

Abstract

Pea enation mosaic virus (PEMV)—a plant RNA virus transmitted exclusively by aphids—causes disease in multiple food crops. However, the aphid-virus interactions required for disease transmission are poorly understood. For virus transmission, PEMV binds to a heavily glycosylated receptor aminopeptidase N in the pea aphid gut and is transcytosed across the gut epithelium into the aphid body cavity prior to release in saliva as the aphid feeds. To investigate the role of glycans in PEMV–aphid interactions and explore the possibility of viral control through blocking a glycan interaction, we synthesized insect N-glycan terminal trimannosides by automated solution-phase synthesis. The route features a mannose building block with C-5 ester enforcing a β-linkage, which also provides a site for subsequent chain extension. The resulting insect N-glycan terminal trimannosides with fluorous tags were used in a fluorous microarray to analyze binding with fluorescein isothiocyanate-labeled PEMV; however, no specific binding between the insect glycan and PEMV was detected. To confirm these microarray results, we removed the fluorous tag from the trimannosides for isothermal titration calorimetry studies with unlabeled PEMV. The ITC studies confirmed the microarray results and suggested that this particular glycan–PEMV interaction is not involved in virus uptake and transport through the aphid.
Introduction
Insect transmission of plant viruses results in widespread crop losses. The major insect vectors are aphids (Aphididae; Hemiptera), who transmit nearly 50% of insect-borne plant viruses.1 For aphids to persistently transmit luteovirids, such as pea enation mosaic virus (PEMV), two events are necessary. First, virions ingested with phloem sap bind a receptor for transcytosis across the mid or hindgut epithelium for release into the hemocoel where the virions circulate in a nonpropagative manner.2 A second receptor-mediated transcytosis event occurs at the accessory salivary glands (ASG) from which virus particles are secreted with saliva to inoculate the plant phloem during subsequent feeding and thereby infect a new plant.3 These interactions between aphids and luteovirids needed for viral transmission are mediated by the viral capsid proteins, which are made of one major coat protein (CP, 22 kDa) and one minor coat protein (read-through domain, CP-RTD, 35–55 kDa).4,5 The RTD is not required for virus particle assembly or for uptake of virus from the gut into the aphid hemocoel, but both CP and RTD are essential for aphid transmission6−8 and are the sole determinants of vector specificity.9 The molecular determinants of this host specificity, however, are still unclear. Pea enation mosaic virus (PEMV) provides an ideal model virus for the study of luteovirid–aphid interactions as it is the only luteovirid that is not phloem-limited and is thus mechanically transmissible to plants.10
Although there are several examples of animal viruses using glycan or proteoglycan receptors for binding to host cells,11,12 relatively little is known about the role of glycans in plant virus–aphid interactions. However, evidence for the involvement of glycans in the uptake of pathogens into insect vectors is increasing.13 The malaria parasite Plasmodium has been shown to require midgut proteoglycans for cell invasion in the mosquito vector14 and antibodies targeting D-mannose residues on the mosquito midgut can block Plasmodium development.15 Protein glycosylation in insects consists primarily of high mannose or paucimannose structures, although more complex glycans have been identified.16,17d-Mannose (α) residues are common in several blood-feeding insects that vector human and livestock disease.18 Mannose residues constitute the most abundant glycans in the aphid gut,19 and mannose-binding lectins targeting glycoprotein receptors on the surface of the insect gut exhibit insecticidal activity against a variety of hemipteran pests.19−23 The requirement for luteovirid interaction with receptor proteins on the aphid gut epithelium and the abundance of glycoproteins in the aphid gut lead to the hypothesis that glycans are involved in luteovirid–aphid interactions.
In contrast to animal-infecting viruses, plant viruses are mostly nonenveloped, and the vast majority of nonenveloped viruses do not contain glycoproteins. There is evidence for the presence of glycosylated residues on structural proteins of Potato virus X (Alphaflexiviridae)24 and Plum pox virus (Potyviridae).25 The PEMV coat protein sequence contains five putative N-glycosylation sites predicted using ScanProsite (http://www.expasy.ch/tools/scanprosite/). A previous study on turnip yellows virus (TuYV, Luteoviridae) showed that deglycosylation of virions diminished aphid transmission of the virus, suggesting that glycosylated structural proteins could be involved in virus recognition and movement in the aphid.26 However, a later study refuted this idea demonstrating that the structural proteins of TuYV and cucurbit aphid-borne yellow virus (CABYV, Luteoviridae) were not glycosylated and argued against the role of glycosylated viral proteins in aphid transmission.27 The glycosylation of PEMV structural proteins themselves and the ability of these viral surface proteins to bind to aphid-associated carbohydrates has not yet been investigated.
To explore the possible role of glycans in virus–aphid interactions, we needed information on the ability of insect protein-associated glycans to bind to PEMV. A gut receptor for PEMV is aminopeptidase N,23b which is heavily glycosylated and bound by the plant lectin Galanthus nivalis agglutinin (GNA).20N-Linked glycans are found on glycoproteins in all eukaryotes, including insects.28 The most common insect N-glycan structures share a terminal trimannoside structure containing a β-mannoside with two α-mannosides attached to the O-3 and O-6 positions.28b,28c We have previously demonstrated the synthesis of the challenging β-mannopyranoside linkage by combining a β-directing C-5 carboxylate methodology29,30b with fluorous tag (F-tag)-assisted automated solution-phase synthesis.30 Herein, we report the development of the first solution-based automated strategy for synthesis of a β-mannoside-containing insect N-glycan terminal branched trimannoside and assays to test the affinity of this branched trisaccharide to PEMV using both fluorous microarray31 and isothermal titration calorimetry (ITC).
Results and Discussion
To synthesize the insect N-glycan terminal trimannoside by automated solution-phase synthesis, we needed two building blocks: a building block32 to construct the two α-linked termini and a branch point building block capable of primarily making the more challenging β-linkage. To obtain the branch point building block, we designed mannuronate building block 6 (Scheme 1) with a 3-O-p-methoxybenzyl (PMB) group as a temporary capping group for the attachment of the α-mannose at the O-3 position, a C-5 carboxylate as the β-directing group,29 and a temporary blocking group for the 6-position branch point. A hydride reduction of the methyl ester should unmask a free 6-OH for the attachment of the other α-mannose. The synthesis of the building block started from the known 4,6-O-benzylidene acetal-protected thiol-linked mannoside 1,33 which could be synthesized from d-mannose in seven steps. After a selective cleavage of the benzylidene acetal by borane tetrahydrofuran complex (BH3·THF) and dibutylboron triflate (Bu2BOTf),34 compound 2 with a free hydroxyl group at the 6-position was formed. The combination of 2,2,6,6-tetramethyl-1-piperidinyloxy free radical (TEMPO) and (diacetoxyiodo)benzene (BAIB) oxidized primary alcohol 2 to mannuronic acid 3.29a After esterification of the carboxylic acid, the anomeric thiophenyl group of the methyl mannuronate 4 was removed by N-bromosuccinimide (NBS) to give 5 with a free anomeric hydroxyl group.35 The subsequent formation of the trichloroacetimidate at the anomeric hydroxyl provided desired donor 6 ready for the automated solution-phase synthesis.
Scheme 1. Synthesis of the Branch Point Mannose Building Block.

We have previously shown that our automated solution phase oligosaccharide synthesis strategy could successfully construct linear oligomers of β-mannuronate and β-mannan up to hexasaccharide.30 Compared to automated solid-phase oligosaccharide synthesis,29e the automated solution-phase methodology had multiple significant advantages due to its homogeneous reaction conditions: (1) lower loadings of the precious glycosyl donor for each glycosylation cycle, (2) real-time monitoring of the reaction by TLC or other means, and (3) intermediate purification any time during the synthesis sequence with the option for reintroduction of the purified compound for additional automated synthesis cycles.
