

Abstract
Background
The explosion of NGS (Next Generation Sequencing) sequence data requires a huge effort in Bioinformatics methods and analyses. The creation of dedicated, robust and reliable pipelines able to handle dozens of samples from raw FASTQ data to relevant biological data is a time-consuming task in all projects relying on NGS. To address this, we created a generic and modular toolbox for developing such pipelines.
Results
TOGGLE (TOolbox for Generic nGs anaLysEs) is a suite of tools able to design pipelines that manage large sets of NGS softwares and utilities. Moreover, TOGGLE offers an easy way to manipulate the various options of the different softwares through the pipelines in using a single basic configuration file, which can be changed for each assay without having to change the code itself. We also describe one implementation of TOGGLE in a complete analysis pipeline designed for SNP discovery for large sets of genomic data, ready to use in different environments (from a single machine to HPC clusters).
Conclusion
TOGGLE speeds up the creation of robust pipelines with reliable log tracking and data flow, for a large range of analyses. Moreover, it enables Biologists to concentrate on the biological relevance of results, and change the experimental conditions easily. The whole code and test data are available at https://github.com/SouthGreenPlatform/TOGGLE.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-015-0795-6) contains supplementary material, which is available to authorized users.
Keywords: NGS, Toolbox, Pipeline, Flexible
Background
Since the appearance of Next Generation Sequencing (NGS) technologies, a large number of bioinformatics softwares and methods have been developed and publicly released to work with these massive sequence data [1, 2]. Depending on the type of data and experiments, multiple steps may be required such as quality control, adapter removal or trimming or both, mapping, variant detection, read counting and so on, and dedicated software may be used for each step. Thus, biologists have to perform a lot of chained manipulations in order to obtain the final information for their research. The vast majority of bioinformatics softwares are command-lines tools, generally designed to work on powerful computers and High-Performance Calculation (HPC) infrastructures. Bioinformaticians have developed dedicated pipelines for specific experiments, using those bioinformatics softwares, as well as combinations of home-made filters (using conditional if) and loops (using the classical while) [3, 4]. However, those scripts are generally monolithic, in the sense that they cannot easily be transferred from one installation to another, or from one experimental subject to the following (different species, different conditions, etc.). Moreover, even if some flexibility is offered to the user by modification of several options before launching, these modifications are generally not trivial, requiring either a long list in the command line, or provision of a complex option file. In addition, writing a new pipeline from scratch is labor-intensive and time-consuming, especially if all safeguards for file format, software execution, or file transfer are implemented each time.
The TOGGLE suite (for TOolbox for Generic nGs anaLysEs) was developed to optimize the creation of new NGS analysis workflows. What we propose here is a set of packages designed for fast implementation of robust and reliable pipelines. Each package represents either a NGS software manipulation or a set of dedicated tools (see Table 1). These packages are written in Perl, with unitary modules the most generic possible. TOGGLE is able to manage hundreds of samples at once, and is not limited to one application. It can be used on DNAseq, RNA-Seq, smallRNA, GBS, and any other sequence data. It can be installed on a variety of infrastructure, from a simple laptop computer to a large HPC cluster. Finally, the TOGGLE assays and options are based on a unique configuration file, that can be easily changed and adapted to each run. The tool and test data are available on GitHub: https://github.com/SouthGreenPlatform/TOGGLE. A Docker image for immediate installation is available (http://bioinfo-web.mpl.ird.fr/toggle/toggle.tgz), as well as an user-space install script (http://bioinfo-web.mpl.ird.fr/toggle/installTOGGLE.sh).
Table 1.
