

Abstract
In the clinical trial process, precise and concise data collection at the source is imperative and requires statistical analysis to be performed to derive the primary and secondary endpoints. The quality of raw data collection has a direct impact on the statistical outputs generated as per the statistical analysis plan. Hence, the data collection tools used for data transcription must be clear, understandable, and precise, which helps the investigator to provide the accurate subject data. Clinical Data Acquisition Standards Harmonization (CDASH) provides guidance to develop the case report form (CRF) for domains that are commonly used for the majority of the clinical trials across the therapeutic areas. This white paper describes the importance of CDASH standards, its advantages and its impact on the efforts and the cost in designing the CRF.
Keywords: Clinical Data Acquisition Standards Harmonization, clinical data interchange standards consortium, data standards, Study Data Tabulation Model
INTRODUCTION
Post the successful completion of laboratory trials, new chemical entities, and new drug formulations are tested in human beings through Phase I–IV studies to assess their safety and efficacy before providing market approval. The study procedures vary from protocol to protocol and prequalified principal investigators conducts the trials at research centers and collect the required data on source documents at multiple visits as per the protocol. The data comprises of subject demographics, habits, medications taken, events, study procedures, test drug usage, etc., These data are called as source data and are a part of hospital records.
The principal investigators transcribe the required trial data onto the case report form (CRF) either in electronic or paper format provided by the sponsor or Clinical Research Organizations (CROs).
The clinical data manager develops the CRF with the help of the statistician, clinical operations team, the medical monitor, and the sponsor before subject enrollment and provides adequate training to the site personnel to complete the CRF as per the requirements.
While designing the CRF, the clinical data manager understands the protocol thoroughly and includes the CRF pages/forms as per the scheduled visits by following sponsor standards or those developed internally. Clinical Data Acquisition Standards Harmonization (CDASH) describes common best practices to develop CRFs instead of following one's own standards. These standards are extensively recommended by regulatory bodies like the Food and Drug Administration (FDA).[1]
CLINICAL DATA ACQUISITION STANDARDS HARMONIZATION
The intent of CDASH is to develop content standards for basic set of global CRF fields. These are global CRF standards that apply for all therapeutic area (TA) across phases.
History
The CDASH standard is part of the clinical data interchange standards consortium (CDISC) initiative. CDSIC initiated work on CDASH standards in October 2006. The teams consisted of clinical data managers, statisticians, medical monitors, and programmers, and they collected data from clinical trial sites with the help of groups like Society of Clinical Data Management, National Cancer Institute, Association of CROs, FDA, CDISC, Critical Path Institute, Cancer Biomedical Informatics Grid, etc., Three International Conference on Harmonization regions – USA, Europe, and Japan have been covered while collecting the data and suggestions. The consolidated first draft has been posted in May 2008 for review.
The CDASH version 1.0 has been released by CDISC in October 2008, subsequently version 1.1 has been posted in January 2011. The version 2.0 is scheduled to be released in 2015.[2]
Clinical Data Acquisition Standards Harmonization standard domains
CDASH classifies different types of data collection fields as:
Highly recommended (HR) - A data collection field that should be on the CRF (e.g. a regulatory requirement)
Recommended/conditional (R/C) - A data collection field that should be on a CRF based on certain conditions (e.g. complete date of birth is preferred but may not be allowed in some regions; AE time should only be captured if there is another data point with which to compare)
Optional (O) - A data collection field that is available for use.[3]
Study Data Tabulation Model (SDTM) has been considered as a base to develop the contents. The latest version CDASH 1.1 describes the contents for 16 standard domains as outlined [Figure 1].[3]
Figure 1.
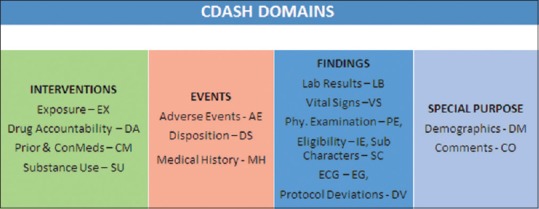
CDASH standard domains
CDASH delivers the field name, prompt, use of the field, guidelines to fill the field, and its recommendation for the 16 domains in the format provided [Table 1].
Table 1.
CDASH domain tables structure

The metadata of CDASH represents the part of Operational Data Model for any regulatory submission.[3]
BENEFITS OF USING CLINICAL DATA ACQUISITION STANDARDS HARMONIZATION STANDARD CASE REPORT FORMS
The design of the database in SDTM is critical for regulatory data submissions. The raw data in SDTM format help in creating the common technical document (CTD) submissions and also helps to create Analysis Dataset Model (ADaM) datasets with minimum programming efforts. As the CDASH standards are a subset of SDTM, initiating the data collection at source smoothens the entire data path, and no redundancy of data collection happens throughout the study.