The automated solution-phase synthesis of the insect glycan (Scheme 2) started from the glycosylation of the allyl C8F17 fluorous tag (F-tag) 13(31a) and donor 6 (3.0 equiv) catalyzed by trimethylsilyl trifluoromethanesulfonate (TMSOTf, 0.1 equiv) at −20 °C. After 45 min, the reaction was quenched by triethylamine (TEA), and the solvent was removed by an evaporation cycle. The PMB group was removed by ceric ammonium nitrate (CAN), and the reaction mixture was purified with fluorous solid-phase extraction (FSPE) to remove nonfluorinated impurities and retain the F-tag-modified product; then, the product was eluted from the FSPE cartridge and transferred out of the synthesis platform for further benchtop purification to isolate the β-mannuronate product from any minor mixtures of anomers. Obtaining pure β-mannuronate at this stage simplified the purification of the final oligosaccharide. A significant advantage of this automated solution-phase approach over more standard solid-phase approaches to biopolymer synthesis is the ability to carry out such off-platform purification protocols prior to the end of the total synthesis without removal of the growing biopolymer chain from the support used for automated purification. By this approach, β-mannuronate analogue 7 was obtained in 78% yield over 2 steps. This fluorous-tagged carbohydrate was then reinjected into the synthesis platform, and the methyl ester was reduced by lithium triethylborohydride (LiTEBH) at 0 °C. After fluorous solid phase extraction (FSPE), the diol was glycosylated with known 2-O-acetyl-mannosyl-trichloroacetimidate donor 8 (6.0 equiv)32 activated by TMSOTf (0.1 equiv) to form two α-mannosidic linkages via neighboring group participation. The crude trisaccharide 9 was transferred out of the synthesis platform; however, the desired trisaccharide was more readily isolated from the mixture after deacetylation under Zemplén conditions. Ultimately, trimannoside 10 was obtained in 50% yield from compound 7 over two automation steps and one benchtop deprotection step.
Scheme 2. Automated Solution-Phase Synthesis of the Insect N-Glycan Terminal Trimannoside.

The final deprotection of the benzyl groups on 10 was facilitated by hydrogenolysis catalyzed by 10% Pd/C under 1000 psi hydrogen at 20 °C for 48 h to provide, in 51% yield, the fully deprotected trimannoside 11 containing a fluorous tag required for adherence of the compound to a fluorous microarray surface31 (Scheme 3). To avoid any possible interactions of the hydrophobic fluorous tag with the viral surface in the measuring of their binding via calorimetry, the additional insect glycan 12 with a short linker to maintain the beta-linkage was made. The synthesis of trimannoside 12 started from the cleavage of the F-tag of 10 by olefin cross-metathesis catalyzed by Grubbs catalyst second generation under ethylene atmosphere to provide the allyl trimannoside. Hydrogenolysis catalyzed by 10% Pd/C and Pd black under a 1000 psi hydrogen atmosphere converted the allyl trimannoside into the fully deprotected n-propyl trimannoside 12 in 91% over two steps for ITC experiments with unlabeled PEMV.
Scheme 3. Deprotection of the Insect N-Glycan Terminal Trimannosides.

With the required insect-associated glycans in hand, microarray experiments were carried out first. The insect N-glycan trimannoside 11 was immobilized using its fluorous tag onto a commercial C8F17-coated glass slide for probing with FITC-labeled PEMV. As a positive control, known F-tag-modified α-mannoside and β-galactoside30a were probed with FITC-labeled mannose binding protein concanavalin A (FITC-ConA). Each of the F-tagged sugars were spotted on the fluorous glass slide by a microarray spotter in a humidified chamber. The resulting slide was then incubated with solutions of FITC-PEMV or FITC-ConA for 1 h. The following rinsing step removed the unbound FITC-PEMV or FITC-ConA; then, the slide was air-dried for fluorescence scanning. From the results of the microarray control experiment of F-tagged α-mannoside, β-galactoside, and 11 incubated with FITC-ConA, the α-mannoside and 11 showed strong binding and the β-galactoside showed no binding as predicted. Interestingly, the FITC-PEMV incubated slide also showed no binding with any of the saccharides, even with elevated FITC-PEMV concentrations (Figure 1, Figure S4).
Figure 1.
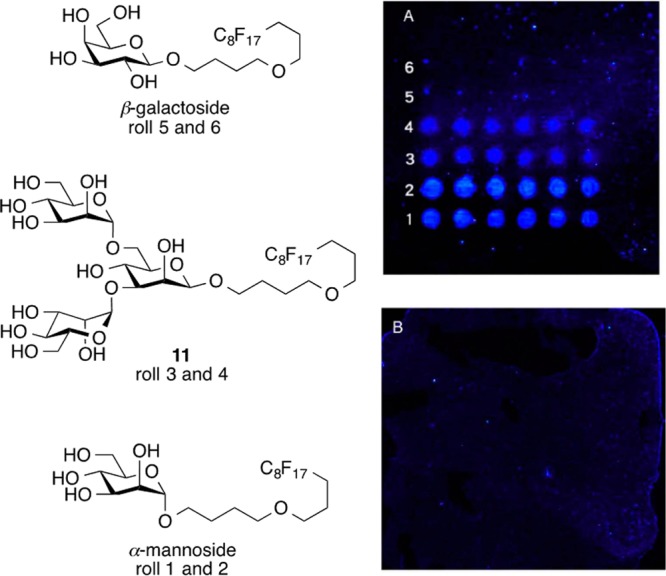
Fluorous microarray of F-tag-attached saccharides incubated with (A) 0.2 μM FITC-ConA or (B) 1.6 μM FITC-PEMV.
To confirm this lack of binding interaction found in the fluorous microarray experiment, we commenced with ITC experiments. n-Propyl trimannoside 12 was used for the ITC experiment with the commercially available methyl α-mannoside as the control group. ConA was the positive control for comparison with the unlabeled PEMV. The ITC experiments involved titration of the ligand (saccharide) solution into the mixing cell with the protein (ConA or PEMV) solution. By comparing the temperature of the mixing cell and the reference cell, the machine inputs an energy pulse to equalize the temperature in the two cells after each injection of the ligand solution. At the end of each titration experiment, Kd, ΔH, ΔS, and N could be extracted from the resulting data. In the ITC experiments with the two saccharides and ConA, there was obvious specific binding (Kd = 4.65 μM for methyl α-mannoside, and Kd = 3.27 μM for 12, Figures S5, S6) as expected based on prior work.36 However, with PEMV, both methyl α-mannoside and 12 did not show any noticeable overall positive binding (Figures S7, S8). This result combined with the fluorous microarray data provides strong evidence that there is no observable specific binding with the insect N-glycan terminal trimannoside and the pea enation mosaic virus. The glycosylation state of the PEMV structural protein was also tested by ITC using ConA. By titrating PEMV into the ConA solution, the result showed weak binding between the PEMV and ConA, which suggests that the PEMV does not contain accessible terminal α-mannoside or α-glucoside moieties (Figure S9).
Conclusions
The first automated solution-phase syntheses of branched N-glycan-related structures using a β-directing mannuronate strategy was successful at producing the insect-associated N-glycan trimannosides 11 and 12. Interestingly, neither fluorous microarray nor ITC experiments showed any evidence for specific binding between the trimannoside motif and the PEMV in a multivalent or monovalent presentation format. This specific case of carbohydrate interaction discussed in this paper is therefore likely not involved in the transmission of PEMV in aphid vectors and, consequently, the development of compounds targeting this interaction for an antiadhesive strategy37 for viral control would be ineffective. In addition, a lectin-binding study with PEMV does not show evidence for accessible terminal α-mannosides or α-glucosides on the viral surface that could be used for interaction with an aphid-associated carbohydrate binding protein. Related studies have now been initiated to test whether the nonglycosylated receptor proteins have any specific binding interaction with PEMV or whether both the peptide and glycan of the receptor are responsible for this interaction.