List of the different packages and main modules. If any other modules exist but are not presented here, multiple dots are shown
| Packages (nb of modules) | Modules | Functions |
|---|---|---|
| bwa (5) | bwaIndex | Indexing the reference Fasta file |
| bwaAln | Create the alignment for FASTQ sequences | |
| bwaSampe | Create the SAM file for pair-end sequences | |
| bwaSamse | Create the SAM file for single-end sequences | |
| ... | ... | |
| cutadapt (2) | CreateConfFile | Create the specific cutadapt configuration file |
| execution | Execute cutadapt command | |
| fastqc (6) | execution | Run FastQC software |
| parse | Parse the FastQC results | |
| ... | ... | |
| gatk (8) | gatkVariantFiltrator | Filter the different SNP based on specific options |
| gatkHaplotypeCaller | Call the haplotype of each individual based on a BAM file | |
| ... | ... | |
| pairing (4) | pairRecognition | Allow the recognition of pairs in a set of FASTQ files |
| repairing | Reorganize in pair two FASTQ files, extract single sequences | |
| picardTools (3) | picardToolsMarkDuplicates | Mark/eliminate different types of duplicate in a BAM file |
| picardToolsCreateSequenceDictionnary | Create the “.dict” file of the reference | |
| picardToolsSortSam | Sort the SAM file | |
| ... | ... | |
| samTools (10) | samToolsSort | Sort the SAM/BAM file |
| samToolsIndex | Index the SAM/BAM file | |
| mergeHeader | Merge the header of multiples SAM/BAM into a single one | |
| ... | ... | |
| toolbox (73) | exportLog | Export information into log files |
| checkFile | Check if a file exists, is readable/writable, and is not empty | |
| existsDir | Check if a directory exists | |
| makeDir | Create a new directory | |
| readDir | Read the content of a directory | |
| extractPath | Extract the complete path of a file | |
| extractName | Create a readgroup from a file name | |
| readFileConf | Read the configuration file and return a hash | |
| extractOptions | Provide the options for a given software from the hash | |
| run | Run the command line given in argument | |
| checkSamOrBamFormat | Check if a file is a true SAM/BAM format | |
| extractHashSoft | Extract specific options for a given tool | |
| checkNumberByWC | Provide the number of sequence in a given FASTQ file | |
| checkEncodeByASCIIcontrol | Check the format of encoding in a FASTQ file | |
| changeEncode | Provide a wrapper to change the encoding of a FASTQ file | |
| ... | ... |
Implementation/results and discussion
Packages and modules
We designed TOGGLE in a suite of more than 10 packages, gathering more than 60 different modules (functions or calling), which can be used independantly to quickly implement pipelines for NGS analyses. Each package represents either the use of a software suite (such as bwa [5] or Genome Analysis ToolKit GATK [6]), or a set of tools dedicated to a given operation or more general tools. Modules are Perl code wrappers for a central command or function, with a validation system for the entry file format, running command, and output (if any). Each module starts generally by calling the file format validation modules from the toolbox.pm package (for FASTQ, SAM and others), and then by sending a specific command line to the toolbox::run module. This module launches the command through the UNIX system, and will return either a given value for correct execution, a warning, or an error.
The toolbox.pm package gathers general tools and functionalities. This package will, among other things, read the software.config.txt file, manage printing of log files printing (see below) and the system command for all modules (Fig. 1). The software.config.txt file can be used to manage the options for the different softwares and modules of TOGGLE. This file is constructed in a basic layout, as shown in Fig. 2, using the option representation specific for each software. If no specific parameters for a given software or module are provided in this file, the default values of the software/module will be used. The software.config.txt file (see Fig. 2) was developed with Biologist users in mind: this file is the only one to change for any option of any step of a given pipeline. The localConfig.pm package will inform of the different softwares paths (for bwa e.g.) on the user’s local machine or server. Users can adapt the paths in this package, and can also test different versions or installations of the same software without having to change the whole pipeline code. Only those three packages/files are mandatory when creating a new script using TOGGLE.
Fig. 1.
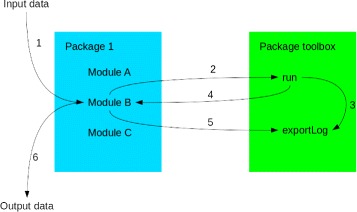
Software execution presentation. (1) Input data are submitted to a given Module. (2) The Module will construct a command line to toolbox::run. (3) The text of the command line and the run report are sent to toolbox::exportLog. (4) The output of the command (ok, error or warning) is sent back to the original Module. (5) The Module will send a report to toolbox::exportLog. (6) The output data are delivered from the Module
Fig. 2.

