at P1 and 3 weeks after birth. Unexpectedly, loss of CIP did not appear to alter the expression of the majority of genes under physiological conditions (data not shown). Next, we asked whether cardiac stress might contribute to the gene expression program in CIP-KO hearts. Indeed, genome-wide transcriptome profiling revealed that 343 genes were significantly upregulated, while 493 genes, including CIP, were significantly downregulated in CIP-KO hearts after TAC (P < 0.05). Gene ontology analysis of upregulated genes revealed that “collagen” and “ribosome” were among the most enriched terms (Figure 6A). This increased expression of collagen-related genes in TAC CIP-KO hearts is consistent with the increased cardiac fibrosis found at 4 weeks after TAC. We confirmed the increased expression of several ribosomal protein genes by qRT-PCR (Figure 6B). Given that an increase in the number of ribosomes is associated with the development of cardiac hypertrophy (24, 25), our results suggested that loss of CIP sensitizes protein translation and therefore promotes cardiac remodeling under pressure overload stress. Next, using ingenuity pathway analysis, we showed that a portion of downregulated genes, including Jun, Pten, and Pml, are regulated by p53 and FOXO1 (Figure 6C). We confirmed the downregulation of the expression of these genes by qRT-PCR (Figure 6D). Further supporting our observation, overexpression of FOXO1 was able to inhibit calcineurin-induced hypertrophic growth (26). Consistent with these reports, we found that the expression of Foxo1 transcripts decreased in stressed CIP-KO hearts and increased in CIP-OE hearts (Figure 6E). Similarly, we observed that expression levels of FOXO1 proteins were lower in CIP-KO hearts (Figure 6F). These results suggested that loss of CIP attenuates the activity of p53 and FOXO1, which partly contributes to the cardiac phenotype.
Figure 6. Signaling pathways that mediate CIP function in regulating cardiac homeostasis.

(A) Bioinformatic analysis of dysregulated genes (fold change >1.25) in CIP-KO and control hearts 2 weeks after TAC operation using DAVID (http://david.abcc.ncifcrf.gov/). (B) qRT-PCR validation of dysregulated genes in the “RIBOSOME” cluster. n = 5 for each group. (C) Ingenuity pathway analysis of the dysregulated genes in CIP-KO hearts 2 weeks after TAC operation. Green ovals indicate that gene expression in CIP-KO hearts is downregulated; blue lines indicate that the upstream gene expression leads to the inhibition of downstream gene expression; blue ovals indicate that gene expression in CIP-KO hearts is predicted to be inhibited; and yellow lines indicate that results are inconsistent with reported data. (D) qRT-PCR validation of dysregulated genes downstream of p53 and FOXO1. n = 5 for each group. (E) qRT-PCR validation of Foxo1 expression in CIP-KO and CIP-OE hearts under cardiac stress. n = 4–5 for each group. (F) Western blot detection of the expression of mouse FOXO1 proteins in CIP-KO and control hearts 2 weeks after TAC operation. GAPDH was used as a control. *P < 0.05, **P < 0.01, 1-way ANOVA with post-hoc Tukey’s test.
FOXO1 mediated the function of CIP in regulating cardiomyocyte hypertrophy.
To further test the hypothesis that the function of CIP in cardiomyocytes is mediated by FOXO1, we turned to isolated cardiomyocytes. CIP was knocked down in neonatal rat cardiomyocytes that were treated with the hypertrophic agonist PE to induce hypertrophy. Whereas FOXO1 expression was induced by PE, knocking down CIP dramatically reduced FOXO1 protein levels in cardiomyocytes (Figure 7A). As noticed previously, knockdown of endogenous CIP alone resulted in modest cardiomyocyte hypertrophy. Loss of CIP accelerated PE-induced hypertrophy (Figure 7B). Adenoviral-mediated overexpression of FOXO1 abolished PE-induced hypertrophy in CIP knockdown samples (Figure 7B), indicating that FOXO1 participates in CIP-dependent hypertrophic growth. Quantification of cardiomyocyte size confirmed the above observations (Figure 7C). We examined the expression of the hypertrophic markers Anp, Bnp, and Acta1. Interestingly, both CIP knockdown and PE treatment induced the expression of hypertrophic marker genes; their expression was completely repressed by FOXO1 (Figure 7D). Our data suggest that FOXO1 is able to, at least in part, mediate CIP-dependent hypertrophy regulation.
