

Abstract
Oncolytic viral (OV) therapy, which uses genetically engineered tumor-targeting viruses, is being increasingly used in cancer clinical trials due to the direct cytolytic effects of this treatment that appear to provoke a robust immune response against the tumor. As OVs enter tumor cells, intrinsic host defenses have the potential to hinder viral replication and spread within the tumor mass. In this report, we show that histone deacetylase 6 (HDAC6) in tumor cells appears to alter the trafficking of post-entry OVs from the nucleus toward lysosomes. In glioma cell lines and glioma-stem–like cells, HDAC6 inhibition (HDAC6i) by either pharmacologic or genetic means substantially increased replication of oncolytic herpes simplex virus type 1 (oHSV). Moreover, HDAC6i increased shuttling of post-entry oHSV to the nucleus. In addition, electron microscopic analysis revealed that post-entry oHSVs are preferentially taken up into glioma cells through the endosomal pathway rather than via fusion at the cell surface. Together, these findings illustrate a mechanism of glioma cell defense against an incoming infection by oHSV and identify possible approaches to enhance oHSV replication and subsequent lysis of tumor cells.
Introduction
Malignant gliomas (such as glioblastoma [GBM]) remain formidable cancers based on their poor prognosis, with a median survivorship of 15 months or less, extensive neurologic morbidity, and cost of care (1, 2). Surgical, radiation-based, and pharmacologic therapies have extended patients’ lives by a few months, but the extensive and complex genetic heterogeneity of these tumors renders therapeutic targeting of a few aberrant signaling networks unlikely to succeed (3, 4). Various immunotherapies have recently been approved by the FDA for the treatment of some cancers and are now also being tested in GBM (5). The theoretical advantage of some immune-based treatments relates to immune cell recognition of any aberrant tumor-associated pathway/molecule and possible immune cell adaptability to the anatomic and temporal heterogeneous nature of the GBM. One form of immunotherapy uses genetically engineered tumor-selective pathogens, such as oncolytic viruses (OVs), to replicate in and kill tumor cells, thereby increasing immune cell recognition of tumor and viral antigens exposed in the lysed tumor “debris” field (6–8).
As OVs are administered into tumors, entry of the agent into the cell, viral replication, cell lysis/death, and release of progeny virions to infect surrounding tumor cells are critical processes that should occur efficiently in order to obtain sufficient tumor cell death to provoke an effective antitumor immune response, resulting in clearance of the neoplasm. Yet, these initial stages of OV action against tumors can still be impeded by a variety of tumor and host factors that limit efficient entry, replication, and intratumoral spread (9, 10). Recognition and identification of these host factors can thus be utilized to try and improve these critical initial phases of OV therapy. One type of OV that has been tested, even in phase III clinical trials (11), is based on genetically engineered herpes simplex virus type 1 (HSV-1). HSV-1 is thought to primarily enter infected cells by fusion of its viral envelope with the cellular membrane and release of the viral capsid into the cell cytosol, after which it travels to the nucleus using the microtubular (MT) apparatus (12, 13). Recently, though, HSV-1 has also been shown to enter some cells through endocytic vesicles that are subsequently fused with viral envelopes to release capsids into the cytosol, suggesting an alternative mechanism of post-entry trafficking of virus capsids from the plasma membrane (PM) into the nucleus (14). Through this alternative mechanism that is widely used by other viruses, such as adenoviruses, incoming viral capsids would need to exit endosomes before these fuse with lysosomes in order to shuttle viral capsids to the nucleus. Sensing of viral infection and recognition of viral nucleic acids also occurs within endosomes (15). Endocytotic materials and cytoplasmic proteins mainly are transported on MT networks, and posttranslational modifications of tubulin control MT function (16, 17). Of particular interest, histone deacetylase 6 (HDAC6) (18), a member of the class IIb histone deacetylases (HDACs), has been characterized as a deacetylase of tubulin and of other cytoplasmic proteins (HSP90 and cortactin) (19) responsible for homeostasis of the cellular MT apparatus (20). In addition, HDAC6 has been shown to be required for selective autophagic processes involving autophagic vesicle fusion with lysosomes, and it is also involved in the process of cellular endocytic uptake (21–23). As a pathogenic defense mechanism, HDAC6 activity has been reported to selectively upregulate type I IFN (24) and prevent HIV-1 envelope–dependent cell fusion and infection (25).
Based on this, we have thus hypothesized that HDAC6 may provide antiviral functions by aiding the initial endocytic entry of oncolytic HSV (oHSV) and subsequent fusion to lysosomes, thus shuttling incoming virions for autophagy/xenophagy rather than to the nucleus for viral replication. In this report, we show for what we believe to be the first time that (a) pharmacologic and genetic inhibition of HDAC6 led to enhanced replication of oHSV, while augmentation of HDAC6 reduced it; (b) the role of HDAC6 in the antiviral effect is linked to the acetylation status of MTs; (c) HDAC6 inhibition (HDAC6i) counteracted the antiviral effect of type 1 IFNs; (d) HDAC6i led to increased shuttling of post-entry oHSV to the nucleus rather than to the lysosome; and (e) HDAC6i also led to improved oHSV replication in a panel of patient-derived spheroid-grown glioma cells. These results thus establish that HDAC6 can be an intrinsic cell mechanism against oHSV and oncolytic tumor therapy.
Results
Pharmacologic inhibition of HDACs enhances oHSV replication.
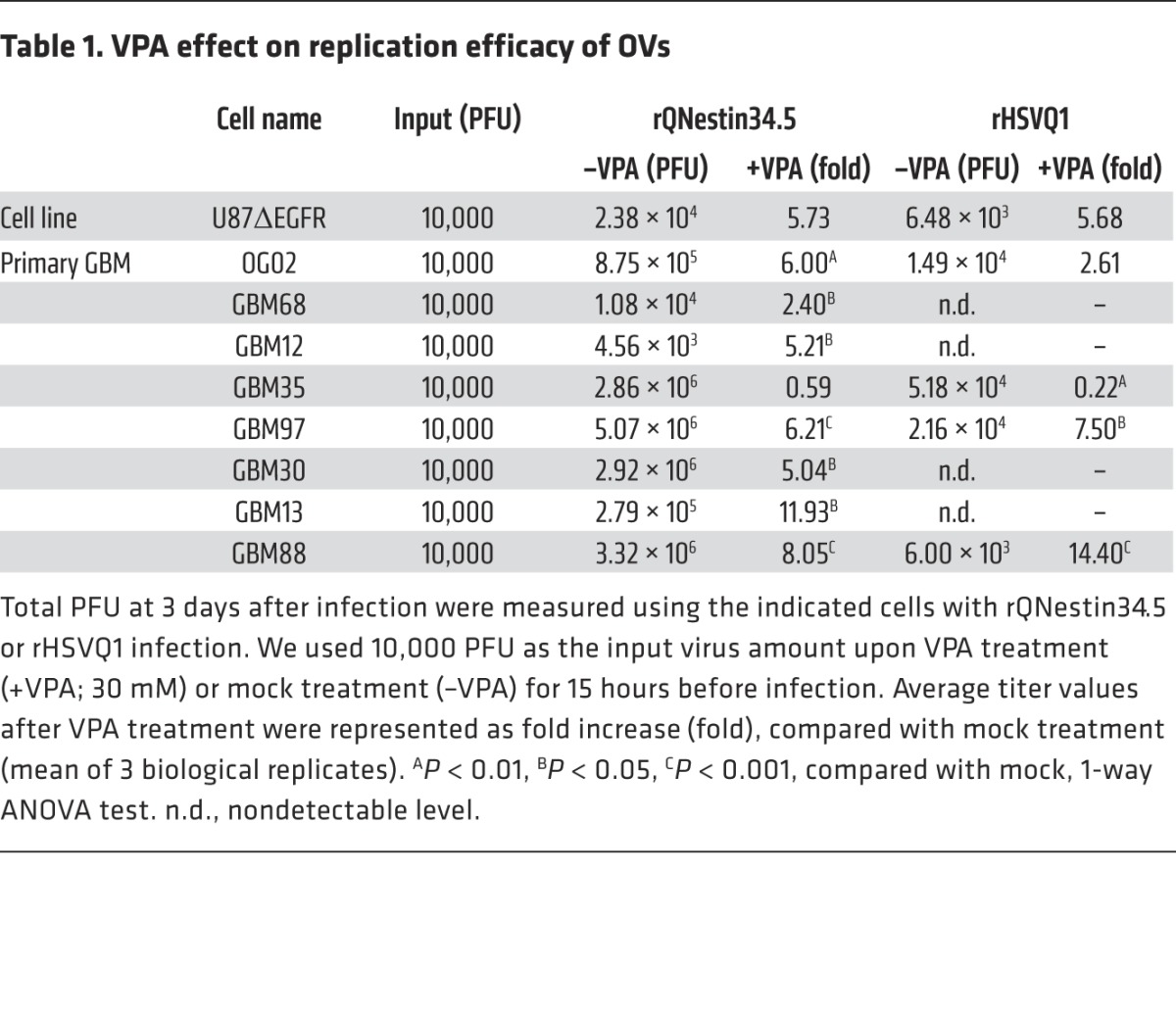
At least 18 HDACs have been identified in cells, and several drugs inhibit the action of multiple types of HDACs. We thus sought to establish whether pan-HDAC inhibition enhanced oHSV replication in several glioma cell lines and primary glioma cells grown to enrich for “stem-like” spheroid-forming properties (glioma-stem-like cells [GSCs]). The data in Table 1 show that valproic acid (VPA), a known pan-HDAC inhibitor, enhanced replication of 2 oHSVs (rQNestin34.5 and rHSVQ1) (26, 27) several fold in both an established glioma line and 7 of 8 tested GSCs. Therefore, these data show that pan-HDAC pharmacologic inhibition leads to enhanced oHSV replication in both established and primary gliomas, confirming previously published observations (28–31).