Softwares configuration file. The lines starting with “$” correspond to the name of the current module called (e.g. bwa aln). The lines just after list the option(s) associated with this call; the list of options is finished with an empty line. Lines starting with “ ♯” are reserved for comments
A large part of the coding effort was to produce a logging system that can be scaled for hundreds of individual analyses run in parallel (Fig. 3). The dedicated toolbox::exportLog module will recover the value transmitted by other modules to generate log information: (i) the module call succeeded; (ii) the module call started correctly but finished incorrectly; (iii) the module was not able to execute the command. The last two types of information will be recovered in the error log file (“.e”). Correct execution of the command will be written to the output log file (“.o”). The log system allows the user to check step by step the progess of the analysis.
Fig. 3.
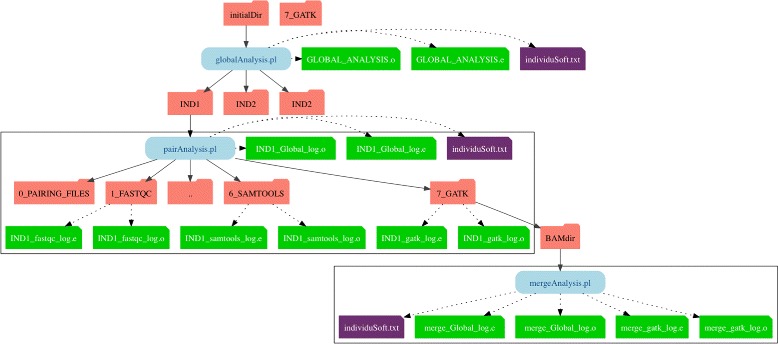
Directories tree structure. Representation of the tree of directories with the log files during the execution of globalAnalysis.pl pipeline, with the example of three individuals representing each possibility. The first one is a paired-end data, the second one is a single-end data, and the last one is a paired-end data which generate single reads during the cleaning step
Adding new modules or packages is relatively simple, with a sample package provided with the complete suite. Linking to the different tools (running and logging system), as well as the replacement of a given module by another in a pipeline is easy to perform. A template for a new pipeline design is also provided.
Example of implementation
All the modules can be arranged to easily generate robust pipelines for NGS analyses. As an example, we provide two functional pipelines: an RNAseq data one (not detailed here) and a DNAseq pipeline for single and paired-end Illumina genomic data for SNP and InDel detection in any eukaryotic diploid species. This latter, that we describe here in details, is based on four scripts: pairAnalysis.pl, singleAnalysis.pl, mergeAnalysis.pl and globalAnalysis.pl (Fig. 4). The first three can be launched independently, while the last, globalAnalysis.pl, is a turn-key pipeline which launches and manages the three others (see Additional file 1).
Fig. 4.
