

Abstract
Owing to the frequent incidence of blast-induced traumatic brain injury (bTBI) in recent military conflicts, there is an urgent need to develop effective therapies for bTBI-related pathologies. Blood-brain barrier (BBB) breakdown has been reported to occur after primary blast exposure, making restoration of BBB function and integrity a promising therapeutic target. We tested the hypothesis that treatment with dexamethasone (DEX) after primary blast injury potentiates recovery of an in vitro BBB model consisting of mouse brain endothelial cells (bEnd.3). DEX treatment resulted in complete recovery of transendothelial electrical resistance and hydraulic conductivity 1 day after injury, compared with 3 days for vehicle-treated injured cultures. Administration of RU486 (mifepristone) inhibited effects of DEX, confirming that barrier restoration was mediated by glucocorticoid receptor signaling. Potentiated recovery with DEX treatment was accompanied by stronger zonula occludens (ZO)-1 tight junction immunostaining and expression, suggesting that increased ZO-1 expression was a structural correlate to BBB recovery after blast. Interestingly, augmented ZO-1 protein expression was associated with specific upregulation of the α+ isoform but not the α− isoform. This is the first study to provide a mechanistic basis for potentiated functional recovery of an in vitro BBB model because of glucocorticoid treatment after primary blast injury.
Keywords: bEnd.3, blood-brain barrier, dexamethasone, primary blast injury, recovery
Introduction
Widespread use of improvised explosive devices in recent military conflicts has been a major source of mortality and morbidity associated with blast-induced traumatic brain injury (bTBI).1, 2 Recent evidence suggests that blood-brain barrier (BBB) disruption occurs after blast exposure,3, 4 and specifically, after primary blast exposure.5, 6, 7, 8, 9 In vivo, the BBB serves as a highly restrictive interface between the systemic circulatory system and the brain,10, 11 making the BBB a promising therapeutic target for the treatment of traumatic brain injury (TBI) caused by explosive blast. There may be clinical utility in developing therapies to enhance repair of the damaged BBB to maintain brain homeostasis and protect neuronal activity and function.12, 13
Glucocorticoids have played an important role in the clinical management of central nervous system disorders associated with a compromised BBB, such as edema, brain tumors, and multiple sclerosis,14, 15 and have been studied extensively in experimental treatment models in vitro and in vivo.10, 13, 16, 17, 18, 19 For example, dexamethasone (DEX) promotes BBB integrity by initiating glucocorticoid receptor-mediated signaling, leading to altered gene expression and upregulation of tight junction proteins including occludin, claudin-5, and zonula occludens (ZO)-1 that strengthen and restore barrier properties.13, 18, 19, 20, 21 Greater than threefold increases in transendothelial electrical resistance (TEER), and similar changes in restriction of water permeability have been attributed to glucocorticoid treatment in epithelial and brain endothelial cell cultures.13, 17, 18, 22
Upregulated expression of ZO-1 has been linked specifically to the proper functioning of tight junctions and heightened resistance to paracellular transport in epithelial and endothelial monolayers.20, 22 ZO-1 is a 220 kDa protein localized to the intracellular surface of the tight junctional complex and is expressed in a wide variety of mammalian tissues.20, 23 ZO-1 is expressed in α+ and α− isoforms, termed ZO-1α+ and ZO-1α−, that result from alternative RNA splicing and can exist in different distributions within a single cell type.23, 24, 25 ZO-1α+ contains an 80-amino acid α domain, which is absent in ZO-1α−.23, 24 Some studies have suggested that expression specifically of ZO-1α+ is associated with more restrictive barrier properties.22, 25
Despite evidence supporting treatment with steroids for mitigating vasogenic edema, raised intracranial pressure, and damage to the BBB after brain injury, their use in the clinic remains controversial.19, 26 Glucocorticoid therapy failed to show substantial benefit in reducing post-injury sequelae in head-injured patients, stroke patients, and in some animal models of TBI.26, 27, 28 However, more recent experimental evidence suggests that concomitantly inhibiting proteasomal degradation of the glucocorticoid receptor and delivering DEX after TBI promoted BBB recovery and improved neurologic outcomes.19 Other in vivo studies clearly indicate that combination therapy with melatonin and DEX synergistically reduced brain injury and promoted BBB recovery after TBI, while minimizing the required dosage and potential side effects of glucocorticoids.16 Taken together, these reports suggest that factors influencing glucocorticoid effectiveness after traumatic insult are numerous and their interactions are complex. As such, positive results obtained by using more refined approaches support further study and development of glucocorticoid therapies in the ongoing search for improved treatments for TBI and other disorders of the central nervous system.