Table 1. VPA effect on replication efficacy of OVs.
Specific HDAC6i enhances oHSV infection and replication.
The finding that pan-HDAC inhibition led to increased oHSV replication suggests that pan-HDAC activity inhibited it (29). We sought to determine whether any one particular HDAC was responsible for this finding and focused on HDAC6, based on the rationale described in the Introduction. We used tubacin, a relatively specific deacetylase inhibitor of HDAC6 (32), to determine whether it affected HSV-1–mediated transgene expression and oHSV replication in U251 human glioma cells. As shown in Figure 1, tubacin did indeed increase luciferase-mediated bioluminescence delivered by an HSV-1 amplicon vector, either in the absence or in the presence of exogenous IFN-β (Figure 1A), and also improved the kinetics of oHSV replication between 24 and 48 hours after infection (Figure 1B). In addition, tubacin partially reversed the antiviral effect of exogenous IFN-β and replicated the effect of VPA (Figure 1C). Next, we attempted to determine whether there was increased cytotoxicity of tumor cells by using tubacin and oHSV. Figure 1D shows cytotoxicity dose-response curves in the presence of 3 different doses of tubacin and 6 MOIs of oHSV: there was a significant reduction of glioma cell survival as these doses increased. As a comparison, we also tried to determine whether relatively specific inhibitors of other HDACs resulted in enhanced cytotoxicity mediated by oHSV, using CI994, an inhibitor of class I HDACs (primarily HDAC1, HDAC2, and HDAC3). As shown in Figure 1E, two doses of CI994 did not significantly increase oHSV cytotoxicity at 8 different MOIs against glioma cells. We also determined that tubacin alone did not affect glioma cell survival at the concentrations used for the study (Figure 1F). These findings thus suggested that HDAC6 could be one of the HDACs responsible for the observed inhibition of oHSV replication in glioma cells.
Figure 1. The HDAC6-specific inhibitor, tubacin, improves HSV-1–mediated gene expression and oHSV replication.

(A) Bioluminescence (measured as RLU) assay was performed 24 hours after infection with a replication-defective HSV-1 encoding a Fluc cDNA of U251 cells (MOI of 3). (B) Replication of rQNestin34.5 (MOI of 0.1) in tubacin-treated (dashed line) and control U251 cells (solid line). The input dose was given at 0 hours. (C) Titration of oHSV-infected (rQNestin34.5-infected) U251 cells (MOI of 0.03) in the presence of IFN-β, with and without VPA or tubacin, 3 days after infection. Doses of tubacin and IFN-β were 5 μM and 1,000 units/ml, respectively. (D) LDH cytotoxicity assay 3 days after infection of U251 cells by rQNestin34.5 in the presence of tubacin (0, 1, 2, and 5 μM; starting at 14 hours before infection). (E) LDH cytotoxicity assay 5 days after infection of U251 cells by rQNestin34.5 in the presence of CI994 (0, 1, and 3 μM; starting at 14 hours before infection). (F) LHD cytotoxicity assay of U251 cells in the presence of tubacin for 5 days. *P < 0.05, **P < 0.01, ***P < 0.001 by 1-way ANOVA test in A–C and F) and 1-way ANOVA with Turkey’s multiple comparisons tests in D and E. Error bars correspond to mean ± SD (n = 3; in A–C and n = 4; in F). Horizontal bars represent the average in A–C. Bars represent the average (n = 4; in D–F) (See also Supplemental Figure 1.).
Genetic modulation of tubulin regulators also changes oHSV replication and HSV-mediated gene expression.
To study the functional roles of HDAC6 in the oHSV life cycle, we used shRNAs against HDAC6. As shown in Figure 2, A and B, shRNA-mediated “knockdown” of HDAC6 in U251 glioma cells significantly increased HSV-1–mediated transgene expression. When exogenous human HDAC6 was transiently overexpressed in U251 glioma cells, HSV-1 amplicon–mediated luciferase gene expression was significantly reduced (Figure 2, C and D). With the knowledge that post-entry HSV-1 capsids traffic to the nucleus to deliver viral DNA via the cellular MT apparatus and that one of HDAC6’s activities is to deacetylate tubulin, we then asked whether the increased acetylation level of α-tubulin changed HSV-mediated gene expression. In fact, transient overexpression of human MEC17 (33), an α-tubulin acetyltransferase (also called TAT1), significantly enhanced the bioluminescence mediated by an HSV-1 amplicon (Figure 2, E and F), thus providing further evidence that HSV-mediated gene expression is enhanced by tubulin acetylation (i.e., counteracting HDAC6 activity). To further ensure that the effect of HDAC6 against HSV-1 infectivity was specifically due to HDAC6 modulation of tubulin acetylation, we used two α-tubulin mutants: a hyperacetylation (K40Q) and an acetylation-resistant (K40R) tubulin mimic. Stable expression of either the K40Q or the K40R mutant resulted in the expected increased or decreased replication of oHSV, respectively (Figure 3). This indicated that HDAC6 activity (i.e., causing tubulin deacetylation) was the specific function that decreased oHSV replication, rather than an “off-target” effect of shRNA or pharmacologic agents.
Figure 2. Functional assays of HSV-1 infection in glioma cells with altered deacetylase or acetyltransferase functions.

(B) Stable HDAC6 gene knockdown mediated by two different HDAC6 targeting shRNAs (referred to as #1 and #2) and (D) transient overexpression of the HDAC6 gene or (F) myc-tagged human MEC17 (an α-tubulin acetyltransferase) in U251 glioma cells were confirmed by Western blots using antibodies against HDAC6, myc, and acetylated and total α-tubulin on parallel gels. Infectivity of replication-defective HSV-1 encoding the Fluc gene at a (A and C) MOI of 3 or (E) other indicated MOIs was measured by luciferase enzymatic activities. (A) Empty vector (Vec) was used as a control and was set as 100 percent for RLU. (C and E) HDAC6 and myc-tagged MEC17 were transiently overexpressed in U251 cells for 48 hours prior to the infection. Horizontal bars and error bars in the plots correspond to average values and mean ± SD, respectively (n = 3; *P < 0.05, ***P < 0.001 by 1-way ANOVA test).
Figure 3. Enhanced and reduced oHSV replication by ectopic acetylation-mimic or -resistant α-tubulin mutant expression, respectively.

(A) mCherry-tagged α-tubulin (WT) and (B) its acetylation-resistant (K40R) and (C) acetylation-mimic (K40Q) mutants were stably expressed in U251 cells. Scale bar: 20 μm. (D) oHSV-infected (rQNestin34.5; MOI of 0.03) U251 cells expressing the indicated mCherry-tubulin fusion genes for 3 days. The significance among 3 groups (n = 3, mean ± SD) was analyzed by a 1-way ANOVA test (P < 0.001), followed by pairwise comparison of groups with Bonferroni-Holm–adjusted P values (**P < 0.01, ***P < 0.001).
Tubacin alters post-entry trafficking of HSV capsid proteins.