Experimental Section
General Synthetic Material and Methods
Dichloromethane (CH2Cl2) for glycosylation was distilled from calcium hydride. Tetrahydrofuran (THF) was collected from a solvent purification system prior to the reactions. All other commercial solvents and reagents were reagent grade and used as received without further purification. The reactions were monitored by thin layer chromatography (TLC) with 250 μm silica gel TLC plates. The hydrogenation reaction under 1000 psi hydrogen was operated in a high pressure reactor. The developed TLC plates were visualized by stain with p-anisaldehyde solution followed by heating on a hot plate. Flash column chromatography was performed with silica gel (40–63 μm particle size). The automated solution phase synthesis was performed in the automated synthesis platform with a hood, 16 reactor vials (13 mL capacity each), and a heating/cooling unit (200 °C to −20 °C) machined to hold the SPE cartridges at the Iowa State University Machine Shop. 1H and 13C NMR spectra were obtained at 400 and 100 MHz and also at 600 and 150 MHz. The C–H coupling constants were measured by the coupled 13C NMR spectra. Chemical shifts (δ) were reported in parts per million (ppm) relative to CDCl3 and CD3OD as internal references. Mass spectra were obtained on a triple quadrupole mass spectrometer fitted with an ESI interface. HPLC traces were obtained from an HPLC system using a 3.9 × 150 mm silica column (4 μm particle size).
General Procedure for Automated Solution Phase Synthesis of Oligosaccharides (Figure S1)
Sample Preparation
Mannuronate donor building block (0.20 g, 0.30 mmol) was dissolved in anhydrous CH2Cl2 (1.6 mL) in the 13 mL vial and placed in the inert reagent rack under an argon atmosphere. A 0.055 M TMSOTf solution (5.0 mL) in anhydrous CH2Cl2 was prepared in an 8 mL vial and placed as indicated in Figure S1 on the inert reagent rack under an argon atmosphere. Acetonitrile (MeCN, 100 mL) was placed in a stock solution bottle at the stock solution station. Toluene (1.0 L) in a stock solution bottle was placed at the reservoir bottle rack with tubing as a reservoir solution for rinsing. Anhydrous CH2Cl2 (20 mL) in the 50 mL vial was placed on the inert reagent rack under an argon atmosphere. Anhydrous THF (10 mL) in the 50 mL vial was placed on the inert reagent rack under an argon atmosphere. An 80% methanol/water (100 mL) stock was prepared in the 100 mL vial and placed on the inert reagent rack. The fluorous-tagged acceptor molecule (50 μmol) was dissolved with anhydrous CH2Cl2 (0.8 mL) in a Wheaton 8 mL E-Z extraction vial (conically bottomed) and flushed with argon, capped with a septa, and placed on the reagent rack. Methanol (MeOH, 8.0 mL) was transferred to an 8 mL-vial capped with septa and placed at the reagent rack. TEA (5.0 mL) was added to an 8 mL vial capped with a septa and placed on the reagent rack. DMSO (8.0 mL) was transferred to an 8 mL vial capped with a septa and placed on the reagent rack. A 0.45 M CAN solution in acetonitrile/water (9:1) (5.0 mL) was prepared in an 8 mL vial capped with a septa and placed on the reagent rack. LiTEBH solution in THF (1.0 M, 3 mL) was transferred to an 8 mL vial capped with a septa and placed on the reagent rack. Water (8.0 mL) was added to an 8 mL vial capped with a septa and placed on the reagent rack. A fluorous solid-phase extraction (FSPE) cartridge (2.0 g, 10 cc) was preconditioned with 80% methanol/water and placed on the machined FSPE block. An empty Wheaton 8 mL E-Z extraction vial was placed underneath the FSPE cartridge (see Figure S1 for the rack positions).
Cleaning Cycle
Before introduction of reagents, the reactor vials were cleaned, dried, and flushed with argon by running the cleaning cycle. During the cleaning cycle, each of the 16 reactor vials (13 mL capacity each) was rinsed with toluene (8.0 mL) and methanol (8.0 mL) 3 times. After the solvent was removed, the reactor vials were dried under vacuum and purged with argon for 45 min. Reagent solutions were prepared by azeotropic removal of water from each building block donor with toluene; the resulting building block donors were then dried under high vacuum. After the cleaning cycles were done, the reagents were transferred into their respective reagent vials, which were then placed on the inert condition reagent rack and general atmosphere reagent rack.
Glycosylation
The needle transferred the acceptor molecule (F-tag) solution (0.80 mL) to the reaction vial, followed by the transfer of the donor molecule solution (0.80 mL). The mixture was vortexed under ambient temperature at 800 rpm for 20 min. Then, the reactor vials were cooled to −20 °C during the 60 min wait time by the heat transfer oil with an 800 rpm vortex rate. The TMSOTf solution (0.10 mL) was transferred into the reactor vial at a 200 rpm vortex rate. After each individual transfer, the needle (inside and outside) was rinsed with toluene (2.0 mL) before operating the next task. The reaction mixture was vortexed at 800 rpm for 45 min at 0 °C under an argon atmosphere. After the reaction time, the needle withdrew 20 μL of the solution from the reaction mixture and placed it into the first well of the microtiter plate for thin layer chromatography monitoring. TEA (0.050 mL) was added to the solution for quenching, and the solvent was evaporated under reduced pressure.
PMB Deprotection
To the dried residue after the glycosylation was added the CAN solution (1.0 mL) in the reactor vial. The reaction mixture was vortexed at 800 rpm for 1 h at ambient temperature. After the reaction time, the needle withdrew 20 μL of the solution from the reaction mixture and placed it into the second well of the microtiter plate for thin layer chromatography monitoring.
Fluorous Solid-Phase Extraction (FSPE)
The reaction mixture (1.2 mL) was carried to the FSPE cartridge at the FSPE block and dispensed at a speed of 1.0 mL/s via the 10 mL syringe. Then, 80% methanol (2.0 mL) was used to rinse the empty reactor vial. The 80% methanol solution was removed from the reactor vial and delivered to the FSPE cartridge. The 80% methanol rinsing and transfer was repeated one more time. An additional 80% methanol solution (4.0 mL, repeated 2 times) was used to rinse the FSPE cartridge. During the 80% methanol rinse, the cartridge was positioned at “SPE waste” for the eluted mixture to be disposed. Acetonitrile (MeCN) (2.0 mL, repeated 3 times) was used to wash the FSPE cartridge for eluting the desired compound. During the task, the FSPE cartridge was positioned as “SPE collect” to be placed right above the 8 mL vial for collection of the sample. After the task, the position of the SPE rack was changed to “SPE direct” for the needle to withdraw the collected sample from the conically bottomed vial and to deliver it to the clean reactor vial for the next reaction.
LiTEBH Reduction
Anhydrous THF (1.0 mL) was added to the sample and vortexed at 800 rpm at 0 °C under an argon atmosphere for 30 min. The LiTEBH (0.30 mL, 1.0 M in THF) was added to the reaction solution, and the mixture was vortexed for 30 min at 800 rpm at 0 °C under an argon atmosphere. After the reaction, the needle withdrew 20 μL of the solution from the reaction mixture and placed it into the fourth well of the microtiter plate for thin layer chromatography monitoring. MeOH (0.50 mL) was added to quench the reaction, and the solvent was removed under reduced pressure.
General Procedure for Benchtop Fluorous Solid-Phase Extraction (FSPE)
Crude product (<0.30 g) was dissolved in 1.0 mL dimethyl sulfoxide (DMSO) and loaded onto the 80% MeOH preconditioned 2 g FSPE cartridge. The cartridge was washed with 80% MeOH (4.0 mL × 3 times). Then, the product was eluted with acetone (12 mL). The solvent was removed under reduced pressure to obtain the desired product.