In the current study, we provide evidence that treatment with DEX after primary blast injury promotes faster recovery of a monolayer of mouse brain microvascular cells (bEnd.3) representing an in vitro model of the BBB. We used a previously described primary blast injury model to precisely control the biomechanical initiators of injury and measure subsequent changes to barrier integrity by methods not possible in vivo.7, 8, 29 We report, for the first time, that treatment with DEX after primary blast exposure potentiated recovery of TEER and hydraulic conductivity of the BBB in vitro, and that glucocorticoid receptor activation accelerated barrier restoration. We further show that DEX-induced BBB recovery was accompanied by increased ZO-1 tight junction immunostaining and expression, specifically associated with upregulation of the ZO-1α+ isoform but not ZO-1α−. These findings hold important implications for development of a potential therapy for restoring BBB integrity and function after bTBI.
Materials and Methods
Blood-Brain Barrier Cell Culture Model
In vitro models of the BBB were generated using monolayers of immortalized mouse brain endothelial cells, bEnd.3 (ATCC, Manassas, VA, USA), as previously described.7, 8 Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; ATCC) supplemented with 10% newborn calf serum (Hyclone Laboratories, Logan, UT, USA). Each Transwell (Corning Life Sciences, Tewksbury, MA, USA) insert (1.12 cm2 surface area) was precoated with poly-l-lysine and seeded with a total of 60 000 bEnd.3 cells. Transwell cultures were grown for 8 days to reach confluency in a cell culture incubator at 37°C and 5% CO2/95% O2. Cells were fed with new medium every 2 to 3 days.
Exposure of Blood-Brain Barrier to Primary Blast Injury
Endothelial cultures were exposed to primary blast injury using an in vitro bTBI model, as described previously in detail.7, 8, 29 Briefly, Transwells were secured within a fluid-filled receiver, designed to mimic the surrounding skull-brain complex, and exposed to a fast-rising pressure transient induced by external shock wave loading. Shock waves were generated using a 76-mm-diameter shock tube with a 25-mm-length driver section pressurized with compressed helium gas, and a 1,240-mm-long-driven section. The bTBI model is capable of isolating interactions between the BBB culture and blast overpressure alone, substantially reducing the influence of inertial loading, thereby enabling precise investigation of primary blast injury.7, 8, 29 Cultures were exposed to one blast condition, characterized by a shock wave with a 571±15 (mean±s.e.m.) kPa peak incident overpressure, 1.06±0.01 ms duration, and 186±1.5 kPa ms impulse in the open-tube configuration.8, 29 The corresponding biomechanical injury parameters measured at the location of the sample in the fluid-filled sample receiver were 1523±91 kPa peak overpressure, 1.48±0.03 ms duration, and 812±29.3 kPa ms impulse.8 This injury severity level was previously reported to cause disruption of the BBB in vitro, and is comparable to exposure to a 105 mm artillery round at a standoff distance of 5 to 10 m.8
Post-Injury Treatment
Dexamethasone is a synthetic glucocorticoid compound known to improve BBB properties by inducing glucocorticoid receptor-mediated signaling activity (Sigma-Aldrich, St Louis, MO, USA).10, 13 RU486 (mifepristone) is an antagonist to the glucocorticoid receptor and inhibits glucocorticoid-induced cell signaling activity (Cayman Chemical, Ann Arbor, Michigan, USA).17 Endothelial cultures were treated with DEX, DEX and RU486, or vehicle (pure ethyl alcohol; Sigma-Aldrich) 30 minutes after exposure to primary blast injury, and treatment was continued once per day after injury. To determine the optimal dosage for postinjury functional recovery, cultures were treated with a range of DEX concentrations (0, 1, 10, and 100 μmol/L) based on concentrations from the literature.10, 18 For all subsequent experiments, cultures were treated with 10 μmol/L DEX; to inhibit effects of DEX, cultures were cotreated with 10 μmol/L DEX and 20 μmol/L RU486.
Measurement of Transendothelial Electrical Resistance
To quantify changes in ion flux through the endothelial monolayer, TEER values across monolayers of endothelial cultures were measured with an Endohm-12 electrode chamber connected to an EVOMX Epithelial Voltohmmeter (World Precision Instruments, Sarasota, FL, USA). TEER of each culture was measured 5 minutes before exposure to blast and 5 minutes after exposure. To assess timecourse changes, TEER was measured in the same cultures once per day for up to 3 days after exposure to blast injury (delivered on day 0). All recorded TEER values were corrected for the TEER of a cell-free Transwell and adjusted to account for the membrane surface area, as previously described.7, 8 Normalized TEER was determined as the ratio of each culture's postinjury TEER value to its pre-injury TEER value. Age-matched and time point–matched sham controls were used for all time course TEER data comparisons. Endothelial cultures were only included in experiments if their preinjury TEER was ⩾10 Ω cm2, indicating a healthy and integral bEnd.3 monolayer.