To determine how HDAC6i improved HSV-1–mediated gene expression (Figure 1A) and replication kinetics (Figure 1B), we asked whether HDAC6i altered the intracellular trafficking of post-entry capsids toward the nucleus for viral genomic release or toward the lysosomes for digestion of large multimolecular particles. We used HSV-1 virions with a GFP-tag located within the VP26 capsid protein, designated K26GFP (34), to infect U251 cells expressing red fluorescent protein–tagged (RFP-tagged) LAMP1, a lysosome membrane–bound protein, in order to enable live monitoring and quantification of HSV-1 capsid trafficking. At 90 minutes after infection, in untreated cells, the majority of green fluorescence localized primarily at the edge of DAPI-stained nuclei (average percentage GFP colocalization at nuclei, 57.27% ± 21.56%) rather than in association with RFP lysosomes (25.41% ± 13.27%) (Figure 4). Tubacin treatment led to a significant increase in GFP-VP26 capsids localizing to the perinucleus (68.45% ± 14.98%) rather than to lysosomes (17.60% ± 8.39%). Conversely, IFN-β treatment led to the opposite effect, with a significant increase in the number of capsids localizing to lysosomes (39.19% ± 18.25%) compared with the number localizing to nuclei (28.07% ± 18.52%). Tubacin treatment reversed this effect (lysosome, 27.17% ± 14.39%, versus perinucleus, 49.42% ± 15.95%). Together, these data indicated that tubacin preferentially routed post-entry HSV-1 capsids to nuclei rather than to lysosomes at early time points after infection, even in the presence of IFN-β.
Figure 4. Percentage colocalization of GFP-VP26 intracellular capsids in RFP-LAMP1–expressing U251 cells.

At 90 minutes after infection, in cells under the indicated treatment conditions (IFN-β, 1,000 units/ml; tubacin, 5 μM), colocalization of GFP fluorescence either (A) with RFP fluorescence (RFP-LAMP1) or (B) with DAPI fluorescence (perinucleus) was counted in each cell after they were fixed with 4% paraformaldehyde and stained with DAPI. The horizontal bars and error bars represent average and mean ± SD, respectively (n = 22–27; *P < 0.05, **P < 0.01, ***P < 0.001 by 1-way ANOVA test). Numbers of cells used for counting GFP dots are as follows: –tubacin/–IFN-β, n = 27; +tubacin/–IFN-β, n = 26; –tubacin/+IFN-β, n = 22; and +tubacin/+IFN-β; n = 23.
We then performed time-lapse microscopic analyses for the localization of the VP26-GFP fusion protein in RFP-LAMP1 cells after K26GFP infection. As reported previously (34), green fluorescence localized throughout the cell as early as 6 hours after infection, and the majority of cells showed an accumulation of GFP fluorescence within large foci (punctae) in the nuclei for up to 20 hours after infection (Figure 5, A and E, for an average of 63.3% at 20 hours after infection [h.p.i.]). Tubacin-treated cells displayed green fluorescence in the cytosol as early as 4 hours after infection, and the temporal kinetics of punctae formation in the nucleus were visibly faster. In addition, tubacin-treated cells exhibited a visibly increased number of GFP-positive punctae in nuclei (Figure 5, B and F, 92% at 20 h.p.i.) when compared with that in nontreated cells. In agreement with the previous observation (Figure 4), IFN-β treatment delayed the temporal kinetics of both GFP expression and perinuclear accumulation of GFP-positive punctae, which were also decreased in number when compared with control cells (Figure 5, C and G, 54% at 20 h.p.i.), but with an increase in GFP signal within RFP-expressing aggregates, indicative of lysosomal accumulation and possible lysosomal degradation, as observed above (Figure 4). Tubacin was able to reverse the effects of IFN-β treatment (Figure 5, D and H, 75% at 20 h.p.i.). Taken together, these results suggested that HDAC6i altered post-entry trafficking of HSV-1 capsids, increasing it toward nuclei rather than lysosomes and reversing the effect of IFN-β that instead routed capsids toward lysosomes rather than nuclei.
Figure 5. Time-lapse imaging after infection of U251 glioma cells with a VP26-GFP–expressing HSV-1 in tubacin-treated and/or IFN-β–treated RFP-LAMP1–expressing U251 cells.

VP26-GFP–expressing HSV-1 (K26GFP) was added to RFP-LAMP1 stably expressing U251 cells after (C, D, G, and H) IFN-β pretreatment and/or (B, D, F, and H) tubacin treatment. GFP/RFP fluorescence signals were captured using time-lapse confocal microscopy. (A–D) The time at which each image was taken after infection is indicated on the top left and the bottom right. Scale bar: 20 μm. (E–H) Blue bars represent the percentages of intranuclear GFP puncta in total GFP-positive cells. Gray bars indicate that the presence of GFP-positive cells is 100%. Total counts of GFP-expressing cells and time (h.p.i.) after the infection are shown at bottom of bar plots. (See also Supplemental Figure 2.)
oHSV capsids can be found in endocytic vesicles in GSCs.
Endosomes can fuse with lysosomes to degrade the uptake of pathogenic particles, and our data presented above showed that HSV-1 capsids colocalized with lysosomes. This would imply that HSV-1 capsids were trafficked into cells via endosomes rather than being trafficked freely into the cytosol immediately upon the viral uptake. HSV-1 entry has not been classically associated with uptake via endosomes, and, in a number of cells, such as EBV-LPD primary cells (35) and Vero cells (ref. 36 and data not shown), oHSVs are taken up by direct fusion of the viral envelope to the cell membrane, releasing capsids into the cytosol. This trafficking pathway thus would not require HDAC6, in contrast with our aforementioned findings. Therefore, we tried to evaluated whether in gliomas entry or uptake of oHSV occurs via the endocytic pathway or via direct fusion of the viral envelope to the PM by analyzing the localization of oHSV capsids in two independent GSCs (GBM326 and GBM83) (37) and U251 cells upon oHSV infection. Transmission electron microscopy (TEM) analysis showed virion uptake occurring via endocytosis (Figure 6, A and B), with occasional fusion of the viral envelope PM (Figure 6C). In addition, virions could be visualized sequestered in endosomal vesicles within the cytosols, with some vesicles showing fusion with virions, suggestive of capsid release from the vesicle (Figure 6D). Quantitative analyses further confirmed that the majority of oHSV uptake in GSCs occurred via endocytosis (Figure 6E). These experiments thus provided further evidence that post-entry oHSV capsids are encapsulated in intracellular vesicles (and thus more likely to be targeted by lysosomes) rather than directly released into the cytosol upon infection in glioma cells.
Figure 6. TEM analysis of oHSV uptake events in GBM83 and GBM326 GSCs and U251 glioma cells.

(A) GBM326 cells at 20 minutes after infection (p.i.) with rQNestin34.5. The inset shows an enveloped-virion–containing vesicle on the PM (arrowhead). N, nucleus. (B) GBM326 cells. The inset shows macropinocytosis of a virion on the PM. (C) GBM83 cells. The inset shows fusions with HSV-1 virions (arrowheads) and the PM. (D) GBM326 cells. The inset shows endocytic vesicles containing virions in the cytoplasm. Arrowheads indicate fusions of virions with vesicle membrane. (E) Summary of TEM analyses in GBM83, GBM326, and U251 cells at 2, 10, and 20 minutes after infection with rQNestin34.5 virus. PM, oHSV on PM; cytosol (vesicle), virions in the cytosolic endocytic vesicle; cytosol (naked), virions in cytosol without vesicles. Scale bar: 500 nm.
In vivo effect of HDAC6i in glioma models.
We next sought to determine whether oHSV replication was also enhanced in vivo in an animal model of glioma. We used human GBM30 cells, a GSC that grows aggressively in the brains of athymic mice. We also used tubastatin A (TA), another highly specific HDAC6 inhibitor (38), because tubacin biodistribution in the brain is limited by the brain-blood barrier. After establishing GBM30 gliomas in the brains of athymic mice and treating them with systemic TA or vehicle, two different types of oHSVs were tested by intratumoral administration. Four days later, brains and tumors were harvested, and oHSVs were titered. As shown in Figure 7A, TA-mediated inhibition of HDAC6 did indeed lead to a significant increase in the in vivo titers of two different oHSVs. To determine whether this translated into a significant anticancer effect, survival of mice with orthotopic GBM30 gliomas was followed after oHSV intratumoral injection, as a function of TA administration. There was a trend toward improved survival for mice treated with TA and oHSV compared with that for mice treated with oHSV alone (Figure 7B). In lieu of detailed pharmacodynamic studies of TA, we analyzed the target of TA action (i.e., HDAC6i hyperacetylated tumors, leading to increased tubulin acetylation in tumors). As shown in Supplemental Figure 3, TA administration led to increased tubulin acetylation in orthotopic tumors in mouse brains. The sum of these results thus suggested that in vivo HDAC6i improved intratumoral replication of oHSV in highly aggressive GSC tumors.
Figure 7. TA effects on oHSV in a GSC orthotopic model.