Figure 7. FOXO1 mediates the function of CIP in cardiomyocyte hypertrophy and cardiac remodeling.

(A) Neonatal rat cardiomyocytes were transfected with siRNA for CIP or control siRNA. Cells were treated or not treated with PE, and the expression of CIP and FOXO1 proteins was examined by Western blotting. GAPDH was used as a control. (B) Immunofluorescence images of α-actinin staining in neonatal rat cardiomyocytes with (AdFOXO1) or without (AdGFP) FOXO1 overexpression. Scale bar: 30 μm. (C) Quantification of cardiomyocyte size. More than 100 cardiomyocytes were measured for each experimental group. (D) qRT-PCR detection of the expression of Anp, Bnp, and Acta1 in cardiomyocytes. Data were obtained from 3 independent experiments. *P < 0.05, **P < 0.01, 1-way ANOVA with post-hoc Tukey’s test. (E) A working model postulating the function of CIP in cardiac remodeling and disease.
Discussion
In this study, we identified a protein in the heart that plays a crucial role during cardiac remodeling in response to stresses. Mice lacking CIP are sensitized to pathological and mechanical signals that lead to the development of cardiomyopathy and heart failure, whereas overexpression of this protein protects the heart from stress-induced adverse remodeling. Our studies uncover CIP, a key component of important regulatory networks for cardiac remodeling and heart failure (Figure 7E).
Loss-of-function studies demonstrate that CIP is dispensable for heart development and normal cardiac function under physiological conditions. However, genetic deletion of CIP sensitizes the heart to remodel in respond to stress. In particular, CIP mutant mice exhibit accelerated progress from cardiac hypertrophy to dilation cardiomyopathy under stress conditions. These observations indicate that CIP may function as a cellular sensor to read mechanical and/or pathophysiological stimuli. Most importantly, overexpression of CIP in cardiomyocytes, either using transgenic or AAV-mediated delivery, protects the heart from stress-induced mal-remodeling, suggesting that it could become a novel therapeutic target for cardiovascular disease.
How does CIP do all these tasks? We suggest that CIP may function as a cellular sensor to read mechanical and/or pathophysiological stimuli in the heart. Previous reports suggested that CIP is a nuclear protein, interacting with transcription factor ISL1 and nuclear envelope protein LMNA (11, 12). Indeed, our results demonstrate a genetic and physical interaction between CIP and LMNA. The latter is a nuclear protein responsible for the integrity of the nuclear envelope. Our results indicate that CIP and LMNA do not appear to directly affect each other’s expression levels. Instead, the physical and functional interaction between these two proteins and their location on the nuclear envelope suggest that they are likely key components of the nuclear envelope that respond to mechanic and/or pathological stresses to modulate cardiac function. Alternatively, CIP proteins are also located in cytoplasm, suggesting that it may function as a signaling molecule to transmit extracellular and cytoplasmic signals into the nucleus to modulate gene expression in response to stress.
Our transcriptome analysis in CIP mutant hearts revealed that signaling pathways involved in stress response and homeostasis, such as p53 and FOXO1, are regulated by CIP. Of particular interest, FOXO1 was previously linked to cardiac hypertrophy through modulation of the CnA signaling pathway (26). We found that FOXO1 transcript and protein expression levels were both decreased in CIP mutant hearts and cardiomyocytes, accompanied by the development of cardiomyocyte hypertrophy. Intriguingly, overexpression of FOXO1 in CIP knockdown cells was sufficient to suppress PE-induced cardiomyocyte hypertrophy, suggesting that FOXO1 functions downstream of CIP in cardiac remodeling. Currently, it is not known how CIP controls the expression of FOXO1; likewise, we do not know whether the CnA pathway is the downstream mediator for CIP function in the heart. Nevertheless, our studies identify CIP as a regulator for cardiac hypertrophy and remodeling. Further investigation to understand the molecular mechanisms by which CIP regulates this important cardiac process will facilitate the development of therapeutic targets to treat human cardiovascular diseases.