Measurement of Hydraulic Conductivity
To quantify changes in water flux through the endothelial monolayer, hydraulic conductivity was measured once for a given sample, at each time point from days 1 to 3 after blast exposure (delivered on day 0).7, 8 Transwell cultures were placed in a custom-built permeability device, as previously described.8, 30 A known hydrostatic pressure of ~20 cm H2O was applied across the endothelial monolayer, and hydrostatically driven fluid flow was tracked using the linear displacement of an air bubble through a calibrated glass tube. Hydraulic conductivity (Lp, cm/s/cm H2O) was calculated using equation (1).8, 30 Cultures were immediately discarded after each Lp measurement.
 |
Where, 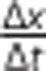 represents the linear displacement of the bubble over time, F the fluid volume of the calibrated tube, S the surface area of the culture, and ΔP the hydrostatic pressure across the culture. Endothelial cultures were only included in experiments if the average Lp of shams was ⩽4 × 1/107 cm/s/cm H2O; otherwise, all samples from the same experimental batch were discarded because of suboptimal culture health.
represents the linear displacement of the bubble over time, F the fluid volume of the calibrated tube, S the surface area of the culture, and ΔP the hydrostatic pressure across the culture. Endothelial cultures were only included in experiments if the average Lp of shams was ⩽4 × 1/107 cm/s/cm H2O; otherwise, all samples from the same experimental batch were discarded because of suboptimal culture health.
Assessment of ZO-1 Tight Junction Morphology
The expression and localization of ZO-1 tight junction protein at the border between adjacent endothelial cells is critical to the function and maintenance of tight junctions.7, 8, 11 One day after exposure to blast injury, endothelial cultures were fixed in 1% formaldehyde and incubated with rabbit anti-ZO-1 polyclonal antibody (Life Technologies, Carlsbad, CA, USA; # 61-7300). The presence of ZO-1 was detected using donkey antirabbit Alexa Fluor 488 secondary antibody (Life Technologies). Cultures were also incubated with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies) to detect cell nuclei to determine cell counts for each sample. Endothelial monolayers were imaged using an Olympus IX81 (Olympus America, Center Valley, PA, USA) fluorescence microscope and analyzed using MetaMorph software (Molecular Devices, Sunnyvale, CA, USA). ZO-1 tight junction immunostaining was quantified by applying an identical threshold for ZO-1 immunofluorescence to five randomly selected images of each endothelial culture. Consistent with methods previously described,7, 8 ZO-1 immunostaining was quantified as the area percentage of an image with fluorescence above a predetermined threshold, normalized to the total number of cells in each image. Immunostaining images were randomly selected from each experimental condition, and an identical quantitative analysis method was applied to all samples.
Western Blot Analysis of ZO-1 Tight Junction Protein
Western blot analysis was performed similarly to our previously published methods,31 but with slight modifications. At day 1 after injury, endothelial monolayers from two to three Transwell cultures representing each experimental condition were washed twice with ice-cold phosphate buffered saline and immediately placed in lysis buffer with inhibitors (40 mmol/L 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, 120 mmol/L sodium chloride, 1 mmol/L EDTA, 1% Triton X-100, 10 mmol/L sodium pyrophosphate decahydrate, 50 mmol/L sodium fluoride, 0.5 mmol/L sodium orthovanadate, 10 mmol/L β-glycerophosphate, 3% sodium dodecyl sulfate, 1% sodium deoxycholate, 1% protease inhibitor cocktail, 1% phosphatase inhibitor cocktails 2 and 3, and 2 mmol/L phenylmethanesulfonyl fluoride; Sigma-Aldrich). Lysed cell samples were collected and sonicated (Sonicator 3000; Misonix, Farmingdale, NY, USA), incubated on ice, then centrifuged to remove cell debris. Approximately, 130 to 150 μg of protein per experimental group was separated by gel electrophoresis on a 4% to 12% Bis-Tris gel (Life Technologies). A semidry blotting apparatus (Fisher Scientific, Pittsburgh, PA, USA) was then used to transfer proteins to a nitrocellulose membrane (Life Technologies). The membrane was blocked in Tris-buffered saline (pH 7.4) with 5% bovine serum albumin, and then incubated overnight at 4°C with rabbit anti-ZO-1 primary antibody (Life Technologies; # 61-7300) and mouse anti-β-tubulin (Life Technologies; # 32–2600) in Tris-buffered saline-T (0.1% Tween 20, pH 7.4; Sigma-Aldrich) with 5% bovine serum albumin. The primary antibody to ZO-1 recognizes amino acids 463 to 1109 of human ZO-1 cDNA, and is known to react with both the α+ and α− isoforms of ZO-1, as previously reported.22, 25 Membranes were then washed, labeled with corresponding secondary antibody (donkey antirabbit Alexa Fluor 488 or goat antimouse Alexa Fluor 647; Life Technologies), and washed. Membranes were imaged using a CRi Maestro 2 Imaging System (PerkinElmer, Waltham, MA, USA). Densitometric analysis of western blots was performed using National Institutes of Health ImageJ software (v. 1.48; NIH, Bethesda, Maryland, USA). Expression of ZO-1α+ and ZO-1α− were each normalized to β-tubulin expression, and experimental groups were further normalized to vehicle-treated sham for each independent experiment.