nu/nu mice with established GBM30 gliomas were injected in tumors with either the oHSV rQNestin34.5 or the oHSV, KNE. TA (1.3 mg per kg body weight) was administered i.p. 3 times every day, starting the day before oHSV injection, and administration continued until tissue harvest. (A) oHSV titers were assayed 4 days later after oHSV injection. n = 5 for rQNestin34.5 with TA; n = 6 for other groups. The horizontal bars and the error bars correspond to average values and mean ± SD, respectively (*P < 0.05, **P < 0.01, 1-way ANOVA test). (B) Survivorship of mice with orthotopic GBM30 gliomas was followed via Kaplan-Meier analysis. Mice with mock treatment (n = 8) (white square) or treatment with the oHSV (rQNestin34.5; 500,000 PFU) stereotactically administered into 5-day-old tumors and then with i.p. vehicle (black triangle, n = 11) or TA (black circle, n = 12) 4, 5, 6, 7, and 8 days after tumor implantation were followed. P = 0.0031, control versus oHSV; P < 0.0001, control versus oHSV plus TA; P = 0.0852, oHSV versus oHSV plus TA, by log-rank test.
Variability of the HDAC6i effect on oHSV replication in a panel of well-characterized GSCs.
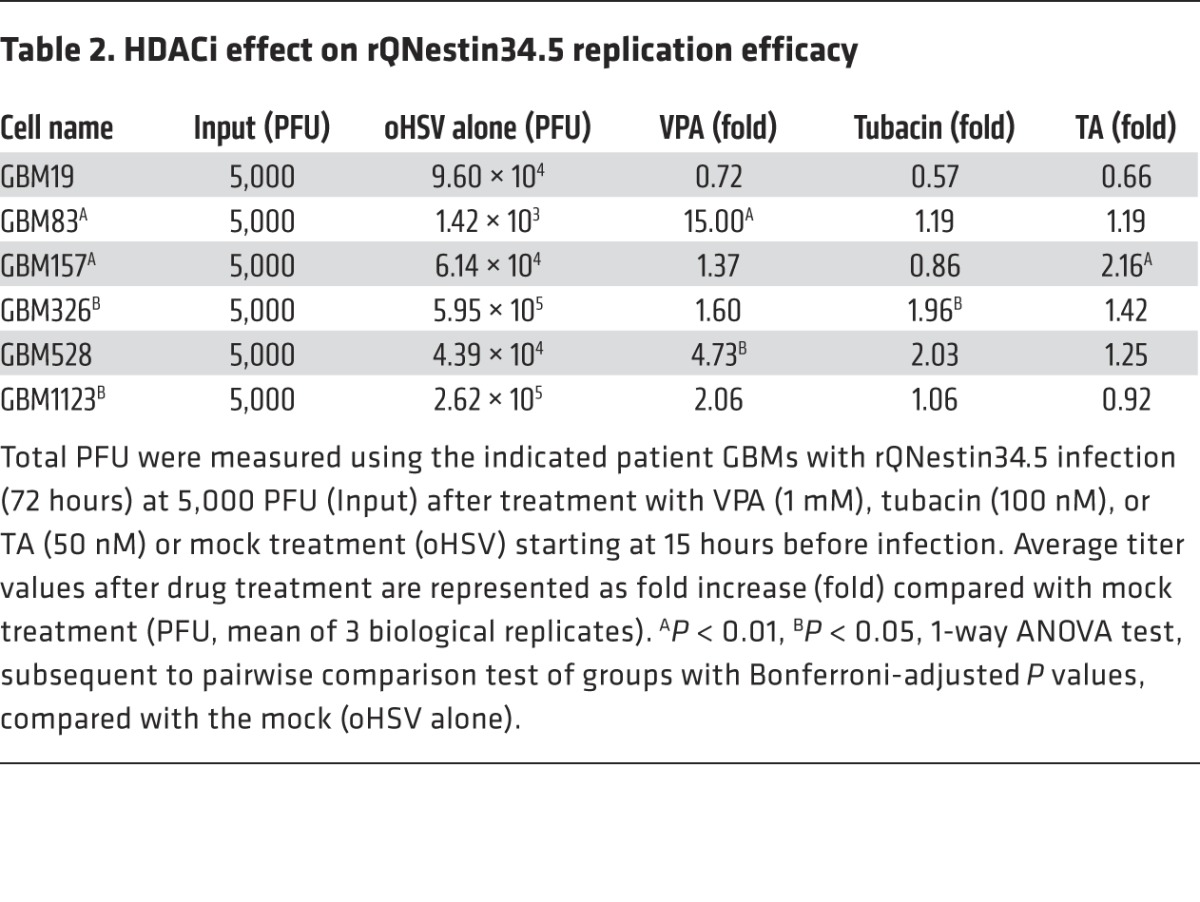
To further study the HDAC6i effects in patient-derived samples, we repeated oHSV replication assays on multiple GSCs that were very well characterized in terms of their molecular profiles (37). As shown in Table 2, there was variability not only in the HDAC6i effect, but also in the VPA-mediated pan-HDACi effect on oHSV replication when assayed on this GSC panel. In fact, in 2 of 6 GSCs (GBM157 and GBM326), either tubacin- or TA-mediated HDAC6i significantly enhanced oHSV replication, a finding that did not occur in 3 GSCs (GBM83, GBM528, and GBM1123); in contrast, in GBM19 cells, HDAC6i reduced oHSV replication. Interestingly, the pan-HDACi effect of VPA was also variable, with significant enhancement in 2 of 6 GSCs (GBM528 and GBM83). Tubacin also significantly augmented oHSV cytotoxicity for GBM326 cells to the same magnitude as pan-HDACi with VPA (data not shown). However, this did not occur with GBM19 cells, in agreement with the viral replication data (Table 2), and did not significantly occur with GBM528 cells (data not shown).
Table 2. HDACi effect on rQNestin34.5 replication efficacy.
To determine whether HDAC6 levels correlated with responses, we evaluated HDAC6 mRNA levels in the tested GSCs. As shown in Figure 8A, all 6 GSCs expressed HDAC6 mRNA, but there was no correlation between HDAC6i sensitivity and HDAC6 mRNA levels: for instance, GBM528 cells expressed the highest levels of HDAC6 mRNA, but HDAC6i had no effect on these cells, while GBM326 cells expressed low to intermediate levels of HDAC6 mRNA and yet were the most sensitive to HDAC6i of all tested GSCs. We also determined that human GBMs expressed HDAC6 mRNA, as shown in Figure 8B. These data thus indicated that there was variability in enhancement of oHSV replication and cytotoxicity by HDAC6i against different GSCs.
Figure 8. HDAC6 mRNA levels.

HDAC6 mRNA levels of (A) cultured GSCs and (B) freshly resected human gliomas and fetal brain tissues. Normalization of HDAC6 mRNA was performed using the housekeeping genes (A) β-actin or (B) GAPDH. Data represent the mean ± SD (n = 3, triplicate analyses of the sample).
Discussion
Eukaryotic cells possess intrinsic defense mechanisms against infections by viruses that directly restrict viral replication and assembly upon infection via ubiquitously existing proteins. Over the last two decades, a variety of genetically engineered viruses have been tested as cytotoxic agents against tumor cells, but little is known about epigenetic mechanisms of the intrinsic defenses that tumors may have against different OVs. We and others have previously shown that pan-HDAC inhibitors lead to enhanced infectivity and replication of different OVs in tumors (28–30, 39). Although it is still possible that the epigenetic effects of the nuclear class of HDACs were responsible for this effect, we have found that the nonnuclear cytosolic HDAC6 can provide antiviral effects in tumor cells exposed to an oHSV. Specifically, we have been able to show that (a) pharmacologic and genetic inhibition of HDAC6 led to enhanced replication of oHSV, while augmentation of HDAC6 function reduced oHSV replication; (b) HDAC6’s role in the antiviral effect is linked to the acetylation status of MTs; (c) HDAC6i counteracted the antiviral effect of type 1 IFNs; (d) HDAC6i led to increased shuttling of post-entry oHSV to the nucleus rather than to the lysosome; and (e) HDAC6i also led to improved oHSV replication in a panel of patient-derived spheroid-grown glioma cells, albeit with some variability. Coupled with the knowledge that HDAC6 is ubiquitously expressed in human GBMs (see Figure 8B), these findings thus indicate that HDAC6 can provide antiviral functions in tumor cells exposed to OVs.