General Materials and Methods for Purification and Labeling of PEMV
Purification of Pea Enation Mosaic Virus
Mechanical infection of pea plants (Pisum sativum) with PEMV was performed as described in Liu et al.38 The method used to purify PEMV from plants was modified from Liu et al.38 Infected pea plants (150 g) were frozen in liquid nitrogen and homogenized in a blender with 0.2 M sodium acetate buffer (pH 6.0) (1 mL/1 g tissue) and an equal volume of chloroform. The homogenized tissue suspension was centrifuged at 3000g for 10 min. The supernatant was transferred to clean tubes and centrifuged at 3000g for 5 min. The supernatant was again transferred to clean tubes and then centrifuged at 17,200g for 2 h. Pellets from this step were saved, and the supernatant was centrifuged at 140,500g for 2.5 h. Virus pellets were soaked in 0.2 M sodium acetate buffer (pH 7.0) overnight at 4 °C and resuspended the following day. After resuspension, the soluble fraction was centrifuged at 145,000g through a 30% sucrose cushion made in the same buffer. The final pellet was washed three times with buffer to remove excess sucrose and resuspended in 0.2 M sodium acetate (pH 7.0). The final virus solution was frozen in liquid nitrogen and stored at −80 °C. The protein concentration of PEMV was determined by densitometric analysis of the Coomassie stained bands with reference to known BSA concentrations resolved by SDS–PAGE.
FITC Labeling of PEMV
Purified PEMV was labeled with fluorescein isothiocyanate (FITC) using a FITC antibody labeling kit following the manufacturer’s protocol. The labeling reaction was completed in 0.2 M sodium acetate buffer (pH 7.0) in place of the recommended 50 mM sodium borate at pH 8.5. This was done to avoid precipitation of the PEMV virions at higher pH. To confirm FITC labeling of PEMV, we separated the virions by SDS-PAGE, and the gel was scanned using a gel imager in the green-excited mode (532 nm).
General Materials and Methods for Fluorous Microarray
Microarrays were printed by a microarray printer equipped with a robotic pin (0.35 mm). The F-tag attached sugar solution with a concentration of 222 μM in MeOH/DMSO/H2O (v:v:v = 1:3.5:1.15) was printed onto the fluorous glass slides. The printed slide was dried in a humid chamber for 20 h and ready for incubation with protein solution. The protein solution was prepared in various concentrations in 1× PBS buffer with 1% bovine serum albumin (BSA). For FITC-ConA, the solution was with 1.0 mg/mL of CaCl2 and 1.0 mg/mL of MnCl2. The incubation was carried out in an incubation chamber for 1 h, and then washed twice with 1× PBS buffer and once with DI water. Then, the slide was dried in air and scanned at the Iowa State University DNA facility using a microarray scanner set at 488 nm.
General Materials and Methods for Isothermal Titration Calorimetry
The ITC experiment was conducted in an ITC calorimeter. The 10 μM protein solution and the ligand solution (300 μM for the saccharides and 100 μM for the PEMV) were both prepared in pH 7.0 NaOAc buffer. For titration experiments with ConA, CaCl2 and MnCl2 (1.0 mg/mL) were added to the buffer for both ConA and ligand solutions. The solutions were degassed prior to the experiment, and the titration was carried out at 26 °C with a 310 rpm stirring rate.
Synthetic Procedures
Phenyl 2,4-Di-O-benzyl-3-O-p-methoxybenzyl-thio-α-d-mannopyranoside (2)
To the phenyl 2-O-benzyl-3-O-p-methoxybenzyl-4,6-O-benzylidene-thio-α-d-mannopyranoside (1) (0.80 g, 1.4 mmol) was added a borane tetrahydrofuran complex 1.0 M solution in tetrahydrofuran (14 mL, 14 mmol) under an argon atmosphere at 0 °C, and the solution was stirred until the starting material was dissolved. Then, a dibutylboryl trifluoromethanesulfonate 1.0 M solution in CH2Cl2 (1.7 mL, 1.7 mmol) was added dropwise, and the reaction mixture was stirred under argon atmosphere at 0 °C for 3 h. Triethylamine (0.30 mL, 2.15 mmol) was added dropwise to the reaction; then, methanol was added slowly to quench the reaction at 0 °C. The solvent was removed under reduced pressure, and the crude mixture was coevaporated with methanol twice. The crude product was purified by flash column chromatography on silica gel using EtOAc/petroleum ether (1:2) as eluent. The product was obtained as a colorless syrup (0.69 g, 1.20 mmol, 86%). Rf = 0.38 (EtOAc/petroleum ether: 1:2). 1H NMR (CDCl3, 600 MHz): δ 7.40–7.26 (m, 17H, Harom), 6.88–6.87 (m, 2H, Harom), 5.50 (d, 1H, J = 1.6 Hz, H-1), 4.96 (d, 1H, J = 10.8 Hz, CHHPh), 4.69–4.64 (m, 2H, CHHPh), 4.57 (s, 2H, CHHPh), 4.12 (m, 1H, H-5), 4.03 (t, 1H, J = 9.2 Hz, H-4), 3.97 (dd, 1H, J = 2.8, 1.6 Hz, H-2), 3.89 (dd, 1H, J = 9.2, 3.2 Hz, H-3), 3.82 (s, 3H, CO2CH3), 3.81–3.75 (m, 2H, 2 × H-6). 13C NMR (CDCl3, 150 MHz): δ 159.4, 138.5, 138.0, 134.1, 131.9, 130.6, 130.3, 129.6, 129.4, 129.2, 128.5, 128.1, 128.0, 127.9, 127.9, 127.9, 127.7, 114.0, 86.1, 79.8, 76.6, 76.6, 75.3, 74.8, 73.4, 72.4, 72.0, 62.2, 55.4. HRMS (ESI): [M + Na]+ calcd for C34H36NaO6S+, 595.2125; found, 595.2119.
Phenyl 2,4-Di-O-benzyl-3-O-p-methoxybenzyl-thio-α-d-mannopyranosiduronic Acid (3)
To a solution of phenyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-thio-α-d-mannopyranoside 2 (0.50 g, 0.87 mmol) in dichloromethane/water (5.8 mL/2.9 mL) were added TEMPO (0.030 g, 0.19 mmol) and (diacetoxyiodo)benzene (0.70 g, 2.17 mmol), and the solution was stirred at ambient temperature. After 45 min, the mixture was diluted with dichloromethane (10 mL) and washed with a 10% Na2S2O3 solution (10 mL) and water (10 mL). The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography on silica gel using EtOAc/petroleum ether (1:2 → 1:0) as eluent. The product was obtained as a light yellow syrup (0.34 g, 0.57 mmol, 66%). Rf = 0.1 (EtOAc/petroleum ether: 1:2). 1H NMR (CDCl3, 600 MHz): δ 7.52 (d, 2H, J = 6.6 Hz, Harom), 7.33–7.24 (m, 13H, Harom), 7.19 (d, 2H, J = 8.4 Hz, Harom), 6.83 (d, 2H, J = 9.0 Hz, Harom), 5.67 (d, 1H, J = 4.8 Hz, H-1), 4.69–4.65 (m, 3H, CHHPh), 4.64 (d, 1H, J = 6.6 Hz, H-5), 4.52 (m, 3H, CHHPh), 4.21 (t, 1H, J = 6.6 Hz, H-4), 3.88 (dd, 1H, J = 4.8, 2.4 Hz, H-2), 3.82 (m, 1H, H-3), 3.78 (s, 3H, CO2CH3). 13C NMR (CDCl3, 150 MHz): δ 174.7, 159.3, 137.8, 137.7, 133.8, 131.4, 129.7, 129.6, 129.0, 128.4, 128.4, 128.4, 128.4, 128.1, 127.9, 127.9, 127.8, 113.8, 75.7, 72.6, 72.3, 72.0, 55.3. HRMS (ESI): [M + Na]+ calcd for C34H34NaO7S+, 609.1917; found, 609.1904.