Methods
Human samples.
LV tissues were taken from patients with terminal-stage heart failure indicated for heart transplantation; surgeries were performed between August 2009 and June 2013 at the Clinic of Heart Surgery, supervised by M. Hulman. In brief, the patient’s heart was removed at the time of transplantation, and LV tissue was subsequently dissected and snap frozen. We used LV samples from healthy hearts that were not implanted (provided by P. Ferdinandy, Semmelweis University, Budapest, Hungary) to serve as controls.
Mice.
CIP-KO and CIP-OE mice were generated in-house. The CIP-KO–targeting vector was generated by replacing the ATG cassette of the CIP locus with the GFP-Cre cassette through a 2-step recombination. In brief, the ATG cassette of CIP was replaced by the GFP-Cre cassette followed by a flped neomycin cassette in the first recombination in a bacterial artificial chromosome (BAC) plasmid. Then, the engineered DNA fragment was retrieved from BAC in the second recombination. The CIP-OE–targeted vector was generated by inserting a flag-tagged CIP transgene in a Rosa26-targeting vector as described previously (27). The recombination of engineered DNA fragments in ES cells was achieved by electroporation. Targeted ES cells were identified by PCR and/or Southern blot analyses and used for blastocyst injection. The resulting chimeric mice were bred to C57BL/6 mice to obtain germline transmission. For CIP-KO mice, the neomycin cassette was subsequently excised by breeding with β-actin–Flp mice. To obtain CIP-OE mice, mice with a Rosa-CIP allele (the stop codon is present and floxed) were bred with cTNT-Cre mice (28) to excise the stop codon and activate the CIP transgene in the heart. Calcineurin transgenic (CnA-Tg) mice (10) and LMNA mutant mice (29) were used in this study. Compound mutant mice on mixed genetic backgrounds were used, and all comparisons used littermates as controls.
Measurement of cardiac function by echocardiography.
Echocardiographic measurements were performed on mice using a Visual Sonics Vevo 2100 Imaging System (Visual Sonics) with a 40-MHz MicroScan transducer (model MS-550D). Mice were anesthetized with isoflurane (2.5% isoflurane for induction and 0.5% for maintenance). Heart rate and LV dimensions, including diastolic and systolic wall thicknesses and LV end-diastolic and end-systolic chamber dimensions, were measured from 2D short-axis views under M-mode tracings at the level of the papillary muscle. LV mass and functional parameters, such as percentage of FS and ejection fraction, were calculated using the above primary measurements and accompanying software.
Electrocardiography recording.
Ambulatory ECG data of 3-month-old CIP-KO mice and their control littermates. All recordings were performed in the morning, between 9 AM and noon. Baseline ECG data were averaged from 1-hour recordings. To see whether there were any β-adrenergic effects, mice were injected with epinephrine (stimulator, 0.4 mg/kg) or propranolol (blocker, 1 mg/kg) at a rate of 1 injection per day. ECG with epinephrine data were analyzed starting 30 seconds after the injection for 10 minutes, while ECG with propranolol data were analyzed starting 10 minutes after the injection for 20 minutes. To record conscious and unrestrained ambulatory ECG data, without any anesthetic effect on ECG parameters, a telemetry device (model TA10-F20, Data Sciences International) was implanted subcutaneously and electric leads were placed to obtain an equivalent to lead I surface ECG. Each mouse was in a regular mouse cage placed on the receiver, and ECG signals were collected while the animal ran freely in the cage.
TAC operation.