Multiple HDACs with diverse functions exist in cells. In fact, at least 18 HDACs have been discovered that not only control deacetylation of histones, thus modifying gene expression, but that also deacetylate other proteins involved in cellular homeostatic functions (40, 41). We and others have previously published results that indicate that inhibition of HDACs with pharmacologic agents, such as VPA, trichostatin A, or SAHA, significantly enhances the OV anticancer activity in vitro and in vivo (28, 31, 39, 42, 43). However, it was not clear whether the effect of pan-HDAC inhibitors was due to inhibition of a specific HDAC protein or multiple HDAC proteins. This study showed that tubacin, a relatively specific HDAC6 inhibitor unlike others that target intranuclear substrates such as histones (32), enhanced HSV-1 vector–mediated transgene expression and improved the kinetics of oHSV replication in glioma cells. It also reversed the antiviral effects of IFN-β (24). We demonstrated that overexpression of tubulin deacetylase HDAC6 and tubulin acetyltransferase MEC17 led to negative and positive effects against HSV-1–mediated gene transduction, respectively. These effects were further supported by overexpression of an acetylation-mimic α-tubulin mutant versus an acetylation-resistant α-tubulin mutant that led to increased versus decreased oHSV replication, respectively. These findings thus suggested that HDAC6, a “stress” surveillance factor (18), may be responsible for attenuating oHSV replication in gliomas. However, this does not imply that other HDACs are not involved. In fact, as shown in Table 2, the findings that the pan-HDAC inhibitor VPA had more pronounced effects than the HDAC6 inhibitor on oHSV replication would support this statement. In fact, while a class 1 HDAC inhibitor did not seem to affect oHSV replication (see Figure 1E), we observed that a HDAC3-specific inhibitor (RGFP966) appeared to enhance it (data not shown). It is thus likely that HDAC6 is not the only HDAC affecting oHSV replication.
The above experiments led us to hypothesize that HDAC6 can provide a host-intrinsic protective mechanism against oHSV by modulating endocytosis of virions and facilitating endosome/autophagosome-lysosome fusion to prevent release of viral genomes into nuclei, while IFN signaling appears to do the opposite (see Figure 5). In fact, recent data show that HDAC6 performs this function with cellular receptors and is responsible for quality-control autophagy (21, 44). In addition, fusion of HIV’s envelope with cellular membranes is regulated by HDAC6 in CD4+ cells (25). HSV-1 entry can occur through macropinocytosis (ATP-requiring) or virion-membrane fusion (ATP-independent) pathways (45). This is important because oHSV entry via endocytosis may target it for lysosomal degradation in GSCs, if it occurs (46). HSV-1 has long been thought to enter cells primarily via fusion of its envelope with the PM, releasing the capsids into the cytosol (47). However, endocytic uptake of HSV-1 has been shown to occur in many cell types (46, 48), and our data also show that oHSVs are taken up into glioma cells primarily via the endocytic pathway. The significance of this relates to HDAC6’s role in endosome/autophagosome fusion with the lysosome. However, additional experiments will be needed to determine whether HDAC6i alters endocytic uptake of oHSV or changes the dynamics of oHSV entry via this pathway while inhibiting endosome/autophagosome fusion.
The MT network plays important roles in nuclear translocation, subcellular localization, and egress of HSV-1 (49). The major HSV-1 tegument protein VP22 localizes on acetylated MTs and affects MT reorganization during viral replication (50, 51). Recent studies have shown that the acetylation status of tubulin does not affect MT polymerization, and either stabilization of tubulin by taxol or destabilization by nocodazole reduces HSV-1 replication and productivity (H. Nakashima and E.A. Chiocca, unpublished observations) (52). Therefore, MT acetylation status may affect the spatial distribution of cellular and viral proteins required for an optimal HSV-1 infection. Our data show that pharmacological inhibition of HDAC6 resulted in more efficient oHSV infectivity than HDAC6 knockdown (compare Figures 1 and 2). Inhibition of HDAC6 enzyme activity may affect MT dynamics by promoting the interaction of HDAC6 with tubulin and its associated proteins (53). HDAC6 is unique among all HDACs, because it can bind ubiquitin and Ub-like proteins via a BUZ domain, and this interaction facilitates functions related to autophagy and to the aggresome (54). HDAC6 inhibitors do not interfere with such protein-protein interactions. Furthermore, we recently reported that HDAC6 can interact with Ub-like ISG15 for lysosomal degradation and tubacin also reduced ISG15 expression in response to IFN treatment in glioma cells (55). Therefore, HDAC6 inhibitors may also support HSV-1 infection by changing subcellular localizations and protein interaction networks of HDAC6-associating proteins. However, the HDAC6-mediated aggresome pathway was recently shown to promote the process of capsid uncoating of influenza A virus (IAV), allowing for release of viral ribonucleoprotein (vRNP) into the nucleus (56): the HDAC6 BUZ domain was shown to play an important role in this process. During HSV-1 infection, the UL36-encoding tegment proteins VP1-2, also known as viral DUB, are proteolytically cleaved at the nuclear pore, triggering a conformational change of the capsid, an essential step for release of the HSV-1 genome (57, 58). Therefore, the nuclear release of IAV vRNP and that of HSV-1 DNA are mechanistically different, and this difference may be a reason for the different outcome that HDAC6 activity has in facilitating versus inhibiting IAV versus HSV-1 replication. However, there may also be other reasons for HDAC6’s different role in IAV and HSV-1 replication kinetics that relate to the differences in IAV versus HSV-1 entry and transport to the nucleus. In fact, HDAC6i seems to enhance trafficking and budding of progeny IAVs (56, 59) as well as trafficking of transfected naked DNA (60, 61).
A possible clinical translation of our findings would be to combine specific pharmacologic HDAC6i with oHSV administration. The rationale for this is that pan-HDAC inhibition may be more toxic due to pleiotropic effects on multiple HDACs. We found that the effect of HDAC6i on a panel of GSCs was variable. Although the reasons for this variability remain to be answered, this should not be too surprising, since it is likely that the degree and magnitude of the HDAC6 effect could depend on the level of intact IFN signaling or the ability to use HDAC6 in endosome/autophagosome fusion or the variability of the trafficking of post-entry oHSVs. These possibilities will require additional experimentation to determine their importance. Nevertheless, the variability of responses does not detract from the finding of a mechanistic link in some tumor cells between HDAC6, IFN signaling, and handling of post-entry oHSV trafficking.
We observed a trend of improved survival in mice with an intracranial GSC treated with oHSV and HDAC6i compared with that of mice treated with oHSV alone, although the difference in survival between these treatment groups did not reach significance, if an α level of 0.05 is used as the cutoff. The P value of 0.0852 shows a trend to significance that would probably be achieved if a larger sample size was used (e.g., a sample size of 24 animals per group will achieve 80% power at a 0.05 significance level to detect a hazard ratio of 0.4 when the proportion surviving in the oHSV treatment group is 0.40 at 30 days, using the log-rank test and 2-sided test). In addition, extensive experimentation may be required to determine if the dose and schedule of TA in vivo were optimal for the intended outcome. Yet, the finding that the tested dose of TA led to significantly increased yields of two different oHSVs in mice with tumors 4 days after injection may indicate that more prolonged administration of the drug would be required to visualize a more significant effect. It is also possible though that HDAC6i by itself may not lead to a sufficient increase in oHSV replication to translate into a significant difference and that additional HDACs would need to be inhibited. In fact, when we used a pan-HDAC inhibitor, such as VPA, there was a highly significant increase in survival of mice with gliomas (28).
In summary, these findings indicate that HDAC6 activity is a factor in the intrinsic intracellular defense of some tumor cells against virotherapy. In light of multiple clinical trials of OVs against cancers, this may be highly relevant to improve treatment efficacy.
Methods
Reagents.
Human IFN-β–1a was obtained from PBL Assay Science; VPA was obtained from Sigma-Aldrich; tubacin used in Figure 1, A–C, Figure 4, and Figure 5 was a gift from Stuart Schreiber (Broad Institute of MIT and Harvard, Boston, Massachusetts, USA) and tubacin used in Figure 1, D–F, and Table 2 was obtained from MedKoo Biosciences; TA was obtained from BioVision Inc.; and CI994 was obtained from Selleck Chemicals. Human IgG was obtained from Talecris Biotherapeutics Inc.
Human specimens.
For each patient, samples of both tumor and brain devoid of gross tumor were resected, aliquoted, and processed for either extraction of total RNA (TRIzol, Invitrogen) or isolation and establishment of patient-derived GSCs (62).
Cell culture.