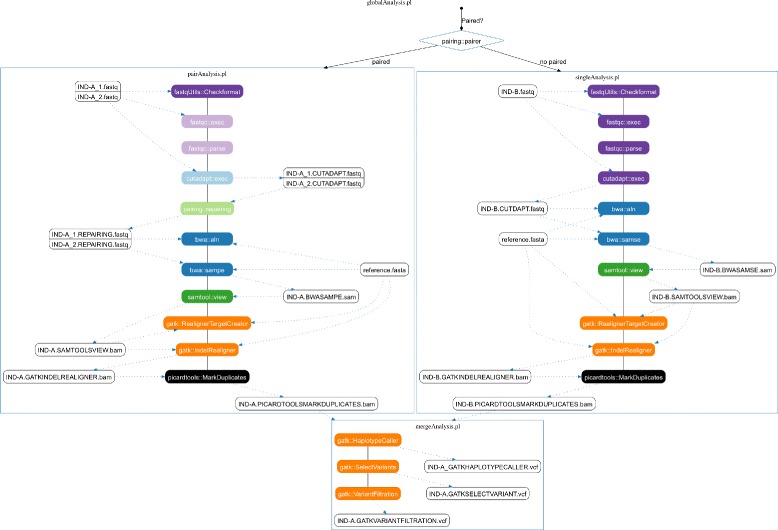
Pipeline DNAseq presentation. Basic overview of the pairAnalysis.pl, singleAnalysis.pl, mergeAnalysis.pl pipelines, and of the wrapping globalAnalysis.pl pipeline. Each colored box represents a given module, and each color a specific package. See text for the corresponding steps. A more complete figure is available on the TOGGLE website
Both start with FASTQ format validation (purple boxes in Fig. 4), followed by a quality control using the FastQC software (yellow boxes in Fig. 4) [7], and a cleaning step performed by the CutAdapt software [8] (sand boxes). For pairAnalysis.pl, an additional “repairing” stage (red boxes in Fig. 4) re-associates the paired-end reads and exports the single-end reads (which may be produced by the cleaning step) to the singleAnalysis.pl script independently from CutAdapt. This step may be excluded when using the last version of CutAdapt and its pair-end mode, but this is not yet implemented in TOGGLE. After cleaning, both scripts follow the same stages again, with specific parameters: mapping using bwa aln (blue boxes; bwa MEM module is also available) [5]; sorting, cleaning and indexing of the BAM files using PicardTools [9] and SAMtools [10] (brown and green boxes, respectively); local realignment with the GATK (orange boxes) [6]; and finally removal of duplicates using PicardTools (brown boxes). The mapping is performed here using the bwa aln/sampe/samse back-track version. It is possible to switch to the bwa mem version by changing the code, as this module is already provided in the bwa.pm package. However, we decided to remain on the back-track version in this pipeline, as it is used in our labs and allows comparison with older mapping data. The data are then treated through the third script mergeAnalysis.pl, for Single Nucleotide Polymorphism (SNP) and Insertion/Deletions (InDels) calling using HaplotypeCaller, followed by the sorting and quality filtering of the obtained variants. All of these steps are performed using GATK, and are multiple sample variant calling. The final Variant Call Format (VCF) file is cleaned and filtered as much as possible through generic Bioinformatics tools, but subsequent specific analyses may be required to optimize it for specific purposes (e.g. SNP identification for CHiP creation, population genetics or breeding).
In order to automate these steps for multiple samples, we created the globalAnalysis.pl script (in currently two versions: one linear and the second one parallelized for SGE type architecture). This script will also organize the data into different directories with a specific tree structure (Fig. 3) defined to facilitate access to intermediate files.
The globalAnalysis.pl script will deal with a unique directory where the whole raw FASTQ data are stored, without correspondance of names between the two paired files (the forward can be named Ulysse and the reverse Enterprise). The script will use the pairing::pairRecognition.pm module (red boxes in Fig. 4) to associate files of paired-end data. The first sequence ID (specific for each run) is used to create the pairs, and we validated this association step of more than 150 paired and single files. It creates independent folders for each sample/individual, on which pairAnalysis.pl or singleAnalysis.pl will be launched according to the data type. After these analyses, globalAnalysis.pl will recover the final BAM file and its index for each sample, and will execute the mergeAnalysis.pl script as described earlier (see Fig. 4).
All those scripts are associated with an indicator file, individuSoft.txt, which inform which step of the analysis has been launched. This indicator file also permits to create the various logs files. Indeed we constructed different level of log files: at global level (GLOBAL_ANALYSIS_date on Fig. 3); at sample/individuals level (indX_global_log on Fig. 3), and at package per individual level (indX_package_log on Fig. 3).
This pipeline is a classical one for DNAseq analyses from FASTQ sequences to BAM files then VCF files, but with a lot of control regarding file structure and format, and is easy to manage in terms of specific and global options through the software.config.txt file. Moreover, it can be used with any reference genome (provided as a FASTA file). A RNAseq pipeline is also provided in the TOGGLE suite, based on a TopHat and HTSeq [11] approach (see Additional file 1).