CDASH highly recommends capturing only key data that are required for statistical analysis. This approach eliminates a few CRF fields; for example, the collection of “yes/no” answers for inclusion/exclusion (IE) criteria CRF. CDASH describes that the IE criteria are at a study level, and it is not necessary to collect this information for each subject. Instead, CDASH recommends collecting only the unmet criterion for which the deviation has been approved by the stakeholders. Figure 2 is the example of nonstandard CRF versus CDASH CRF.[3]
Figure 2.
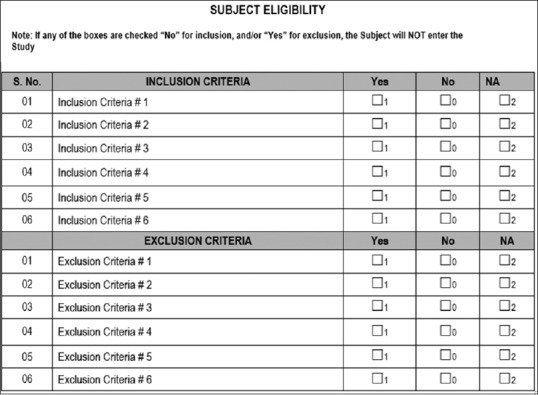
Traditional case report form for inclusion and exclusion criteria

Similarly, CDASH recommends to collect the physical examination test status instead of collecting the data for each body system as any abnormality should be either part of “medical history” or “adverse event” CRF. Thus, CDASH standards avoid duplicate data collection by eliminating some variety of CRFs [Figure 3].
Figure 3.
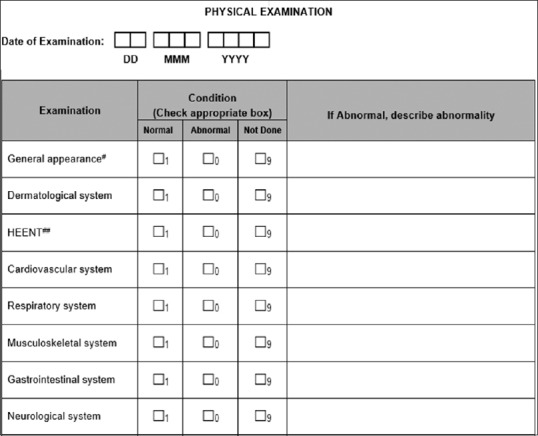
Traditional physical examination page

The above CRF:
Eliminates collection and reconciliation of duplicate data by capturing abnormal data in one central location
Reduces number of queries, thus reducing workload for data managers and site personnel
Supports consistency and standardization for data reporting purposes
Reduces coding needs (if PE abnormalities are coded).
CDASH Standards improve the common understanding across the clinical trial stakeholders and yield better quality data, reduce data queries and facilitate efficient SDTM mapping for regulatory submissions.[4]
Challenges in implementing Clinical Data Acquisition Standards Harmonization standards
The majority of the Clinical Data Management System (CDMS)/Electronic Data Capture (EDC) tools such as Medidata Rave, Oracle Clinical and Oracle Remote Data Capture, inform, and ClinPlus support CDASH electronic CRF development. Sometimes, the implementation may need customized programming, especially while data integration between lab data, electronic patient reported outcome, interactive web response system, and EDC. The requirements vary based on the complexity and the study procedures.[5]
Adapting to CDASH standards for long-term trials that are on-going is a potential challenge. A customized approach is needed to map the data points. For example, shifting mid-ways during the study to CDASH standards on a 5-year trial that is being conducted per sponsor standards is challenging During this migration, a lot of effort would need to be invested in document generation, mapping of fields with currently available fields and various other activities. So it is recommended that the decision on which data standards should be used should be made up front.
Similarly, mapping of legacy trial data to CDASH standards also requires a customized approach.
CLINICAL DATA ACQUISITION STANDARDS HARMONIZATION IMPLEMENTATION: IMPACT ON COST AND ANALYSIS
The accurate and targeted design of simple to use CRFs with clear CRF completion guidelines is crucial as a part of the study start-up activities for a protocol. The draft CRF is reviewed by the study stakeholders including clinical research associates, data managers, medical monitors, and statisticians. The approved CRF is the source for the programmers to configure the CRF in the EDC tools or CDMS tools that are deployed for the study protocol. Usually, it takes 2–3 months to design the CRF and make the database live for data entry.[6]
The CDASH domain CRFs can be used across studies, across the industry with no limitation of TAs. One time efforts in generating the repository of CDASH defined CRFs can be utilized for multiple studies: thus time and efforts are minimized which in turn enables a reduction of the operational cost with respective to study start up activities.