African green monkey Vero kidney cells (and the derivative 7b cell line) (63), human U251 glioma cells (28) and their derivative cell lines, human Gli36 glioma cells and their derivative cell lines (G16-9, Gli36ΔEGFR), and U87ΔEGFR and U373 cells were cultured on adhesive culture dishes containing DMEM (Invitrogen) supplemented with 2% or 10% FBS (Sigma-Aldrich), 100 μg/ml penicillin/streptomycin (Invitrogen), and 10 mM HEPES (Invitrogen) at 37°C in a humidified incubator at 5% CO2. For passage, trypsin (Invitrogen) was used as a dissociation reagent. Primary GSCs were maintained as nonadhesive spheroids in flasks containing neurobasal medium supplemented with B27 (Invitrogen), 100 μg/ml penicillin/streptomycin, GlutaMAX (Invitrogen), and 50 μg/ml of both human EGF and FGF-2 (both from R&D Systems). Spheres were dissociated using TrypLE (Invitrogen).
DNA constructs.
Human MEC17 cDNA (BC047303) was obtained from a mammalian gene collection cDNA library and amplified by PCR (forward: 5′-AGATCTGTCGACATGTGGTTGACCTGGCCTTTCTG-3′; reverse: 5′-GGATCCGCGGCCGCTCACCAAGGCCTGGTGCTGCGACGTT-3′) in the pCR4Blunt-TOPO vector (Life Technologies), followed by engineering a site to join the BglII/NotI fragment to the SalI site of pCMV-Myc (Sigma-Aldrich) with a Myc-epitope tag at the N-terminus (called pCMV-myc–human MEC17). HDAC6 flag (plasmid #13823), pmCherry_a_tubulin_IRES_puro2 (plasmid#21043), and LAMP1-RFP (plasmid #1817) were obtained from Addgene. The K40Q and K40R mutants of α-tubulin were generated using the QuikChange XL Site-Directed mutagenesis Kit (Agilent Technologies), following the manufacturer’s protocol. The correct identity of all mutant constructs was verified by DNA sequencing.
Gene transfer and knockdown.
Gene transfer was performed using Lipofectamine (Life Technologies), following the manufacturer’s protocol. To isolate stably expressing transfectants, cells were treated with G418/geneticin (500 μg/ml, Life technologies), and individual G418-resistant clones (for LAMP1-RFP and mCherry–α-tubulin cells) were selected, followed by analysis using fluorescence microscopy (Nikon). To knockdown the HDAC6 gene in U251 cells, we used the pGIPZ vector containing HDAC6-targeting sequences (V2LHS_71187 for clone 1 and V2LHS_71188 for clone 2 in Figure 2A, OpenBiosystems). Lentiviral vectors (including control shRNA vectors) were packaged in 293FT cells. Infected cells were cultured in the presence of puromycin (2 μg/ml, Sigma-Aldrich) prior to use.
HSV-1 viruses and titration.
K26GFP virus was obtained from P. Desai (Johns Hopkins University School of Medicine, Baltimore, Maryland, USA) (34). rQNestin34.5, rHSVQ1, and KNE viruses used in this study have been reported previously (26, 64). In brief, rQNestin34.5 recombinant virus contains a deletion of the UL39 gene (encoding ICP6) and both copies of the γ134.5 genes, with the addition of one copy of γ134.5 gene under the control of nestin promoter inserted into the deleted UL39 locus. KNE virus contains a mutated form of glycoprotein D, with residues 2 to 24 deleted and a single amino acid change at residue 38 (Y38C) in the KOS strain that eliminates entry via HVEM and nectin-1, with insertion of a single-chain antibody (scFv) directed against EGFR in the deleted position. Titration assays followed a standard plaque formation assay with minor modifications. Briefly, the virus was serially diluted 1:3 (25 μl into 50 μl DMEM containing 2% FBS) across the 96-well plate toward the end columns, with changing of pipette tips between each well. Vero cells in 100 μl medium were then added at a final density of 20,000 cells in each well, and the plates were incubated at 5% CO2, 34°C, overnight for 20 hours before adding 50 μl of 0.4% human IgG solution diluted in DMEM containing 2% FBS for the next 3 days.
oHSV purification.
All oHSV viral stocks were prepared using the 7b cell line (63) in 5- or 10-layer Nunc Cell Factories (Thermo Fisher Scientific) at a MOI of 0.005 to 0.01 at 37°C in serum-free media (Life Technologies). After the 2-hour adsorption period, media with fetal bovine serum (Sigma-Aldrich) were added to a final concentration of 2%. At 24 hours after infection, cultures were shifted to 33°C and checked daily for the appearance of cytopathic effect (CPE). When cultures reached approximately 80% CPE, the cultures were exposed to an additional 0.45 M in NaCl (Sigma-Aldrich) and 100 μg/ml in dextran sulfate (Sigma-Aldrich) as previously described (65). Twenty-four hours later, the cultures were spun at 660 g in a table-top refrigerated centrifuge (Thermo-Fisher), and the supernatants were purified under GLP-like conditions as previously described (65). Virus was resuspended in D-PBS (Sigma-Aldrich), and sterile glycerol (Sigma-Aldrich) was added to a final concentration of 10%, aliquoted into cryovials (Corning), and stored at –80°C. Titers were determined in triplicate in 7b cells according to standard protocols, and the number of viral genomes was determined by qPCR (65).
Replication-defective HSV-1 amplicon vector.
The engineering and packaging of the pHGCagY-Luc vector, which expresses the firefly luciferase (Fluc) under the control of a CAG promoter and the EGFP reporter gene under the control of a HSV-1 IE4/5 promoter, was previously described (66). Briefly, pHGCagY-Luc plasmid DNA was cotransfected with fHSVΔpac in Vero 2-2 cells using Lipofectamine (Life Technologies). After 3 days, transfected cells were scraped into the medium, the suspension was frozen and thawed 3 times, and the cell debris was removed by centrifugation (10 minutes, 3,000 g). To concentrate the virus, the supernatant was added over the layer of 25% sucrose in HBSS in a centrifuge tube, followed by ultracentrifugation using SW-41 rotor (2.5 hours, 106,000 g). The pellet was dissolved in HBSS, and the solution was frozen by dry ice/ethanol and stored at –80°C until use. Titration was assayed in G16-9 human glioma cells and calculated as GFP transduction unit per ml (TU/ml). Luciferase assays were performed using the Luciferase Assay Kit (Promega), and luciferase activities in each well were measured using a microplate reader (POLARStar Omega, BMG Labtech) to obtain RLU.
Viral infection assay.
Human U251 glioma cells and derivative clones were plated at 500,000 cells per 6-well plate containing DMEM supplemented with 2% FBS (DMEM/2F) the day before infection. In some experiments, cells were treated with VPA, tubacin, or TA for 15 hours prior to the infection, and IFN-β (1,000 units per ml) was added in cultured media for 14 hours before the infection. VPA was removed from media prior to infection because prolonged exposure generated cytotoxicity, while the HDAC6 inhibitors (tubacin and TA) were replenished during infection. On the day of infection, cultured media were replaced with 1 ml of DMEM/2F containing the diluted virus stock at 37°C. For some experiments, the media also contained fresh doses of tubacin or TA. After 1 hour of infection with oHSV or 4 hours of infection with the HSV-1 amplicon at 37°C in a 5% CO2 incubator, cells were washed with acidic glycine saline buffer (pH 3.0) and warmed to 37°C to remove input viruses that failed to enter during the 1-hour absorption period from cell surfaces. Cells were then carefully washed with D-PBS warmed to 37°C before adding 2 ml of DMEM/2F, warmed to 37°C. Cells were scraped into the medium that was collected in 1.5-ml microcentrifuge tubes or 15-ml conical tubes, followed by 3 repeated cycles of freezing in dry ice–ethanol and thawing in a 37°C water bath for virus stock titration.
For patient-derived primary GBM cells, 12-well plates were coated overnight with fibronectin (EMD Millipore, 2.5 μg/ml for GBM19, GBM157, and GBM528), poly-l-lysine (Sigma-Aldrich, 2 μg/ml for GBM83 and GBM1123), or poly-d-lysine (Sigma-Aldrich, 2 μg/ml for GBM326), each diluted in D-PBS. Plates were washed twice with D-PBS before cells were seeded at a density of 500,000 cells per well in 12-well plates in growth medium. Cells were treated with 1 mM VPA, 100 nM tubacin, or 50 nM TA for 15 hours before infection with rQNestin34.5 in 1 ml of growth medium without drug. One hour after infection, cells were washed with medium and kept in growth medium supplemented with the respective HDAC inhibitor. Cells were scraped in their medium 72 hours after infection, subjected to 3 freeze/thaw cycles, and stored at –80°C before titration.
Cytotoxicity assay.