Mice were anesthetized with isoflurane (3%–4% isoflurane for induction, 1%–2% isoflurane for maintenance). The chest was shaved and cleaned with alcohol. A suture was placed around the front upper incisors and pulled taut so that the neck was slightly extended. The tongue was retracted and held with forceps, and a 20-gauge catheter was inserted into the trachea. The catheter was then attached to the mouse ventilator via a Y-shaped connector. Ventilation was performed with a tidal volume of 220 to 240 μl for a 25–30 g mouse and a respiratory rate of 130 to 140 breaths per minute. 100% oxygen was provided to the inflow of the ventilator. The chest was opened through a left second intercostal thoracotomy. The 26-gauge needle without its sharp tip was put on the ascending aorta. The needle and the ascending aorta were tightly ligated together using a 7-0 nylon suture (Ethicon) at the position between the brachiocephalic artery and the common carotid artery, and the 26-gauge needle was removed immediately after ligation. In the sham operation, no ligation was performed. Isoflurane was stopped, and the lungs were slightly overinflated to assist in removal of air in the pleural cavity. Dissected intercostal space and chest skin were closed using a 6-0 silk suture (Ethicon). All manipulations were performed by an operator without knowledge of the genotype. Four weeks after the TAC operation, mice developed significant cardiac hypertrophy. Ten weeks after the TAC operation, mice developed dilated cardiomyopathy.
Generation and administration of AAV (serotype 9) and adenovirus.
GPF-tagged mouse CIP cDNA and GFP were separately cloned into ITR-containing AAV plasmid (Penn Vector Core P1967) harboring the chicken cardiac TNT promoter to yield constructs pAAV9.cTnT::CIPGFP and pAAV9.cTnT::GFP, respectively. AAV was packaged using AAV9:Rep-Cap and pAd:deltaF6 (Penn Vector Core) as described previously (30). AAV9 was packaged in 293T cells with AAV9:Rep-Cap and pAd deltaF6 and then purified and concentrated by gradient centrifugation. AAV9 titer was determined by quantitative PCR. AAV9 virus (3 × 1011 virus genome per animal) was injected into P2 CnA-Tg pups and their control littermates by subcutaneous injection. Hearts were harvest at the age of 4 weeks. GPF-tagged mouse CIP cDNA was also cloned into pENTR3C vector (Life Technologies) and then subcloned into pAd/CMV/V5-DEST (Life Technologies) vector through recombination. Ad-CIP-GFP virus was packaged in 293AD cells with the pAd-CIP-GFP and purified with the Fast-Trap Adenovirus Purification and Concentration Kit (EMD Millipore). Ad-FOXO1 virus (SL100754) was purchased from SignaGen Laboratories.
Immunochemistry and β-gal staining.
Cells were washed with PBS 3 times and then fixed with 4% PFA for 10 minutes. After washing with PBS, cells were then blocked in 5% serum/PBS and subjected to immunostaining. Mouse heart tissue and embryos were dissected out, collected, and fixed in 4% PFA at 4°C for 4 hours. After washing in PBS, embryos were treated in 15% and 30% sucrose for 2 hours each and embedded in OCT. Approximately 5- to 8-μm cryostat sections were collected on positively charged slides. Sections were washed in PBS, blocked in 5% serum/PBS, and subjected to immunostaining. Antibody sources were as follows: α-actinin (Sigma-Aldrich, catalog A7811); LMNA (Cell Signaling Technology, catalog 4777); GFP (Life Technologies, catalog A21311); and Alexa Fluor 488 and 594 secondary antibodies (Life Technologies). Fluorescently stained cells were counterstained with DAPI and imaged with an FV1000 confocal microscope (Olympus). To quantify the cross-sectional area of cardiomyocytes, heart sections were deparaffined and stained with Wheat Germ Agglutinin, Alexa Fluor 647 Conjugate (Life Technologies, catalog W32466). Stained sections were examined with an epifluorescence microscope, and the cross-sectional area of cardiomyocytes was quantified with ImageJ software. For β-gal staining, tissues were fixed with 4% PFA for 2 hours at 4°C, washed with PBS for 3 times, and then stained with a solution containing 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 1 mg/ml X-gal substrate at 37°C for 12 hours after fixation.
H&E staining and fast green/Sirius red collagen staining.
Mouse heart tissues were dissected out, rinsed with PBS, and fixed in 4% PFA (pH 8.0) overnight. After dehydration through a series of ethanol baths, samples were embedded in paraffin wax according to standard laboratory procedures. For routine histological examination with a light microscope (SteREO Discovery.V8, Zeiss), 5-μm sections were stained with H&E. For Sirius red/fast green collagen staining, sections were fixed with prewarmed Bouin’s solution at 55°C for 1 hour and then washed in running water. Sections were stained in 0.1% fast green solution for 10 minutes and then washed with 1% acetic acid for 2 minutes. After rinsing in tape water, sections were stained in 0.1% Sirius resolution for 30 minutes. After staining, sections were dehydrated and cleared with xylene. The images were examined with a light scope (SteREO Discovery.V8, Zeiss) and quantified with ImageJ software.