CDASH guidelines specify details of domain and its corresponding variable definitions. These comprise of variable name, definition, instructions to clinical sites, implementation, and core configuration. The core configuration describes whether the variable is highly recommended, recommended/conditional or optional. However, CDASH does not specify length, order, and contents of variables. CDASH does not specify code lists but refers to SDTM terminology, and they recommend terms where code lists are not listed in SDTM Implementation guide.[7]
The impact of Clinical Data Acquisition Standards Harmonization on full-time equivalent utilization and cost: Estimate and analysis
Assume that there are 10 multiple TA studies with 60 CRF pages per study for each subject, of which 25 are unique CRFs. The CRFs and edit check specifications can be finalized in accordance with client specific standards, CRO standards (SOPs) or CDASH standards.
By implementing the CDASH standards, the number of hours spent for CRF development, edit check specifications, programming screens, edits, and SDTM mapping will be reduced. The review efforts on CRFs and completion guidelines by other study stakeholders are also minimized with the CDASH standards implementation. There will be a significant reduction (~60%) in full-time equivalent utilization which in turn reduces the operational cost and improvise the efficiency proportionately. Savings reaped, the cost of implementation and return on investment of standards will vary depending on various factors.
Existing use of proprietary standards
-
Stage of implementation.
- Start-up stage (~70–90% savings)
- Study conduct (~40% savings)
- Analysis and reporting (~50% savings)
- Overall (~60% savings).[8]
Regulatory submission requirements
The reviewers at regulatory bodies analyze the clinical trial data submitted by the pharma and biotech companies. It takes approximately 18 months for the review of nonstandard data submission models. The FDA recommends submitting the data in CDSIC standardized formats. This fastens the review process as the regulators can reuse their programs if the data is present in a standard datasets.[4]
As per the regulatory requirements, the implementation of CDISC standards from the initial stages is highly beneficial for pharma and biotech companies and also helps in the faster review process by FDA.[4]
The CDISC standardization path:
CDASH → SDTM → ADaM → TLFs → CSR → eCTD
CONCLUSION
There are many benefits to be gained if the clinical research industry adapts to data standards especially CDISC standards. Technology companies providing data collection tools should leverage the benefits of data standards and should optimize operational cost. Pharma companies spend more money on following their own standards while developing the CRFs and designing the databases in the EDC/CDMS tools. This approach again needs data conversion techniques for regulatory submissions. As a common goal, it is highly recommended to initiate and implement the CDISC standardization path that provides the benefits of cost, time and high accuracy of data. At the same time, CDASH standards act as a catalyst to reduce the turnaround time for the review process and helps companies to launch drugs in the markets. In the case of orphan drugs and fast track drugs, the implementation of standards is imperative in reducing the time required in bringing the drug to the market.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
I would like to express my sincere thanks to Dr. Nimita Limaye, VP, CDM, Medical Writing and Risk Based Monitoring, Tata Consultancy Services for reviewing the article ad providing guidance and insights.
REFERENCES
- 1.FDA. [Last accessed on 2015 May 10]. Available from: http://www.fda.gov/
- 2.CDISC. [Last accessed on 2015 May 10]. Available from: http://www.cdisc.org/cdash .
- 3.CDASH Version 1.1 Implementation Guide. [Last accessed on 2015 May 10]; [Google Scholar]
- 4.Ben Vali MS. The Importance of CDASH. [Last accessed 2015 Jun 2]. Available from: http://www.cdisc.org/system/files/all/generic/application/pdf/vali_the_importance_of_cdash.pdf .
- 5.Linger RS. Implementing CDASH Standards into Data Collection and Database Design. [Last accessed 2015 Jun 2]. Available from: http://www.portal.cdisc.org/CDISC20User20Networks/North20America/Bay20Area/Presentations/Meeting2010110920-20Implementing20CDASH20Standards20-20Robert20Stemplinger.pdf .
- 6.De Bondt J. CDASH: A Must for EDC Trials. [Last Accessed 2015 Jun 2]. Available from: http://www.sgs.com/~/media/Global/Documents/Technical%20Documents/SGS-Clinical-CDASH-EN-11.pdf .
- 7.Price J. A Practical Introduction to Clinical Data Acquisition Standards Harmonization (CDASH) [Last Accessed 2015 Jun 2]. Available from: http://www.bioclinica.com/assets/Uploads/a-practical-introduction-to-cdash.pdf .
- 8.Business Case for CDISC Standards: Summary; PhRMAGartner-CDISC Project; September, 2006. [Last accessed 2015 May 10]. Available from: http://www.cdisc.org/system/files/all/article/application/pdf/businesscasesummarywebmar09.pdf .