Cells were plated on a 96-well plate the day before infection in the presence of drugs (tubacin, CI996) or vehicle. Serially diluted aliquots of oHSV were prepared in a ultralow attachment 96-well plate and then transferred to the plates with tumor cells. After 3 days, intracellular and extracellular lactate dehydrogenase (LDH) levels were measured using a Pierce LDH Cytotoxicity Assay Kit (Life Technologies), following the manufacture’s protocol. Cytotoxicity was calculated with following formula: percentage cytotoxicity = {(LDH of uninfected cells) – (intracellular + extracellular LDH of sample cells)}/(LDH of uninfected cells) × 100.
Western blot analyses.
Cell lysates were prepared in lysis buffer (50 mM Tris-Cl [pH 7.4], 150 mM NaCl, 2.5 mM EDTA, 0.5% Triton X-100, 40 μM MG132, 5 mM DTT, and protease inhibitor cocktail) and were sonicated before centrifugation at 20,000 g for 10 minutes at 4°C to collect the supernatants. For Western blot, commercially available antibodies were used for the detection of HDAC6 (sc-28386; Santa Cruz), α-tubulin (T6074; Sigma-Aldrich), acetylated α-tubulin (T6793; Sigma-Aldrich), FLAG M2 (F3165; Sigma-Aldrich), and myc 9E10 (11667149001; Roche). ECL anti-mouse or anti-rabbit IgG and HRP-linked whole antibodies (NA931V and NA934V, respectively; GE Healthcare) were used for the detection of primary antibodies. Detection of HRP was performed by incubating blots with ECL Plus reagent (GE Healthcare) and subsequently exposing them to film.
Fluorescence microscopy analysis.
To observe living cells under confocal microscopy (LSM-510META or LSM-710, Zeiss), cells were seeded onto glass-bottom dishes (In Vitro Scientific) containing DMEM/2F, the day before analysis. Infection of K26GFP was performed on ice or in a 4°C refrigerator for 1 hour, and unattached virus particles were washed out using D-PBS, followed by the addition of DMEM warmed to 37°C containing 2% FBS. The dish was then set up in the 5% CO2 chamber on the stage of the enclosure microscope at 37°C. The z-axis stack images were taken every 15 minutes and assembled by ImageJ 1.48 or an earlier version of the ImageJ software (NIH) with the function of Z project (standard deviation type); we started using time stamps after the temperature shift to 37°C. To analyze the colocalization of GFP-VP26 with either LAMP1-RFP or the periphery region of DAPI-stained nuclei, cells were seeded on the 12-well plate containing a German glass round coverslip (#1 thickness) the day before K26GFP infection. At 1.5 h.p.i. (temperature shift from 4°C to 37°C), cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS (pH 7.4) at room temperature, and excess paraformaldehyde was removed using 50 mM NH4Cl for 5 minutes at room temperature. After permeabilization with 50 μg/ml digitonin (EMD), cellular nuclei were stained with DAPI before the coverslips were mounted with VectaShield (Vector Laboratories) on glass slides. Fluorescent images were acquired with a Zeiss LSM510 META or LSM710 confocal microscopy system and processed using ImageJ. For quantitative analysis of colocalizing GFP/RFP/DAPI signals, we fixed the cells at 90 minutes after virus-cell fusion and quantified the number of individual GFP-tagged VP26 capsids present in RFP-LAMP1 lysosomes versus nuclei/perinuclei as a function of IFN-β and/or tubacin treatment. GFP dots were manually counted using each individual z-stack sectioned image, and total colocalized GFP dot numbers in a single cell were summed from whole z-stack images and plotted in Figure 5.
Virus uptake through TEM analysis.
Cells were incubated with rQnestin34.5 at a high MOI (MOI = 50) on ice for 1 hour to facilitate attachment of numerous viruses to single cells for TEM. The temperature of the cultures was then shifted to 37°C for up to 30 minutes to trigger virus uptake. Cell pellets were collected, washed 3 times with PBS, and then fixed in 2.5% glutaraldehyde (catalog 16220, Electron Microscopy Sciences) in cacodylate buffer for 4 hours at 4°C. Cells were then washed 3 times with cacodylate buffer followed by post-fixation in 1% OSO4 (catalog 0972A, Polysciences) in cacodylate buffer for 1 hour at 4°C. After 3 cacodylate buffer washes, the pellets were then dehydrated through graded alcohol, embedded in Epon/Araldite, and polymerized at 60°C overnight. Blocks were then sectioned at 70 to 80 nm and collected on the grids. The grids were first stained with 1% aqueous uranyl acetate (catalog NC0740462, Fisher Scientific) for 12 minutes, followed by a ddH2O rinse. The grids were then stained with Reynolds lead citrate (catalog 17900, Electron Microscopy Sciences) for 5 minutes in a moisture-free chamber. After the rinse with ddH2O, the grids were set to dry before TEM analysis via Hitachi H-7650 SEM/TEM Hitachi S-4800.
Animal experiments.
Female athymic nu/nu mice at 6 to 8 weeks olds were purchased from Charles River or Harlen Laboratories. GBM30 cells (100,000 cells) in 5 μl HBSS buffer were injected intracranially to establish the xenograft tumor in the mouse brain. After 14 days (in Figure 7A) or 5 days (in Figure 7B), 3 μl of oHSV in HBSS buffer was injected intratumorally. TA or vehicle diluted in DMSO was administrated at a dose of 1.3 mg per kg body weight. oHSV types, their administration doses, and TA administration conditions are described in the legend of Figure 7.
Statistics.
One-way ANOVA and pair-wise comparisons with adjusted P values were used for the statistical analyses, using RStudio software run with the R language (R Core Team; http://www.R-project.org) and R package “stats” version 2.15.3. Kaplan-Meier survival curves were compared using the log-rank test, using JMP Pro version 9 (SAS Institute Inc.). P values of less than 0.05 were considered significant.
Study approval.
The protocols for collection of human specimens and use of microbe agents were approved by the IRBs of The Ohio State University and Brigham and Women’s Hospital. Informed consent was acquired from participants who provided specimens. All animal studies were reviewed and approved by the IACUC of The Ohio State University and Harvard Medical School.
Author contributions
HN and EAC developed the concept, designed experiments, and wrote the manuscript. EAC oversaw and supervised all analyses and strategies. HN and TN performed in vitro experiments using cell lines, and JKK and KO performed in vitro experiments using primary gliomas. TN, MCS, and HN conducted an in vivo experiment. PYW performed TEM and flow cytometry analyses, and TPC supervised these analyses and interpretation. AO performed an experiment using HDAC1 knockdown. HN and TN constructed the DNA used in this study. SF supervised statistical data analyses. IN supervised analysis and preparation of primary gliomas. WFG performed analyses of oHSV products. PG and JCG supervised oHSV vectors. KK and SL provided the critical review and technical expertise.
Supplementary Material
Acknowledgments
This work was supported by NCI grant P01CA163205.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(11):4269–4280. doi:10.1172/JCI80713.
Johanna K. Kaufmann’s present address is: Genocea Biosciences Inc., Cambridge, Massachusetts, USA.