Parallelization
TOGGLE can manage parallelized jobs, and may be implemented on HPC cluster machines. Concurrent jobs can be launched using the globalAnalysisSGE.pl script for example, which will launch all individual pairAnalysis.pl (or singleAnalysis.pl) instances in different jobs. The specific SGE parameters for all individual jobs can be specified in the option file. For the moment, the main level of the paralellization is based on the sample level (each individual) only. We are conscious this is a main drawback compared to system such as HugeSeq [4] or Churchill [3] (see below). However, TOGGLE was designed to requested a very limited number of mandatory softwares/configuration outside of bioinformatics one. Thus it could be deployed on any kind of infrastructure. Nevertheless, in future versions we plan to develop a finer approach using a MapReduce/embarrassingly parallel approach, splitting each sample in multiples subsamples at the different levels (FASTQ, SAM/BAM, VCF), in a similar way to Churchill [3].
Still, the current scripts do not manage the parallelization based on specific options. If a given parameter in the option file specifies a certain number of parallel threads (e.g. bwa aln “-t” option), and if the SGE options provided do not require the same number of cores, more than one thread per core will be launched, which could have a major impact on the general efficiency of the job (up to server crash) if too much threads per core are requested.
Comparison with already available pipeline tools
Different pipelines or pipeline development tools exist for NGS data. The graphical systems (such as Galaxy [12], Taverna [13], Tavaxy [14]) are limited in their options or the available tools, and are generally not able to reach the scale of thousands of samples. The command-line based systems are more versatile (see Table 2), but harder to use.
Table 2.
Comparison with the different pipelining systems available
| Function | TOGGLE | GATK-Queue | HugeSeq | Churchill | GotCloud | bcbio-nextgen |
|---|---|---|---|---|---|---|
| Raw FASTQ Data | Y | N | N | N | N | Ya |
| Reference indexing | Y | N | N | N | N | Ya |
| FASTQ to final Data | Y | N | Y | Y | Y | Y |
| Multiple Samples Calling | Y | Y | Y | Y | Y | Y |
| Modularity | Y | Y | N | N | Y | Y |
| Between Sample Paralellization | Y | Y | Y | Y | Y | Y |
| Within Sample Parallelization | Y/Nb | Yb | Y | Y | Y | Y |
| Structural Variant Analysis | N | N | N | Y | Y | N |
| Annotation | Ya | Y | N | Y | Y | Y |
| Scalability | Y | Y | N | Y | Y | Y |
| Evolutivity | Y | Y | Y | Y | Y | Y |
Y signals that a feature is present, N is absent. Evolutivity means the possibility to add future module or functions to the system
aAvailable as separated steps
bDepending the considered step
GATK-Queue [6] is a scripting framework for the Genome Analysis ToolKit, which treats SAM/BAM or VCF files using GATK tools. GATK-Queue is based on the Scala language, and can create simple scripts. However, it is limited to GATK tools and will start only after mapping.
The HugeSeq pipeline [4] uses a MapReduce-like approach on HPC clusters to optimize the calculation of genomic variants from a set of a few already cleaned FASTQ data. The scripts are fixed, and parameters can be changed only in hard code modifications or through an extensive and complex command-line. Moreover, a lot of external commands have to be performed before analyses (e.g. creation of reference index and dictionary).
Churchill [3] also uses an embarassingly parallel approach in order to achieve MapReduce performance. It is dedicated mainly to identification of genomic variants, and scripts are hard coded and can not be modified. It is possible to switch between different mappers and variant callers using a dedicated option file. However, the index and dictionaries for the references have to be created independently, and pair recognition is based only on file names. The whole path for all FASTQ files must be provided, and not all options are available for all softwares. Finally, Churchill is freely available for academic purposes, as the other ones, but is not open-source.
GotCloud [15] is a set of pipelines dedicated to massive NGS analyses, from already cleaned sequence data to association results. It is dedicated to detection of genomics variations, and can switch between mappers or callers. The options system is also based on a single file, but requires that the user knows the GotCloud version for each software option. Moreover, it runs only on clusters or in the Cloud (an Amazon EC2 instance is possible), and the name of all FASTQ files has to be provided.