To detect and verify the identity of the ZO-1α+ isoform, western blotting was performed with rabbit anti-ZO-1α+ polyclonal antibody (Hycult Biotech, Plymouth Meeting, PA, USA; # HP9044). The rabbit polyclonal antibody, raised against amino acids 886 to 995 of human ZO-1, only recognizes the ZO-1α+ isoform.24, 25
Statistical Analysis
Repeated-measures analysis followed by Bonferroni post hoc tests were used to determine the overall effect of blast on the dose-dependent response and time course of TEER after exposure. Hydraulic conductivity, tight junction immunostaining, and western blotting data were analyzed by one-way analysis of variance followed by Bonferroni post hoc tests. (SPSS v. 22, IBM, Armonk, NY, USA; significance *P<0.05).
Results
Potentiated Transendothelial Electrical Resistance Recovery with Dexamethasone Treatment after Blast
In vitro BBB cultures were exposed to blast with a 571 kPa peak overpressure, 1.06 ms duration, and 186 kPa ms impulse.8 Following blast exposure (day 0), TEER of injured cultures decreased acutely to ~80% of preexposure levels compared with 102%±1% in sham controls (Figure 1). One day following blast exposure (day 1), TEER increased in a dose-dependent manner after DEX treatment at a concentration range of 0 to 10 μmol/L, with no significant increase after a 100 μmol/L treatment (Figure 1). TEER decreased to 80%±4% acutely after blast, and after treatment with 10 μmol/L DEX, TEER recovered to 117%±5% one day after injury (Figure 1). Administration of 10 μmol/L DEX produced a maximal TEER increase (of the treatment concentrations tested) in the in vitro BBB culture, which was consistent with published results.10 Therefore, 10 μmol/L DEX was selected as the treatment concentration for all subsequent experiments.
Figure 1.

Dose-dependent effect of dexamethasone (DEX) on transendothelial electrical resistance (TEER) after blast injury. Cultures were exposed to blast with a 571 kPa peak overpressure, 1.06 ms duration, and 186 kPa ms impulse. Postinjury treatment with 10 μmol/L DEX resulted in the greatest TEER recovery 1 day after blast exposure compared with all other treatment concentrations. *P<0.05; ±s.e.m.; n⩾9 per group.
TEER of vehicle-treated injured cultures required 3 days to fully recover spontaneously, compared with 1 day in DEX-treated (10 μmol/L) injured cultures, showing faster BBB recovery because of treatment (Figure 2). TEER of vehicle-treated injured cultures increased from 75%±2% to 85%±2% one day after exposure, whereas TEER of DEX-treated injured cultures increased from 78%±2% to 115%±2% at the same time point (Figure 2). Vehicle-treated shams maintained consistently high TEER levels at ~100% for 3 days after sham exposure. TEER of DEX-treated shams increased to 119%±2% one day after sham exposure, which was a level comparable to that of DEX-treated injured cultures at the same time point, and was sustained for 3 days (Figure 2). These results support that in addition to potentiated TEER recovery after blast exposure, DEX treatment resulted in overall strengthening of the barrier and a more differentiated BBB phenotype.10, 18 Absolute preinjury TEER values were 13±1.3 Ω cm2 (mean±s.d.), which were representative of TEER values previously reported for bEnd.3 cultures.30 In addition, absolute TEER decreased to 10±1.9 Ω cm2 on day 0 postinjury, and recovered to 15±1.1 Ω cm2 at day 1 in DEX-treated injured cultures.
Figure 2.

Potentiated transendothelial electrical resistance (TEER) recovery induced by dexamethasone (DEX) treatment (10 μmol/L) after blast injury. Transendothelial electrical resistance of vehicle-treated injured cultures required 3 days to fully recover spontaneously, whereas TEER of DEX-treated injured cultures fully recovered 1 day after blast exposure. Effects of DEX treatment were sustained for at least 3 days after injury. *P<0.05; ±s.e.m.; n⩾10 per group.
Reduced Hydraulic Conductivity with Dexamethasone Treatment after Blast
Hydraulic conductivity was significantly decreased—indicating a tighter barrier—in DEX-treated injured cultures and sham cultures compared with vehicle-treated injured cultures from days 1 to 3 after blast. Hydraulic conductivity was elevated to 3.51±0.89 × 1/107 cm/s/cm H2O in vehicle-treated injured cultures compared with 1.74±0.15 × 1/107 cm/s/cm H2O in vehicle-treated shams 1 day postinjury (Figure 3; not significant). Hydraulic conductivity remained elevated at 4.10±0.94 × 1/107 cm/s/cm H2O in vehicle-treated injured cultures compared with 1.76±0.46 × 1/107 cm/s/cm H2O in vehicle-treated shams 2 days postinjury (Figure 3; not significant). Vehicle-treated injured cultures spontaneously recovered to 2.18±0.44 × 1/107 cm/s/cm H2O by 3 days postinjury, which was very close to 2.00±0.52 × 1/107 cm/s/cm H2O in vehicle-treated shams (Figure 3). However, Lp of DEX-treated injured cultures fully recovered to 1.44±0.22 × 1/107 cm/s/cm H2O by day 1 after blast, and remained significantly decreased compared with vehicle-treated injured cultures from days 1 to 3 after injury (Figure 3). Overall, these Lp results support the postinjury time course of TEER recovery (Figure 2), showing potentiated and sustained recovery of BBB integrity in DEX-treated injured cultures lasting for at least 3 days after blast.