Cardiomyocyte culture.
Neonatal mouse and rat cardiomyocytes were prepared as previously described (17). Briefly, neonatal cardiomyocytes were isolated by enzymatic disassociation of 1-day-old neonatal mouse or rat hearts with the Neonatal Cardiomyocyte Isolation System (Cellutron Life Technology). Cardiomyocytes were plated for 2 hours to remove fibroblasts. Cells were then plated on 1% gelatin-coated plates in medium containing 10% horse serum and 5% fetal calf serum. Eighteen hours after plating, cells were changed into serum-free medium and infected with adenovirus (25 MOI) for 24 hours. For the treatment of siRNA, 50 nM siRNA-targeting CIP transcript and control siRNA (from Dharmacon) was transfected into cardiomyocytes by using Lipofectamine RNAiMAX transfection reagent. Six hours later, media with transfection reagent were removed. Cells were then treated with the hypertrophic agent PE (20 μM) by changing the PE-contained serum-free medium. Cells were harvested 24 hours after PE treatment for RNA isolation or 48 hours after PE treatment for immunochemistry.
Quantitative RT-PCR and Western blot analysis.
Total RNA was isolated from cells and tissue samples using TRIzol Reagent (Life Technologies). For quantitative RT-PCR, 2.0 μg RNA samples were reverse transcribed to cDNA by using random hexamers and MMLV reverse transcriptase (Life Technologies) in 20 μl reaction system. In each analysis, a 0.1 μl cDNA pool was used for quantitative PCR. The relative expression of genes of interest was normalized to the expression of 18S rRNA or GAPDH. For Western blot analyses, cell extractions were cleared by 10,000 g centrifugation for 10 minutes. Samples were subsequently analyzed by SDS-PAGE and transferred to PVDF membranes that were incubated with Odyssey Blocking Buffer (LI-COR) and anti-CIP (1:2,000, 21st Century Biochemicals, customized); anti–β-tubulin (1:20,000, Sigma-Aldrich, catalog T0198); anti-GAPDH (1:10,000, Fitzgerald, catalog 10-1501); anti-FOXO1 (1:500, Abcam, catalog ab52857); anti-Flag (1:5,000, Sigma-Aldrich, catalog F7425); and anti-His (1:2000, Sigma-Aldrich, catalog M6693) overnight at 4°C and then washed 3 times with PBS buffer before adding IgG secondary antibody. Specific protein bands were visualized by Odyssey CLx imager (LI-COR).
Coimmunoprecipitation assays.
HEK293 cells were transiently transfected with plasmids encoding Flag-tagged CIP and His-tagged mouse LMNA using FuGENE6 (Promega). Cells were harvested 48 hours after transfection in lysis buffer composed of PBS containing 0.5% Triton X-100, 1 mM EDTA, 1 mM PMSF, and complete protease inhibitors (Roche). After a brief sonication and removal of cellular debris by centrifugation, Flag-tagged CIP proteins were precipitated with anti-Flag antibodies and protein A/G beads and analyzed by Western blotting with anti-His antibodies.
Statistics.
Values are reported as mean ± SEM unless indicated otherwise. Statistical significance was determined with ANOVA. For multiple group comparisons, a post-hoc Tukey’s test was performed when ANOVA reached significance. P values of less than 0.05 were considered statistically significant.
Study approval.
All mouse experiments were approved by the IACUC of Boston Children’s Hospital. The human study was performed according to the principles of the Declaration of Helsinki. The Institutional Ethics Committee of the National Institute of Cardiovascular Diseases, Bratislava, Slovakia, approved the study protocol. Patients provided written informed consent.