Methyl (Phenyl 2,4-di-O-benzyl-3-O-p-methoxylbenzyl-thio-α-D-mannopyranoside)uronate (4)
To a solution of phenyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-thio-α-d-mannopyranosiduronic acid 3 (0.50 g, 0.85 mmol) in anhydrous DMF (4.0 mL) were added K2CO3 (0.12 g, 0.85 mmol) and iodomethane (0.30 g, 2.13 mmol). The reaction mixture was stirred at ambient temperature under argon atmosphere for 6 h. The mixture was diluted with EtOAc (10 mL) and washed with water (10 mL). The aqueous portion was separated and extracted with EtOAc (10 × 2 mL). The combined organic layer was dried over Na2SO4. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography on silica gel using EtOAc/petroleum ether (1:3) as eluent. The product was obtained as a light yellow syrup (0.47 g, 0.78 mmol, 92%). 1H NMR (CDCl3, 600 MHz): δ 7.52 (d, 2H, J = 7.2 Hz, Harom), 7.35–7.25 (m, 13H, Harom), 7.19 (d, 2H, J = 7.8 Hz, Harom), 6.83 (d, 2H, J = 8.4 Hz, Harom), 5.67 (d, 1H, J = 4.8 Hz, H-1), 4.66–4.62 (m, 4H, CHHPh, H-5), 4.52–4.46 (m, 3H, CHHPh), 4.21 (t, 1H, J = 7.2 Hz, H-4), 3.88 (dd, 1H, J = 4.8, 3.0 Hz, H-2), 3.81 (m, 1H, H-3), 3.78 (s, 3H, OCH3). 13C NMR (CDCl3, 150 MHz): δ 169.7, 159.4, 138.1, 137.9, 134.0, 131.5, 130.0, 129.6, 129.6, 129.0, 128.5, 128.5, 128.1, 127.9, 127.9, 127.8, 127.3, 113.8, 76.0, 73.1, 72.4, 72.2, 55.3, 52.3. HRMS (ESI): [M + Na]+ calcd for C35H36NaO7S+, 623.2074; found, 623.2071.
Methyl (2,4-Di-O-benzyl-3-O-p-methoxylbenzyl-α-d-mannopyranose)uronate (5)
To a solution of methyl (phenyl 2,4-di-O-benzyl-3-O-p-methoxylbenzyl-thio-α-d -mannopyranoside) uronate 4 (0.50 g, 0.83 mmol) in 10% water/acetone (10 mL) was added N-bromosuccinimide (0.44 g, 2.49 mmol). The reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was diluted with EtOAc (30 mL) and washed with a saturated NaHCO3 solution (30 mL). The organic layer was dried over Na2SO4. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography on silica gel using EtOAc/petroleum ether (2:3) as eluent. The product was obtained as a pale yellow syrup (0.38 g, 0.75 mmol, 90%). Rf = 0.24 (EtOAc/petroleum ether, 1:2). 1H NMR (CDCl3, 400 MHz): δ 7.36–7.20 (m, 12H, Harom), 6.85 (d, 2H, J = 8.4 Hz, Harom), 5.43 (t, 1H, J = 4.8 Hz, H-1), 4.75 (d, 1H, J = 12.0 Hz, CHHPh), 4.69 (m, 3H, CHHPh), 4.51 (s. 2H, CHHPh), 4.47 (d, 2H, J = 6.4 Hz, H-5), 4.22 (t, 1H, J = 6.8 Hz, H-4), 3.91 (dd, 1H, J = 7.2, 2.8 Hz, H-3), 3.80 (s, 3H, CO2CH3), 3.71 (dd, 1H, J = 4.4, 2.8 Hz, H-2), 3.65 (s, 3H, OCH3), 3.18 (d, 1H, J = 4.8 Hz, OH). 13C NMR (CDCl3, 100 MHz): δ 170.3, 159.3, 138.4, 138.2, 130.3, 129.5, 129.5, 128.5, 128.5, 128.5, 128.4, 128.0, 127.9, 127.7, 113.8, 93.0 (JC1–H1 = 168.8 Hz, C-1), 75.9, 75.5, 74.0, 72.9, 72.4, 72.2, 55.4, 52.4. HRMS (ESI): [M + Na]+ calcd for C29H32NaO8+, 531.1989; found, 531.1985.
Methyl (2,4-Di-O-benzyl-3-O-p-methoxylbenzyl-α/β-d-mannopyranose)uronate Trichloroacetimidate (6)
To a solution of methyl (2,4-di-O-benzyl-3-O-p-methoxylbenzyl-α-d-mannopyranose)uronate (5) (0.50 g, 0.98 mmol) in dichloromethane (35 mL) was added trichloroacetonitrile (0.85 g, 5.88 mmol) at 0 °C under an argon atmosphere. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (0.030 g, 0.2 mmol) was then added, and the reaction mixture was stirred at 0 °C under argon atmosphere for 3 h. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography on silica gel using EtOAc/petroleum ether/triethylamine (1:2:0.1) as eluent. The product was obtained as a colorless syrup and a mixture of anomers (α/β = 4:1, 0.59 g, 0.93 mmol, 95%). Rf = 0.56 (EtOAc/petroleum ether, 1:2).
1H NMR (CDCl3, 400 MHz): δ 9.32 (s, 1H, HNβ), 8.62 (s, 1H, HNα), 7.41–7.26 (m, 10H, Harom), 7.21 (d, 2H, J = 8.8 Hz, Harom), 7.16 (d, 2H, J = 8.4 Hz, Harom), 6.83 (d, 2 H, J = 8.4 Hz, Harom), 6.40 (d, 1H, J = 2.8 Hz, Hα-1), 5.96 (d, 1H, J = 7.6 Hz, Hβ-1), 4.85–4.74 (m, 3H, CHHPh), 4.65–4.48 (m, 4H, CHHPh, H-5), 4.39 (d, 1H, J = 8.4, Hα-4), 4.28 (t, 1H, J = 8.4 Hz, Hα-3), 4.17 (dd, 1H, J = 4.4, 2.4 Hz, Hβ-3), 3.90–3.87 (m, 1H, Hβ-2), 3.83–3.81 (m, 1H, Hα-2), 3.80 (s, 3H, CO2CH3 3.71 (s, 3H, OCH3), 3.64 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ 169.4, 169.1, 160.4, 159.5, 159.5, 160.0, 138.0, 137.9, 137.2, 130.0, 129.8, 129.4, 128.7, 128.6, 128.6, 128.5, 128.3, 128.2, 128.1, 128.1, 128.0, 128.0, 128.0, 113.9, 113.9, 95.8, 95.2, 90.9, 77.3, 75.6, 75.4, 75.2, 75.0, 74.3, 74.1, 74.0, 73.7, 73.6, 73.6, 73.6, 73.0, 72.7, 72.6, 72.3, 55.4, 52.7, 52.6. HRMS (ESI): [M + Na]+ calcd for C31H32Cl3NNaO8+, 674.1086; found, 674.107.