Figure 3.

Decreased hydraulic conductivity (Lp) owing to dexamethasone (DEX) treatment after blast injury. Hydraulic conductivity was significantly reduced—indicating a tighter barrier—in DEX-treated cultures compared with vehicle-treated injured cultures from days 1 to 3 after injury. Elevated Lp of vehicle-treated injured cultures spontaneously recovered to vehicle-treated sham levels by day 3 postinjury. *P<0.05; ±s.e.m.; n⩾7 per group.
Inhibition of Dexamethasone-Induced Potentiated Blood-Brain Barrier recovery
Cotreatment with DEX and the glucocorticoid receptor antagonist, RU486, inhibited potentiated TEER recovery attributed to DEX treatment alone (Figure 4A). DEX-treated injured cultures recovered 1 day after injury, compared with 3 days in injured cultures receiving both DEX and RU486. TEER of DEX-treated injured cultures increased from 79%±3% to 119%±2% one day after exposure, whereas TEER of DEX and RU486-treated injured cultures increased from 77%±3% to 88%±2% at the same time point (Figure 4A). The time course for TEER recovery in injured cultures receiving cotreatment of DEX and RU486 matched the spontaneous recovery time course in vehicle-treated injured cultures (Figure 2). In addition, sham cultures treated with RU486 maintained high TEER levels at ~100% for the duration of the time course, confirming that RU486 alone did not negatively affect TEER. Cotreatment with DEX and RU486 also inhibited the reduction in Lp (tightening of the barrier) effected by DEX treatment alone (Figure 4B). Hydraulic conductivity of DEX-treated injured cultures recovered 1 day after injury to 1.12±0.08 × 1/107 cm/s/cm H2O, whereas Lp of DEX and RU486-treated injured cultures was significantly elevated to 3.03±0.62 × 1/107 cm/s/cm H2O, which was similar to Lp of vehicle-treated injured cultures 1 day postinjury (Figure 3).
Figure 4.

Inhibition of dexamethasone (DEX)-induced potentiated BBB recovery. (A) Cotreatment with DEX and RU486 inhibited potentiated transendothelial electrical resistance (TEER) recovery attributed to DEX treatment alone. TEER of DEX-treated injured cultures recovered 1 day after injury, compared with 3 days in injured cultures cotreated with DEX and RU486. (B) Cotreatment with DEX and RU486 inhibited the reduction in hydraulic conductivity (Lp) attributed to DEX treatment alone. Hydraulic conductivity of DEX-treated injured cultures recovered 1 day after injury, whereas Lp of injured cultures receiving DEX and RU486 was significantly elevated compared with sham and DEX-treated injured groups. *P<0.05; ±s.e.m.; n⩾9 per group in A and B.
Increased ZO-1 Tight Junction Immunostaining with Dexamethasone Treatment after Blast
Characteristic morphology and localized staining of the tight junction protein, ZO-1, were visible in vehicle-treated shams (Figure 5A). Bearing close resemblance to integral endothelial monolayers described in our previous publications,7, 8 cells from vehicle-treated shams formed a characteristically elongated, spindle-shaped morphology with widespread expression of ZO-1 protein at the cytoplasmic membrane surface and at junctions between adjacent cells. ZO-1 staining was visibly compromised in vehicle-treated injured cultures, consistent with our previously reported results (Figure 5B).7, 8 ZO-1 immunofluorescence was visibly stronger and recovered at the cell periphery in DEX-treated injured cultures, suggesting that increased ZO-1 at tight junctions is a structural correlate to potentiated recovery of the BBB after blast exposure (Figure 5C). Cotreatment with DEX and RU486 inhibited DEX-induced increases in ZO-1 after injury (Figure 5D). The area percentage of ZO-1 immunostaining per cell significantly decreased to 0.09%±0.01% in vehicle-treated injured cultures compared with 0.14%±0.01% in vehicle-treated shams, consistent with our previous study (Figure 5E).7 ZO-1 staining in DEX-treated injured cultures significantly recovered to 0.15%±0.01%, whereas staining remained significantly depressed at 0.09%±0.01% in injured cultures cotreated with DEX and RU486 (Figure 5E). Therefore, the quantified area percentage of ZO-1 immunostaining per cell confirmed qualitative trends observed in the immunofluorescence images acquired 1 day after blast exposure, further supporting the critical role of ZO-1 in mediating DEX-induced functional recovery of the BBB.
Figure 5.