The bcbio-nextgen framework [16], written in Python, is highly modular, and is similar to TOGGLE. Different mappers and callers are already available, and others can be add easily. The framework can be used for germline variants, somatic variants and RNA-Seq, as for TOGGLE. However, the reference sequences are limitated to a fixed set, and even if it is possible to add new items, the system requests those references to be listed.
Compared to the similar frameworks currently available, the current implementations of TOGGLE scripts work on DNA and RNA, and begin with a set of raw FASTQ in a given folder. It will automate all steps, from identification of pairs (if any), creation of indexes and dictionaries for the reference, data cleaning and so on. The end-users only have to modify the basic option file software.config.txt, in which the very same options as for the command line softwares need to be written. TOGGLE can be extended easily for new bricks and software plugins, and new scripts for dedicated analyses can be assembled quickly. Moreover, TOGGLE can be installed in the user space (without admin rights) as well as at the system level, from a simple laptop to a HPC cluster or in the Cloud.
Installation
Power users can install TOGGLE following the manual provided on GitHub https://github.com/SouthGreenPlatform/TOGGLE. We are aware that installing all dependencies and working versions of a such large number of softwares is generally problematic and complex. Therefore, we provide alternative methods for installation, a Docker image (available at http://bioinfo-web.mpl.ird.fr/toggle/toggle.tgz) and a bash script (available on the GitHub and at http://bioinfo-web.mpl.ird.fr/toggle/TOGGLEinstall.sh). The Docker image, based on the basic Ubuntu 14.04 image from Docker Hub, is ready to run Out of the box any NGS data transferred in the container. The bash script can be run at user space level and will download all the versions that work with the current version of TOGGLE, and will adapt the different paths for TOGGLE to work. Nevertheless, for an optimized and stable installation, we recommend a more precise installation as detailed on GitHub.
Conclusion
TOGGLE is a combination of tools for large-scale NGS analyses of DNA and RNAseq data. Users can easily modify the different options of the numerous softwares using the software.config.txt file, without going through the code for each step. Users who want to use the general scripts provided in the suite just have to launch the globalAnalysis.pl pipeline on the raw FASTQ files (in a single folder) and wait to get the BAM and VCF files. The efficient log files provide information and statistics on the data and pipeline progress quickly, as well as on potential errors or warnings. Thus, users can concentrate on the biological relevance of the results rather than on technical aspects, even if using a huge amount of data.
Bioinformaticians can rapidly write new packages and modules useful for Biologists using a highly reliable system. The addition of new modules is fast, and they can be easily linked to the logging system. A lot of tools are available already, and we will add new modules for structural variant analyses, with softwares such as NovelSeq [17] or BreakDancer [18] in the near future.
Availability and requirement
Project name: TOGGLE
Project home page:https://github.com/SouthGreenPlatform/TOGGLE
Operating system: Linux
Programming Language:Perl
Other requirements:Java 1.7, bwa 0.7.2 or higher, SAMtools 0.1.18 or higher, CutAdapt 1.2.1 or higher, FastqC 0.11.1 or higher, PicardTools 1.124 or higher, GATK 3.3x or higher, Perl modules: Data::Dumper, Data::Translate, Test:More, Test::Deep, Capture::Tiny.
License: GNU GPLv3/CeCill-C
Acknowledgements
Authors want to thank Ndomassi Tando for his great help for various software installations, the different β-testers that perform the first true data tests, the other members of the SouthGreen platform (http://www.southgreen.fr/) for their help, and Dr Tomas Harrop for his input in manuscript correction. We would like also to greatly thanks our reviewers for their very helpful comments in improving the mansucript. CM was supported by a grant from the French National Research Agency ANR (AfriCrop project ♯ ANR-13-BSV7-0017). AK was supported by a grant from the Agropolis Fondation (This project is supported by AgropolisFondationunder the reference ID 1403-026 through the ≪ Investissementsd’avenir ≫ programme(LabexAgro:ANR-10-LABX-0001-01)). MA was supported by a “FloriPalm” Specific Research Agreement between CIRAD and a FASSB and “PalMarkers” research contract between CIRAD and PalmElit SAS. SA was supported by the “PalMarkers” research contract between CIRAD and PalmElit SAS.