Methyl (cis-4-(1H,1H,2H,2H,3H,3H-Perfluoroundecyloxy)-2-butenyl-2,4-di-O-benzyl-β-d-mannopyranoside)uronate (7)
The crude product solution was transferred out of the ASW1000 after the FSPE step (10th step) of the first cycle. The solvent was removed under reduced pressure, and the product was purified by flash column chromatography on silica gel using EtOAc/petroleum ether (1:3.5) as eluent. The product was obtained as a colorless syrup (72 mg, 0.078 mmol, 78% over 2 steps). Rf = 0.57 (EtOAc/petroleum ether, 1:2). 1H NMR (CDCl3, 400 MHz): δ 7.35–7.23 (m, 10H, Harom), 5.74 (m, 2H, HC=CH), 4.96 (d, 1H, J = 12.0 Hz, CHHPh), 4.77 (d, 1H, J = 11.2 Hz, CHHPh), 4.63 (d, 1H, J = 1.6 Hz, H-1), 4.61 (d, 1H, J = 12.0 Hz, CHHPh), 4.60 (d, 1H, J = 11.2 Hz, CHHPh), 4.47 (dd, 1H, J = 12.8, 4.8 Hz, O–CHHC=C), 4.24 (dd, 1H, J = 12.8, 6.4 Hz, O–CHHC=C), 4.04 (m, 2H, C=CCH2-O), 3.99 (t, 1H, J = 7.6, H-4), 3.90 (d, 1H, J = 8.0, H-5), 3.81 (d, 1H, J = 2.4, H-2), 3.78 (m, 1H, H-3), 3.72 (s, 3H, CO2CH3), 3.49 (m, 2H, O–CH2CH2), 2.69 (d, 1H, J = 9.6, 4-OH), 2.23 (m, 2H, CH2CF2), 1.88 (m, 2H, O–CH2CH2). 13C NMR (CDCl3, 100 MHz): δ 169.4, 138.2, 138.2, 130.6, 128.7, 128.6, 128.4, 128.3, 128.2, 128.1, 128.1, 128.0, 100.3 (JC1–H1 = 157.7 Hz, C-1), 78.0, 76.0, 74.5, 74.3, 72.3, 69.0, 66.7, 65.1, 52.5, 28.4 (t, JC–F = 22.0 Hz), 21.0. HRMS (ESI): [M + Na]+ calcd for C36H35F17NaO8+, 941.1953; found, 941.1959.
4.6.7. cis-4-(1H,1H,2H,2H,3H,3H-Perfluoroundecyloxy)-2-butenyl-2,4-di-O-benzyl-3,6-di-O-(2-O-acetyl-3,4,6-tri-O-benzyl-α-d-mannopyranosyl)-β-d-mannopyranoside (9)
The crude mixture was transferred out of the synthesis platform after the third cycle (33rd step of automated synthesis), diluted with CH2Cl2 (20 mL), and washed with satd NH4Cl(aq). The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. Then, the crude product was purified by flash chromatography on silica gel EtOAc/petroleum ether (1:3). The disaccharide byproduct could not be separated at this stage, and the crude mixture was carried to the next step without further purification.
Rf = 0.47 (EtOAc/petroleum ether, 1:2). HRMS (ESI): [M + Na]+ calcd for C93H95F17NaO19+, 1861.6088; found, 1861.6118.
cis-4-(1H,1H,2H,2H,3H,3H-Perfluoroundecyloxy)-2-butenyl 2,4-di-O-benzyl-3,6-di-O-(3,4,6-tri-O-benzyl-α-d-mannopyranosyl)-β-d-mannopyranoside (10)
Crude 9 (58 mg) from the automated synthesis was dissolved in MeOH (2.0 mL), and Na (0.50 mg, 0.22 mmol) was added. The mixture was stirred at ambient temperature for 1 h. The reaction was added by Dowex 50WX8 200 acidic resin until the pH paper showed neutral. The solvent was removed under reduced pressure, and the crude product was purified by flash column chromatography on silica gel using EtOAc/petroleum ether (4:5 → 1:0) as eluent. The product was obtained as a colorless syrup (44 mg, 0.025 mmol, 50% over 2 automated synthesis steps and 1 benchtop step).
Rf = 0.34 (EtOAc/petroleum ether, 1:1). COSY and HSQC were used for characterization (see Supporting Information). 1H NMR (CDCl3, 600 MHz): δ 7.41–7.14 (m, 40 H, Harom), 5.67 (m 2H, HC=CH), 5.23 (d, 1H, J = 1.2 Hz, Hα-1), 5.08 (d, 1H, J = 1.2 Hz, Hα-1), 4.99 (d, 1H, J = 12.0 Hz, CHHPh), 4.83 (2 × d, 2H, J = 10.8 Hz, CHHPh), 4.72 (d, 1H, J = 12.6 Hz), 4.66–4.59 (m, 5H, CHHPh), 4.55–4.46 (m, 7H, CHHPh), 4.39 (s, 1H, Hβ-1), 4.35 (dd, 1H, J = 12.6, 4.8 Hz, O–CHHC=C), 4.11 (d, 1H, J = 1.8 Hz, Hα-2), 4.09 (dd, 1H, J = 13.2, 6.6 Hz, O–CHHC=C), 4.02–3.95 (m, 3H, Hα-2, C=CCH2–O), 3.93 (d, 1H, J = 3.0 Hz), 3.88–3.84 (m, 5H), 3.83 (dd, 1H, J = 9.0, 3.6 Hz, Hα-3), 3.79–3.74 (m, 4H), 3.71 (dd, 1H, J = 10.8, 4.2 Hz), 3.65–3.60 (m, 3H), 3.41 (m, 2H, OCH2CH2), 3.33 (m, 1H), 2.33 (d, 1H, J = 1.8 Hz, 2-OH), 2.30 (d, 1H, J = 2.4 Hz, 2-OH), 2.18 (m, 2H, CH2CF2), 1.83 (m, 2H, O–CH2CH2). 13C NMR (CDCl3, 150 MHz): δ 139.2, 138.7, 138.7, 138.5, 138.4, 138.1, 138.0, 130.0, 128.7, 128.6, 128.6, 128.5, 128.4, 128.4, 128.3, 128.1, 128.1, 128.1, 128.0, 128.0, 128.0, 128.0, 127.9, 127.9, 127.9, 127.8, 127.8, 127.7, 127.7, 127.5, 101.8 (JC1–H1 = 165.0 Hz, C-1), 100.8 (JC1–H1 = 151.8 Hz, C-1), 100.2 (JC1–H1 = 170.1 Hz, C-1), 81.2, 80.2, 79.8, 78.1,75.5, 75.3, 75.3, 75.2, 75.0, 74.6, 74.5, 74.4, 73.6, 73.5, 72.4, 72.1, 71.6, 71.4, 69.4, 69.1, 68.9, 68.9, 68.1, 66.8, 66.4, 64.9, 28.4 (t, JC–F = 22.4 Hz), 21.0. HRMS (ESI): [M + Na]+ calcd for C89H91F17NaO17+, 1777.5877; found, 1777.5875.
3-(Perfluorooctyl)propanyloxybutanyl 3,6-Di-O-(α-d-mannopyranosyl)-β-d-mannopyranoside (11)
Cis-4-(1H,1H,2H,2H,3H,3H-perfluoroundecyloxy)-2-butenyl 2,4-di-O-benzyl-3,6-di-O-(3,4,6-tri-O-benzyl-α-d-mannopyranosyl)-β-d-mannopyranoside 10 (44 mg, 0.025 mmol) was dissolved in MeOH (3.0 mL), and 10% Pd/C (10 mg) was added. The mixture was stirred under 1000 psi H2 atmosphere at 20 °C. After 48 h, the mixture was filtered through a short pad of Celite, and the solvent was removed under reduced pressure. The desired product was collected as a white foam (13.2 mg, 0.013 mmol, 51%). COSY was used for characterization (see Supporting Information). 1H NMR (CDCl3, 600 MHz): δ 5.08 (d, 1H, J = 1.2 Hz, H-1), 4.83 (d, 1H, J = 1.2 Hz, H-1), 4.50 (s, 1H, H-1), 4.10 (d, 1H, J = 3.0 Hz, H-2), 3.98 (dd, 1H, J = 3.0, 1.2 Hz, H-2), 3.95–3.91 (m, 2H), 3.88–3.84 (m, 3H), 3.83–3.79 (m, 3H), 3.77 (d, 1H, J = 9.6 Hz), 3.74–3.72 (m, 2H), 3.71–3.67 (m, 2H), 3.65–3.63 (m, 2H), 3.60–3.56 (m, 3H), 3.52 (t, 2H, J = 18.0 Hz, −OCH2CH2CH2CF2), 3.49 (m, 2H, CH2CH2O−), 3.37 (m, 1H), 2.28 (m, 2H, CH2CF2), 1.87 (m, 2H, OCH2CH2CH2CF2), 1.67 (s, 4H, −OCH2CH2CH2CH2O−). 13C NMR (CDCl3, 150 MHz): δ 104.1, 101.8, 101.6, 83.0, 77.0, 75.1, 74.5, 72.8, 72.6, 72.2, 72.2, 72.1, 71.9, 70.6, 70.2, 69.1, 68.8, 67.7, 67.4, 63.2, 63.1, 29.2 (t, JC–F = 21.9 Hz), 27.6, 27.5, 22.1. HRMS (ESI): [M + Na]+ calcd for C33H45F17NaO17+, 1059.2278; found, 1059.2269.