Increased tight junction immunostaining owing to dexamethasone (DEX) treatment 1 day after blast injury. (A) Characteristic staining of the zonula occludens (ZO)-1 tight junction protein was present in vehicle-treated shams. (B) Reduced ZO-1 staining was evident in vehicle-treated injured cultures after blast exposure. (C) Stronger ZO-1 tight junction staining in DEX-treated injured cultures. (D) Reduced ZO-1 staining in injured cultures treated with DEX and RU486. (E) Quantified ZO-1 staining per cell was consistent with qualitative changes depicted in immunofluorescence images, confirming recovered ZO-1 tight junction immunostaining in DEX-treated injured cultures. *P<0.05; ±s.e.m.; n⩾6 cultures (⩾30 images) per group; ZO-1 immunostaining (green); cell nuclei (blue); and scale bar, 50 μm.
Increased ZO-1 Tight Junction Protein Expression with Dexamethasone Treatment after Blast
The influence of DEX treatment on ZO-1 protein expression 1 day after injury was assessed by western blot analysis. ZO-1 expression was detected by two immunoreactive bands corresponding to the α+ and α− isoforms of ZO-1, as previously reported.22, 23, 24, 25, 32 DEX treatment in injured cultures increased the intensity of the ZO-1α+ band compared with all other experimental conditions, but did not affect the intensity of the ZO-1α− band (Figure 6A). As determined by densitometric analysis, normalized ZO-1α+ expression was significantly increased in DEX-treated injured cultures to 573%±189% compared with 100%±25% in vehicle-treated shams, 104%±30% in vehicle-treated injured cultures, and 159%±11% in DEX and RU486-treated injured cultures (Figure 6B). However, there were no significant changes in normalized ZO-1α− levels owing to DEX treatment (Figure 6C). By using an antibody specific to ZO-1α+, only the protein band of higher molecular mass corresponding to ZO-1α+ was detected, verifying identity of the isoform (Supplementary Figure 1).
Figure 6.

Upregulation of zonula occludens (ZO)-1α+ tight junction protein with dexamethasone (DEX) treatment 1 day after blast injury. (A) Representative western blot of ZO-1 expression after blast or sham exposure, showing immunoreactive bands for the α+ and α− isoforms of ZO-1, and associated β-tubulin expression. Dexamethasone treatment in injured cultures increased intensity of the ZO-1α+ band compared with all other experimental conditions, but did not affect intensity of the ZO-1α− band. (B) Significant upregulation of ZO-1α+ with DEX treatment after blast injury, as quantified by western blot analysis. The expression of ZO-1α+ was normalized to β-tubulin expression, and experimental groups were further normalized to vehicle-treated sham. (C) No significant changes to ZO-1α− after blast injury, as quantified by western blot analysis. The expression of ZO-1α− was normalized to β-tubulin expression, and experimental groups were further normalized to vehicle-treated sham. *P<0.05; ±s.e.m.; n⩾5 independent experiments per group in B and C.
Discussion
Treatment with DEX promotes BBB formation and functional recovery after injury by upregulating synthesis of tight junction proteins ZO-1, occludin, claudin-5, and other cell junction–related proteins.13, 16, 17, 18, 19, 21 Increased protein expression at the tight junctional complex induced by glucocorticoid treatment correlates with reduced paracellular permeability and augmented electrical resistance in epithelial and endothelial cells.10, 13, 17, 18, 20, 21, 22 At the molecular level, ZO-1 acts as an intracellular scaffolding protein that critically regulates the integrity of tight junctions by anchoring transmembrane proteins, occludin, and claudin-5, to the cytoskeleton.11 Together, these findings described in the literature strongly support results of our current study, which is the first to report that increased ZO-1 with DEX treatment after exposure to primary blast injury is linked to potentiated functional recovery of an in vitro BBB model.
Effects of glucocorticoid treatment are generally mediated by glucocorticoid receptor signaling activity and modulated expression of target genes.13, 17, 18, 33 To confirm glucocorticoid receptor-mediated upregulation of ZO-1 and potentiated recovery of the in vitro BBB model, we blocked DEX treatment effects with RU486, a potent and specific glucocorticoid receptor antagonist. Specifically, cotreatment with DEX and RU486 after injury eliminated the effects of potentiated recovery of TEER and hydraulic conductivity owing to DEX treatment alone, and inhibited DEX-induced increases in ZO-1 immunostaining and protein expression. These results provide a mechanistic basis for signaling pathways affected by DEX treatment that facilitate functional BBB recovery after blast injury. While entering the cell, glucocorticoids can act in three ways: (1) complex with the glucocorticoid receptor to directly initiate transcription of target genes; (2) complex with the glucocorticoid receptor and interact with transcription factors including nuclear factor-κB; or (3) initiate nongenomic signaling pathways through membrane-associated receptors and second messengers.33 It is unlikely that restoration of BBB integrity after injury was influenced by nongenomic pathways, as related studies strongly suggest the prominent role of glucocorticoid receptor signaling in initiating transcriptional and translational activity to regulate tight junction–related proteins and BBB integrity.13, 17, 19 More detailed biochemical and molecular investigation of these signaling mechanisms will help to identify promising targets for regulating recovery of the BBB after blast exposure.