Additional file
Complete scheme of the globalAnalysis.pl script. Colored boxes correspond to analysis steps, with color related to a given package (see text for more information). White and black boxes correspond to input/output files. The globalAnalysis.pl script will determine the state of each sample (single or pair), then launch the corresponding subscript, and finally will gather all corrected BAM files to the mergeAnalysis.pl script that will perform the multiple sample calling. (PDF 60 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CM: blueprint, code of modules and packages, code of pipelines, coordination, writing of the manuscript. CTD: blueprint, code of modules and packages, code of pipelines, correction of the manuscript. AK: code of modules and packages, code of pipelines. MS: code of modules and packages. CF: code of modules and packages. MA: code of modules and packages. JOB: code of modules and packages. SR: code of modules and packages. SA: code of modules and packages, code of RNAseq pipeline. EOA: code of modules and packages, installation scripts. FS: initiator of the program, code of modules and packages, Docker image, coordination, correction of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Cécile Monat, Email: cecile.monat@ird.fr.
Christine Tranchant-Dubreuil, Email: christine.tranchant@ird.fr.
Ayité Kougbeadjo, Email: ayite.kougbeadjo@cirad.fr.
Cédric Farcy, Email: cedric.farcy@cirad.fr.
Enrique Ortega-Abboud, Email: enrique.ortega-abboud@cirad.fr.
Souhila Amanzougarene, Email: souhila.amanzougarene@ird.fr.
Sébastien Ravel, Email: sebastien.ravel@cirad.fr.
Mawussé Agbessi, Email: mawusse.agbessi@ird.fr.
Julie Orjuela-Bouniol, Email: julie.orjuela@adnid.fr.
Maryline Summo, Email: maryline.summo@cirad.fr.
François Sabot, Email: francois.sabot@ird.fr.
References
- 1.Bao S, Jiang R, Kwan W, Wang B, Ma X, Song Y. Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011;56:406–14. doi: 10.1038/jhg.2011.43. [DOI] [PubMed] [Google Scholar]
- 2.Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, et al. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2014;15(2):256–78. doi: 10.1093/bib/bbs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelly BJ, Fitch JR, Hu Y, Corsmeier DJ, Zhong H, Wetzel AN, et al. Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics. Genome Biol. 2015;16(1):6. doi: 10.1186/s13059-014-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lam HYK, Pan C, Clark MJ, Lacroute P, Chen R, Haraksingh R, et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol. 2012;30(3):226–9. doi: 10.1038/nbt.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation dna sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.FASTQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 8.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 9.PicardTools. http://broadinstitute.github.io/picard/index.html.
- 10.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and samtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goecks J, Nekrutenko A, Taylor JEA. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oinn T, Addis M, Ferris J, Marvin D, Senger M, Greenwood M, et al. Taverna: a tool for the composition and enactment of bioinformatics workflows. Bioinformatics (Oxford, England) 2004;20(17):3045–54. doi: 10.1093/bioinformatics/bth361. [DOI] [PubMed] [Google Scholar]
- 14.Abouelhoda M, Issa SA, Ghanem M. Tavaxy: Integrating Taverna and Galaxy workflows with cloud computing support. BMC Bioinformatics. 2012;13(1):77. doi: 10.1186/1471-2105-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jun G, Wing MK, Abecasis GR, Kang HM. An efficient and scalable analysis framework for variant extraction and refinement from population scale DNA sequence data. Genome Res. 2015:176552–114. doi:10.1101/gr.176552.114. [DOI] [PMC free article] [PubMed]
- 16.Bcbio-nextgen. https://github.com/chapmanb/bcbio-nextgen.
- 17.Hajirasouliha I, Hormozdiari F, Alkan C, Kidd J, Birol I, Eichler E, et al. Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics. 2010;26:1277–83. doi: 10.1093/bioinformatics/btq152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen K, Wallis J, McLellan M, Larson D, Kalicki J, Pohl C, et al. Breakdancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]