n-Propyl 3,6-Di-O-(α-d-mannopyranosyl)-β-d-mannopyranoside (12)
Cis-4-(1H,1H,2H,2H,3H,3H-perfluoroundecyloxy)-2-butenyl 2,4-di-O-benzyl-3,6-di-O-(3,4,6-tri-O-benzyl-α-d-mannopyranosyl)-β-d-mannopyranoside 10 (55 mg, 0.031 mmol) was dissolved in dry CH2Cl2 (4.0 mL), and Grubbs catalyst second generation (5.4 mg, 6.4 μmol) was added. The solution was stirred at 20 °C, and ethylene gas was bubbled through for 30 min; then, an ethylene balloon was attached, and the mixture was stirred for 24 h. The solvent was evaporated, and the mixture was loaded onto a short silica gel column for purification (CH2Cl2 → EtOAc/petroleum ether, 1:1). The product fraction was collected; the solvent was evaporated, and the crude product was purified by FSPE following the general procedure for benchtop fluorous solid-phase extraction. After the acetone fraction was collected, and the solvent was removed under reduced pressure, the product was dissolved in MeOH (3.0 mL) and 10% Pd/C (25 mg) was added. The mixture was stirred at 20 °C under 1000 psi H2 atmosphere. After 24 h, another portion of Pd black (20 mg) and AcOH (0.30 mL) was added, and the mixture was stirred at 20 °C under 1000 psi H2 atmosphere for an additional 24 h. The mixture was filtered through a small pad of Celite, and the Celite pad was washed with MeOH (10 × 3 mL). After the solvent was removed under reduced pressure, the residue was washed with CH2Cl2 (3 × 1.0 mL), and the fully deprotected product was obtained as a white foam (15.6 mg, 0.028 mmol, 91% over 2 steps). COSY was used for characterization (see Supporting Information). 1H NMR (CD3OD, 600 MHz): δ 5.1 (d, J = 1.2 Hz, 1H, H-1), 4.83 (d, 1H, J = 1.8 Hz, H-1), 4.50 (s, 1H, H-1), 4.10 (d, 1H, J = 3.0 Hz, H-2), 3.98 (dd, 1H, J = 3.0, 1.8 Hz, H-2), 3.95 (dd, J = 10.8, 5.4 Hz), 3.88–3.80 (m, 6H), 3.77–3.71 (m, 3H), 3.70–3.60 (m, 4H), 3.59–3.56 (m, 2H), 3.51 (m, 1H), 3.37 (m, 1H), 1.64 (m, 2H, CH2CH3), 0.96 (t, 3H, J = 7.2 Hz, CH3). 13C NMR (CD3OD, 150 MHz): δ 104.1, 101.8, 101.5, 83.0, 76.9, 75.1, 74.5, 72.7, 72.6, 72.5, 72.2, 72.1, 72.1, 69.0, 68. 7, 67.6, 67.3, 63.2, 63.0, 24.0, 11.0. HRMS (ESI): [M + Na]+ calcd for C21H38NaO16+, 569.2052; found, 569.2040.
Acknowledgments
This work was sponsored in part by grants from the US National Institutes of Health (1R01GM090280) to N.L.P. and by the US Department of Agriculture (USDA AFRI 2008-03996) to B.C.B.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.5b01428.
Automated solution-phase synthesis protocol, microarray results, ITC results, and spectral data (PDF)
Author Present Address
§ S.-L.T.: Energy Biosciences Institute, University of California, Berkeley, CA 94720
Author Present Address
¶ L.B.L.: Douglas Scientific, Alexandria, MN 56308
The authors declare no competing financial interest.
Supplementary Material
References
- Dedryver C. A.; Le Ralec A.; Fabre F. C. R. Biol. 2010, 333, 539–553 10.1016/j.crvi.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Gray F.; Gildow F. E. Annu. Rev. Phytopathol. 2003, 41, 539–566 10.1146/annurev.phyto.41.012203.105815. [DOI] [PubMed] [Google Scholar]
- Brault V.; Uzest M.; Monsion B.; Jacquot E.; Blanc S. C. R. Biol. 2010, 333, 524–538 10.1016/j.crvi.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Mayo M. A.; Ziegler-Graff V. Adv. Virus Res. 1996, 46, 413–460 10.1016/S0065-3527(08)60077-9. [DOI] [PubMed] [Google Scholar]
- Miller W. A.; Liu S.; Beckett R. Mol. Plant Pathol. 2002, 3, 177–183 10.1046/j.1364-3703.2002.00112.x. [DOI] [PubMed] [Google Scholar]
- Reavy B.; Mayo M. A.. Advances in Botanical Research; Academic Press, 2002, Vol. 36, pp 21–46. [Google Scholar]
- Brault V.; van den Heuvel J. F.; Verbeek M.; Ziegler-Graff V.; Reutenauer A.; Herrbach E.; Garaud J. C.; Guilley H.; Richards K.; Jonard G. EMBO J. 1995, 14, 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chay C. A.; Gunasinge U. B.; Dinesh-Kumar S. P.; Miller W. A.; Gray S. M. Virology 1996, 219, 57–65 10.1006/viro.1996.0222. [DOI] [PubMed] [Google Scholar]
- Rochow W. F. Science 1970, 167, 875–878 10.1126/science.167.3919.875. [DOI] [PubMed] [Google Scholar]
- Demler S. A.; de Zoeten G. A.; Adam G.; Harris K. F. Plant Viruses 1996, 5, 303–344 10.1007/978-1-4899-1772-0_12. [DOI] [Google Scholar]
- Schneider-Schaulies J. J. Gen. Virol. 2000, 81, 1413–1429 10.1099/0022-1317-81-6-1413. [DOI] [PubMed] [Google Scholar]
- Haywood A. M. J. Virol. 1994, 68, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinglasan R. R.; Jacobs-Lorena M. Infect. Immun. 2005, 73, 7797–7807 10.1128/IAI.73.12.7797-7807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinglasan R. R.; Alaganan A.; Ghosh A. K.; Saito A.; van Kuppevelt T. H.; Jacobs-Lorena M. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15882–15887 10.1073/pnas.0706340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinglasan R. R.; Fields I.; Shahabuddin M.; Azad M.; Sacci J. B. Jr. Infect. Immun. 2003, 71, 6995–7001 10.1128/IAI.71.12.6995-7001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels K.; Van Damme E. J.; Smagghe G. Arch. Insect Biochem. Physiol. 2010, 73, 193–212 10.1002/arch.20351. [DOI] [PubMed] [Google Scholar]
- Vandenborre G.; Smagghe G.; Ghesquière B.; Menschaert B.; Nagender Rao R.; Gevaert K.; Van Damme E. J. M. PLoS One 2011, 6, e16682. 10.1371/journal.pone.0016682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinglasan R. R.; Valenzuela J. G.; Azad A. F. Insect Biochem. Mol. Biol. 2005, 35, 1–10 10.1016/j.ibmb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Rahbe Y.; Sauvion N.; Febvay G.; Peumans W. J.; Gatehouse A. M. R. Entomol. Exp. Appl. 1995, 76, 143–155 10.1111/j.1570-7458.1995.tb01956.x. [DOI] [Google Scholar]