Although DEX treatment after blast injury induced significant changes in TEER and hydraulic conductivity in the endothelial cultures, bEnd.3 cells may be limited in their ability to form tight monolayers relative to other cell lines and primary cell culture preparations. Transendothelial electrical resistance of the BBB in vivo is expected to range from ~2,000 to 5,000 Ω cm2, whereas TEER of immortalized brain capillary endothelial cells used to generate in vitro models of the BBB range from ~50 to 100 Ω cm2.17 Other studies have used an immortalized human brain microvascular endothelial cell line, hCMEC/D3, which has been reported to retain critical morphologic and brain endothelial markers, and form tight monolayers.34 Treatment of hCMEC/D3 cells with the glucocorticoid, hydrocortisone, resulted in an increase in TEER from ~70 to 200 Ω cm2.34 Experimental use of the human-derived cell line would be highly advantageous to the potential translation of findings from in vitro studies to the human condition. However, it is important to note that a recent study reported that the maximum TEER of hCMEC/D3 (and other) human brain endothelial cells was only ~12 Ω cm2 under representative culture conditions,35 suggesting potentially wide variability in culture quality and glucocorticoid responsiveness in hCMEC/D3 cells. In the current study, absolute TEER values of bEnd.3 cultures were close to the typical reported range of 20 to 30 Ω cm2, and had similar DEX-induced changes in TEER as a comparable endothelial cell line.10, 30 We also note that the measured time course of TEER was strongly supported by the parallel time course of hydraulic conductivity, showing sustained recovery to sham levels by 1 day postinjury (Figure 3). The twofold to threefold decrease in hydraulic conductivity (barrier tightening) owing to DEX treatment was strongly supported by previous studies describing glucocorticoid-mediated regulation of endothelial fluid-flow resistance and tight junction formation.22
Administration of DEX as early as 30 minutes after TBI in mice has been shown to result in therapeutic effects on BBB integrity, neurologic function, and secondary brain damage by 24 hours postinjury.16, 19 Other studies have also reported increases in tight junction proteins and associated BBB recovery 24 hours after DEX treatment in vitro and in vivo,13, 19 which supports our findings of potentiated recovery of TEER, hydraulic conductivity, and ZO-1 expression 1 day after treatment. Our DEX treatment regimen was also prolonged, with a new dose administered once per day after injury. DEX-induced changes in TEER and hydraulic conductivity were permanent for at least 3 days after injury, the last time point tested. Enhanced tight junction protein expression and barrier properties have been observed in endothelial cells with continual DEX treatment lasting for weeks or months, without adverse effects on culture health.17, 22, 25 However, DEX is described as a long-acting glucocorticoid with biologic half-life of 36 to 54 hours and potency greater than other glucocorticoids including hydrocortisone and methylprednisolone,36 so it may be possible that a reduction in the DEX treatment regimen (concentration and duration) would have yielded similar effects on BBB recovery. Future studies will investigate a therapeutic time window for glucocorticoid delivery to help optimize dosing strategy to rapidly restore BBB integrity and reduce treatment duration.
For over 30 years, treatment with corticosteroids was a standard clinical practice for patients with head injuries and cerebral edema.28 However, early results of the multicenter corticosteroid randomization after significant head injury trial, released in 2004, reported that 48 hours of infusion of methylprednisolone within 8 hours of head injury increased the risk of mortality in enrolled patients.28 Despite the unfavorable outcome, the exact cause and mechanism of increased mortality associated with methylprednisolone treatment remain unclear, as multiple complicating factors related to trauma were present.28 Furthermore, enrollment criteria of the trial were broadly defined, inclusive of patients with mild to severe TBI and a Glasgow coma score of 14 or less, making it difficult to realize the influence of different severities and etiologies of head injury on outcome. Unlike head injuries sustained by patients of the corticosteroid randomization after significant head injury (CRASH) trial, the current study investigated effects of DEX treatment in the context of blast-induced TBI, which is a fundamentally different biomechanical injury mechanism that warrants further study of the potential therapeutic benefits of corticosteroids after blast exposure.
Owing to potent antiinflammatory and immunosuppressant effects, glucocorticoids have been widely used in the clinic to treat brain edema, autoimmune disorders like rheumatoid arthritis, and disorders characterized by a leaky BBB like multiple sclerosis.10, 19, 21 In some cases, however, steroidal treatments have been ineffective in restoring the damaged BBB in human trauma and stroke patients.26 Dexamethasone treatment at high doses has also been reported to exacerbate cognitive deficits in a spatial acquisition task after TBI in rodents.27 In addition, treatment with DEX or other glucocorticoids has been hampered by reports of toxicity and side effects including osteoporosis, candidiasis, insulin resistance, hypertension, increased intraocular pressure, and gastrointestinal bleeding, especially after long-term treatment.10, 13, 36, 37, 38, 39 It is important to note, nonetheless, that the toxicity of glucocorticoids is dependent on the dosage and duration of treatment, with higher dosages and longer therapies associated with greater toxicity.36 Therefore, efforts to elucidate molecular mechanisms underlying the effects of glucocorticoids on the BBB may contribute to the identification of novel therapeutic targets and treatment strategies to help circumvent known side effects.