Author contributions
Z-PH and D-ZW conceived the project, designed and analyzed the experiments, and wrote the manuscript. Z-PH performed molecular biology experiments. JK contributed to human sample acquisition and cDNA library preparation. Z-PH, LM, and BZ generated and characterized CIP-KO and CIP-OE mice. MK performed TAC surgery. Z-PH and JC contributed to the echocardiographic data acquisition and analysis. Z-PH, GW, JD, CZ, and ZL contributed to AAV preparation and administration. HW performed electrocardiographic data acquisition and analysis. Z-PH, MN, JL, XH, and Z-LD contributed to the histological and immunofluorescent data acquisition and analysis. CES and JGS supervised the echocardiographic data acquisition and reviewed the manuscript. WTP supervised the CIP-KO mouse generation and reviewed the manuscript.
Supplementary Material
Acknowledgments
We thank members of the Wang laboratory for advice and support. Work in the Wang laboratory is supported by the March of Dimes Foundation, the Muscular Dystrophy Association, and the NIH (HL085635, HL116919). M. Kataoka was supported by Banyu Life Science Foundation International. Z.-P. Huang is supported by NIH grant T32HL007572.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(11):4122–4134. doi:10.1172/JCI82423.
References
- 1.Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115(3):518–526. doi: 10.1172/JCI24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358(13):1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 3.Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194(3):355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koitabashi N, Kass DA. Reverse remodeling in heart failure--mechanisms and therapeutic opportunities. Nat Rev Cardiol. 2012;9(3):147–157. doi: 10.1038/nrcardio.2011.172. [DOI] [PubMed] [Google Scholar]
- 5.Houser SR, et al. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012;111(1):131–150. doi: 10.1161/RES.0b013e3182582523. [DOI] [PubMed] [Google Scholar]
- 6.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313(5795):1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Cell Biol. 2013;14(8):529–541. doi: 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad F, et al. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005;112(20):3140–3148. doi: 10.1161/CIRCULATIONAHA.105.550806. [DOI] [PubMed] [Google Scholar]
- 9.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 10.Molkentin JD, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93(2):215–228. doi: 10.1016/S0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang ZP, et al. CIP, a cardiac Isl1-interacting protein, represses cardiomyocyte hypertrophy. Circ Res. 2012;110(6):818–830. doi: 10.1161/CIRCRESAHA.111.259663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmady E, et al. Identification of a novel muscle A-type lamin-interacting protein (MLIP) J Biol Chem. 2011;286(22):19702–19713. doi: 10.1074/jbc.M110.165548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yutzey KE, Robbins J. Principles of genetic murine models for cardiac disease. Circulation. 2007;115(6):792–799. doi: 10.1161/CIRCULATIONAHA.106.682534. [DOI] [PubMed] [Google Scholar]
- 14.Hill JA, et al. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101(24):2863–2869. doi: 10.1161/01.CIR.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 15.Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen GM, et al. Large-scale gene trapping in C57BL/6N mouse embryonic stem cells. Genome Res. 2008;18(10):1670–1679. doi: 10.1101/gr.078352.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xing W, et al. Myocardin induces cardiomyocyte hypertrophy. Circ Res. 2006;98(8):1089–1097. doi: 10.1161/01.RES.0000218781.23144.3e. [DOI] [PubMed] [Google Scholar]
- 18.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 19.Nikolova V, et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. 2004;113(3):357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5(9):1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 21.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109(13):1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 22.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18(1):43–52. doi: 10.1038/gt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5(3):285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 24.Fanburg BL. Experimental cardiac hypertrophy. N Engl J Med. 1970;282(13):723–732. doi: 10.1056/NEJM197003262821306. [DOI] [PubMed] [Google Scholar]
- 25.Clarke K, Ward LC. Protein synthesis in the early stages of cardiac hypertrophy. Int J Biochem. 1983;15(10):1267–1271. doi: 10.1016/0020-711X(83)90217-3. [DOI] [PubMed] [Google Scholar]
- 26.Ni YG, et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 2006;114(11):1159–1168. doi: 10.1161/CIRCULATIONAHA.106.637124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou P, et al. Interrogating translational efficiency and lineage-specific transcriptomes using ribosome affinity purification. Proc Natl Acad Sci U S A. 2013;110(38):15395–15400. doi: 10.1073/pnas.1304124110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiao K, et al. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17(19):2362–2367. doi: 10.1101/gad.1124803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan T, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147(5):913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1(3):1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.