- Fitches E.; Wiles D.; Douglas A. E.; Hinchliffe G.; Audsley N.; Gatehouse J. A. Insect Biochem. Mol. Biol. 2008, 38, 905–915 10.1016/j.ibmb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Rao K. V.; Rathore K. S.; Hodges T. K.; Fu X.; Stoger X.; Sudhakar D.; Williams S.; Christou P.; Bharathi M.; Bown D. P.; Powell K. S.; Spence J.; Gatehouse A. M.; Gatehouse J. A. Plant J. 1998, 15, 469–477 10.1046/j.1365-313X.1998.00226.x. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S.; Roy A.; Das S. Plant Sci. 2001, 161, 1025–1033 10.1016/S0168-9452(01)00507-6. [DOI] [Google Scholar]
- a Fitches E.; Wiles D.; Douglas A. E.; Hinchliffe G.; Audsley N.; Gatehouse J. A. Insect Biochem. Mol. Biol. 2008, 38, 905–915 10.1016/j.ibmb.2008.07.002. [DOI] [PubMed] [Google Scholar]; b Linz L. B.; Liu S.; Chougule N. P.; Bonning B. C. J. Virol. 2015, n/a. 10.1128/JVI.01479-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratova L. A.; Fedorova N. V.; Dobrov E. N.; Lukashina E. V.; Kharlanov A. N.; Nasonov V. V.; Serebryakova M. V.; Kozlovsky S. V.; Zayakina O. V.; Rodionova N. P. Eur. J. Biochem. 2004, 271, 3136–3145 10.1111/j.1432-1033.2004.04243.x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez M. R.; Camafeita E.; Bonay P.; Mendez E.; Albar J. P.; Garcia J. A. J. Biol. Chem. 2002, 277, 135–140 10.1074/jbc.M106883200. [DOI] [PubMed] [Google Scholar]
- Seddas P.; Boissinot S. Arch. Virol. 2006, 151, 967–984 10.1007/s00705-005-0669-8. [DOI] [PubMed] [Google Scholar]
- Revollon S.; Strub J. M.; Fitchette A. C.; Wiss L.; Gomord V.; Van Dorsselaer A.; Brault V. Virology 2010, 402, 303–314 10.1016/j.virol.2010.03.037. [DOI] [PubMed] [Google Scholar]
- a Schmaltz R. M.; Hanson S. R.; Wong C.-H. Chem. Rev. 2011, 111, 4259–4307 10.1021/cr200113w. [DOI] [PubMed] [Google Scholar]; b Rendić D.; Wilson I. B.; Paschinger K. Croat. Chem. Acta 2008, 81, 7–21. [Google Scholar]; c Shi X.; Jarvis D. L. Curr. Drug Targets 2007, 8, 1116–1125 10.2174/138945007782151360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van den Bos L. J.; Dinkelaar J.; Overkleeft H. S.; van der Marel G. A. J. Am. Chem. Soc. 2006, 128, 13066–13067 10.1021/ja064787q. [DOI] [PubMed] [Google Scholar]; b Codee J. D. C.; van den Bos L. J.; de Jong A.-R.; Dinkelaar J.; Lodder G.; Overkleeft H. S.; van der Marel G. A. J. Org. Chem. 2009, 74, 38–47 10.1021/jo8020192. [DOI] [PubMed] [Google Scholar]; c Dinkelaar J.; de Jong A. R.; van Meer R.; Somers M.; Lodder G.; Overkleeft H. S.; Codee J. D. C.; van der Marel G. A. J. Org. Chem. 2009, 74, 4982–4991 10.1021/jo900662v. [DOI] [PubMed] [Google Scholar]; d Walvoort M. T. C.; Lodder G.; Mazurek J.; Overkleeft H. S.; Codee J. D. C.; van der Marel G. A. J. Am. Chem. Soc. 2009, 131, 12080–12081 10.1021/ja905008p. [DOI] [PubMed] [Google Scholar]; e Walvoort M. T. C.; van den Elst H.; Plante O. J.; Kröck L.; Seeberger P. H.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Angew. Chem., Int. Ed. 2012, 51, 4393–4396 10.1002/anie.201108744. [DOI] [PubMed] [Google Scholar]
- a Pohl N. L. ACS Symp. Ser. 2008, 990, 272–287 10.1021/bk-2008-0990.ch013. [DOI] [Google Scholar]; b Tang S.-L.; Pohl N. L. B. Org. Lett. 2015, 17, 2642–2645 10.1021/acs.orglett.5b01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mamidyala S. K.; Ko K.-S.; Jaipuri F. A.; Park G.; Pohl N. L. J. Fluorine Chem. 2006, 127, 571–579 10.1016/j.jfluchem.2006.01.001. [DOI] [Google Scholar]; b Jaipuri F. A.; Collet B. Y. M.; Pohl N. L. Angew. Chem., Int. Ed. 2008, 47, 1707–1710 10.1002/anie.200704262. [DOI] [PubMed] [Google Scholar]; c Ko K.-S.; Jaipuri F. A.; Pohl N. L. J. Am. Chem. Soc. 2005, 127, 13162–13163 10.1021/ja054811k. [DOI] [PubMed] [Google Scholar]; d Edwards H. E.; Nagappayya S. K.; Pohl N. L. B. Chem. Commun. 2012, 48, 510–512 10.1039/C1CC16022B. [DOI] [PubMed] [Google Scholar]
- Song E.-H.; Osanya A. O.; Petersen C. A.; Pohl N. L. B. J. Am. Chem. Soc. 2010, 132, 11428–11430 10.1021/ja103351m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D.; Li W.; Li H. J. Am. Chem. Soc. 2004, 126, 15081–15086 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Chan C.-H. Tetrahedron Lett. 1998, 39, 355–358 10.1016/S0040-4039(97)10599-8. [DOI] [Google Scholar]
- Liew S.-T.; Wei A. Carbohydr. Res. 2002, 337, 1319–1324 10.1016/S0008-6215(02)00124-6. [DOI] [PubMed] [Google Scholar]
- a Dam T. K.; Roy R.; Das S. K.; Oscarson S.; Brewer C. F. J. Biol. Chem. 2000, 275, 14223–14230 10.1074/jbc.275.19.14223. [DOI] [PubMed] [Google Scholar]; b Casas-Solvas J. M.; Ortiz-Salmerón E.; García-Fuentes L.; Vargas-Berenguel A. Org. Biomol. Chem. 2008, 6, 4230. 10.1039/b809542f. [DOI] [PubMed] [Google Scholar]
- a Sinclair H. R.; Kemp F.; de Slegte J.; Gibson G. R.; Rastall R. A. Carbohydr. Res. 2009, 344, 1968–1974 10.1016/j.carres.2009.06.038. [DOI] [PubMed] [Google Scholar]; b Lane J. A.; Mariño K.; Rudd P. M.; Carrington S. D.; Slattery H.; Hickey R. M. J. Microbiol. Methods 2012, 90, 53–59 10.1016/j.mimet.2012.03.017. [DOI] [PubMed] [Google Scholar]; c Lane J. A.; Mariño K.; Naughton J.; Kavanaugh D.; Clyne M.; Carrington S. D.; Hickey R. M. Int. J. Food Microbiol. 2012, 157, 182–188 10.1016/j.ijfoodmicro.2012.04.027. [DOI] [PubMed] [Google Scholar]
- Liu S. J.; Sivakumar S.; Wang Z. H.; Bonning B. C.; Miller W. Arch. Virol. 2009, 154, 469–479 10.1007/s00705-009-0327-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.