References
- 1.Ahluwalia MS, Chang SM. Medical therapy of gliomas. J Neurooncol. 2014;119(3):503–512. doi: 10.1007/s11060-014-1495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostrom QT, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16(7):896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas AA, Brennan CW, DeAngelis LM, Omuro AM. Emerging therapies for glioblastoma. JAMA Neurol. 2014;71(11):1437–1444. doi: 10.1001/jamaneurol.2014.1701. [DOI] [PubMed] [Google Scholar]
- 4.Sturm D, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14(2):92–107. doi: 10.1038/nrc3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reardon DA, et al. Immunotherapy advances for glioblastoma. Neuro Oncol. 1458;16(11):1441–1458. doi: 10.1093/neuonc/nou212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiocca EA, Blair D, Mufson RA. Oncolytic viruses targeting tumor stem cells. Cancer Res. 2014;74(13):3396–3398. doi: 10.1158/0008-5472.CAN-14-0290. [DOI] [PubMed] [Google Scholar]
- 7.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2(4):295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol. 2014;16(3):334–351. doi: 10.1093/neuonc/not310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiocca EA. The host response to cancer virotherapy. Curr Opin Mol Ther. 2008;10(1):38–45. [PubMed] [Google Scholar]
- 10.Kurozumi K, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst. 2007;99(23):1768–1781. doi: 10.1093/jnci/djm229. [DOI] [PubMed] [Google Scholar]
- 11.Donnelly O, Harrington K, Melcher A, Pandha H. Live viruses to treat cancer. J R Soc Med. 2013;106(8):310–314. doi: 10.1177/0141076813494196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garner JA. Herpes simplex virion entry into and intracellular transport within mammalian cells. Adv Drug Deliv Rev. 2003;55(11):1497–1513. doi: 10.1016/j.addr.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Heldwein EE, Krummenacher C. Entry of herpesviruses into mammalian cells. Cell Mol Life Sci. 2008;65(11):1653–1668. doi: 10.1007/s00018-008-7570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stiles KM, Krummenacher C. Glycoprotein D actively induces rapid internalization of two nectin-1 isoforms during herpes simplex virus entry. Virology. 2010;399(1):109–119. doi: 10.1016/j.virol.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brencicova E, Diebold SS. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front Cell Infect Microbiol. 2013;3: doi: 10.3389/fcimb.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janke C. The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol. 2014;206(4):461–472. doi: 10.1083/jcb.201406055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6(17):2152–2160. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]
- 18.Matthias P, Yoshida M, Khochbin S. HDAC6 a new cellular stress surveillance factor. Cell Cycle. 2008;7(1):7–10. doi: 10.4161/cc.7.1.5186. [DOI] [PubMed] [Google Scholar]
- 19.Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18(6):291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Hubbert C, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 21.Gao YS, Hubbert CC, Yao TP. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem. 2010;285(15):11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mak AB, et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012;2(4):951–963. doi: 10.1016/j.celrep.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34(3):259–269. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 24.Nusinzon I, Horvath CM. Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol Cell Biol. 2006;26(8):3106–3113. doi: 10.1128/MCB.26.8.3106-3113.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valenzuela-Fernandez A, et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol Biol Cell. 2005;16(11):5445–5454. doi: 10.1091/mbc.E05-04-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34. Cancer Res. 2005;65(7):2832–2839. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- 27.Kambara H, Saeki Y, Chiocca EA. Cyclophosphamide allows for in vivo dose reduction of a potent oncolytic virus. Cancer Res. 2005;65(24):11255–11258. doi: 10.1158/0008-5472.CAN-05-2278. [DOI] [PubMed] [Google Scholar]
- 28.Otsuki A, et al. Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses. Mol Ther. 2008;16(9):1546–1555. doi: 10.1038/mt.2008.155. [DOI] [PubMed] [Google Scholar]
- 29.Poon AP, Liang Y, Roizman B. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. J Virol. 2003;77(23):12671–12678. doi: 10.1128/JVI.77.23.12671-12678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu TC, Castelo-Branco P, Rabkin SD, Martuza RL. Trichostatin A and oncolytic HSV combination therapy shows enhanced antitumoral and antiangiogenic effects. Mol Ther. 2008;16(6):1041–1047. doi: 10.1038/mt.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen TL, et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A. 2008;105(39):14981–14986. doi: 10.1073/pnas.0803988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100(8):4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akella JS, et al. MEC-17 is an α-tubulin acetyltransferase. Nature. 2010;467(7312):218–222. doi: 10.1038/nature09324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Desai P, Person S. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J Virol. 1998;72(9):7563–7568. doi: 10.1128/jvi.72.9.7563-7568.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang PY, et al. Expression of HSV-1 receptors in EBV-associated lymphoproliferative disease determines susceptibility to oncolytic HSV. Gene Ther. 2013;20(7):761–769. doi: 10.1038/gt.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuller AO, Spear PG. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc Natl Acad Sci U S A. 1987;84(15):5454–5458. doi: 10.1073/pnas.84.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mao P, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110(21):8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132(31):10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White MC, Frampton AR., Jr The histone deacetylase inhibitor valproic acid enhances equine herpesvirus type 1 (EHV-1)-mediated oncolysis of human glioma cells. Cancer Gene Ther. 2013;20(2):88–93. doi: 10.1038/cgt.2012.89. [DOI] [PubMed] [Google Scholar]
- 40.Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19(9):1049–1066. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107(4):600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cody JJ, Markert JM, Hurst DR. Histone deacetylase inhibitors improve the replication of oncolytic herpes simplex virus in breast cancer cells. PLoS One. 2014;9(3): doi: 10.1371/journal.pone.0092919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DR, et al. Combination therapy of conditionally replicating adenovirus and histone deacetylase inhibitors. Int J Mol Med. 2012;29(2):218–224. doi: 10.3892/ijmm.2011.831. [DOI] [PubMed] [Google Scholar]
- 44.Lee JY, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29(5):969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol. 2005;79(11):6655–6663. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nicola AV, Straus SE. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol. 2004;78(14):7508–7517. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koyama AH, Uchida T. The mode of entry of herpes simplex virus type 1 into Vero cells. Microbiol Immunol. 1987;31(2):123–130. doi: 10.1111/j.1348-0421.1987.tb03075.x. [DOI] [PubMed] [Google Scholar]
- 48.Nicola AV, McEvoy AM, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol. 2003;77(9):5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyman MG, Enquist LW. Herpesvirus interactions with the host cytoskeleton. J Virol. 2009;83(5):2058–2066. doi: 10.1128/JVI.01718-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elliott G, O’Hare P. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J Virol. 1998;72(8):6448–6455. doi: 10.1128/jvi.72.8.6448-6455.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yedowitz JC, Kotsakis A, Schlegel EF, Blaho JA. Nuclear localizations of the herpes simplex virus type 1 tegument proteins VP13/14, vhs, and VP16 precede VP22-dependent microtubule reorganization and VP22 nuclear import. J Virol. 2005;79(8):4730–4743. doi: 10.1128/JVI.79.8.4730-4743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kotsakis A, Pomeranz LE, Blouin A, Blaho JA. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J Virol. 2001;75(18):8697–8711. doi: 10.1128/JVI.75.18.8697-8711.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zilberman Y, Ballestrem C, Carramusa L, Mazitschek R, Khochbin S, Bershadsky A. Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J Cell Sci. 2009;122(pt 19):3531–3541. doi: 10.1242/jcs.046813. [DOI] [PubMed] [Google Scholar]
- 54.Kovacs JJ, Hubbert C, Yao TP. The HDAC complex and cytoskeleton. Novartis Found Symp. 2004;259:170–177. doi: 10.1002/0470862637.ch12. [DOI] [PubMed] [Google Scholar]
- 55.Nakashima H, Nguyen T, Goins WF, Chiocca EA. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J Biol Chem. 2015;290(3):1485–1495. doi: 10.1074/jbc.M114.593871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banerjee I, et al. Influenza A virus uses the aggresome processing machinery for host cell entry. Science. 2014;346(6208):473–477. doi: 10.1126/science.1257037. [DOI] [PubMed] [Google Scholar]
- 57.Abaitua F, Hollinshead M, Bolstad M, Crump CM, O’Hare P. A Nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J Virol. 2012;86(17):8998–9014. doi: 10.1128/JVI.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jovasevic V, Liang L, Roizman B. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J Virol. 2008;82(7):3311–3319. doi: 10.1128/JVI.01919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Husain M, Cheung CY. Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J Virol. 2014;88(19):11229–11239. doi: 10.1128/JVI.00727-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vaughan EE, et al. Microtubule acetylation through HDAC6 inhibition results in increased transfection efficiency. Mol Ther. 2008;16(11):1841–1847. doi: 10.1038/mt.2008.190. [DOI] [PubMed] [Google Scholar]
- 61.Barua S, Rege K. The influence of mediators of intracellular trafficking on transgene expression efficacy of polymer-plasmid DNA complexes. Biomaterials. 2010;31(22):5894–5902. doi: 10.1016/j.biomaterials.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 62.Peruzzi P, et al. MicroRNA-128 coordinately targets Polycomb Repressor Complexes in glioma stem cells. Neuro Oncol. 2013;15(9):1212–1224. doi: 10.1093/neuonc/not055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krisky DM, et al. Deletion of multiple immediate-early genes from herpes simplex virus reduces cytotoxicity and permits long-term gene expression in neurons. Gene Ther. 1998;5(12):1593–1603. doi: 10.1038/sj.gt.3300766. [DOI] [PubMed] [Google Scholar]
- 64.Uchida H, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther. 2013;21(3):561–569. doi: 10.1038/mt.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goins WF, Huang S, Cohen JB, Glorioso JC. Engineering HSV-1 vectors for gene therapy. Methods Mol Biol. 2014;1144:63–79. doi: 10.1007/978-1-4939-0428-0_5. [DOI] [PubMed] [Google Scholar]
- 66.Saeki Y, Fraefel C, Ichikawa T, Breakefield XO, Chiocca EA. Improved helper virus-free packaging system for HSV amplicon vectors using an ICP27-deleted, oversized HSV-1 DNA in a bacterial artificial chromosome. Mol Ther. 2001;3(4):591–601. doi: 10.1006/mthe.2001.0294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.