A limitation of this study is that treatment-induced regulation of other proteins essential for tight junction formation and function of the BBB, such as occludin and claudin-5,11 were not examined. In addition to ZO-1, glucocorticoid treatment has been reported to result in heightened expression of occludin and claudin-5, ultimately enhancing barrier restriction.17, 21 However, it should be noted that cell type plays an important role in determining the specific biologic signaling activity triggered by glucocorticoid treatment.18 For example, specific increases in occludin, but not claudin-5, were observed 24 hours after DEX treatment in a mouse brain endothelial cell line.13 Interpretation of these results therefore requires careful consideration of multiple factors, including cell type and species, when assessing the molecular and functional effects of glucocorticoid treatment. Despite the variable influence of treatment on tight junction protein regulation, it is remarkable that DEX-induced tightening of the BBB was abolished by inhibiting ZO-1 expression with antisense phosphorothioate oligonucleotides—supporting the critical role of ZO-1 in the formation and functional maintenance of tight junctions.22
Recent evidence supports that BBB breakdown occurs after blast injury, but detailed understanding of this phenomenon and subsequent barrier recovery remain limited. BBB damage after blast exposure has been observed by IgG and Evans blue extravasation,3, 4, 6, 9, 40 along with reduced immunostaining and protein expression of ZO-1, occludin, and claudin-5.5, 7, 8 On the contrary, others have reported BBB damage with no significant reductions in occludin or ZO-1 after blast injury in rats; however, upregulation of the same proteins with bryostatin-1 treatment resulted in restoration of barrier integrity.40 In our current study, ZO-1 immunostaining was compromised by blast exposure while ZO-1 protein expression was unaltered by blast. It is interesting that in the absence of blast-induced changes to protein levels, upregulation of ZO-1 with DEX treatment in injured cultures resulted in potentiated BBB recovery. To place our results in context of recent studies, it is possible that blast injury disrupted barrier integrity by modifying localization or cellular distribution of ZO-1, without altering total protein expression. Our results are supported by published findings that BBB breakdown in rats exposed to blast-triggered acute upregulation of the protein kinase C α (PKCα) isozyme, which has been reported to influence the redistribution of tight junction proteins and brain endothelial permeability.40 Taken together, these results emphasize that blast injury is a complex phenomenon likely associated with subtle structural changes to tight junctions that underlie BBB breakdown. Broader neuropathologic implications of blast-induced changes in BBB integrity include increased oxidative and nitrosative stress, astrocyte reactivity, and microglial activation.4, 5 Future studies will mechanistically examine molecular alterations to tight junction proteins responsible for compromised BBB integrity after blast exposure.
Although ZO-1 tight junction immunostaining recovered in DEX-treated injured cultures, changes in ZO-1 protein expression provided additional mechanistic insight that upregulation of the ZO-1α+ isoform critically contributed to potentiated BBB recovery. Specific and dramatic upregulation of ZO-1α+ effected by DEX treatment has previously been showed in human trabecular meshwork endothelial cells that express both isoforms of ZO-1.25, 37 Our results strongly agree with the literature that ZO-1α+ may play a key role in the restrictive permeability of endothelial barriers.22, 25, 37
The α+ and α− isoforms of ZO-1 are known to be differentially expressed, depending on cell type.22, 23, 25, 37 Consistent with this, we observed relatively less expression of ZO-1α+ compared with ZO-1α− in uninjured cells. Differential distribution of the two ZO-1 isoforms may provide a molecular distinction between two functional classes of tight junctions.23 Although the specific function of the 80-amino acid α domain is unclear, ZO-1α+ is predominantly expressed in epithelial cells, while ZO-1α− is more widely expressed in endothelial cells.23, 32, 37 Some studies suggest that ZO-1α+ plays a distinct role in tight junction assembly and stabilization, and may be associated with higher-resistance junctions.23, 25, 32 In addition, later expression of the ZO-1α+ isoform, after ZO-1α− expression, is reported to be a final step in tight junction synthesis and sealing during mouse blastocyst formation.32
Treatment with DEX in vivo, especially when administered as part of a combination therapy with other supplements, can exert neuroprotection after TBI.16, 19 The use of DEX as part of a combination therapy may help to enhance neuroprotection and minimize unwanted side effects by decreasing the required dosage and duration of treatment.16 For example, combination treatment with melatonin (10 mg/kg) and DEX (0.025 mg/kg) 1 and 6 hours after injury reduced the overall degree of brain injury caused by controlled cortical impact in mice.16 It is important to note that DEX treatment alone also significantly attenuated infarct size, mitigated motor deficits, and prevented expression of apoptosis proteins, inducible nitric oxide synthase, matrix metalloproteinase-2, and -9.16 Combined treatment with DEX (10 mg/kg) and bortezomib (0.2 mg/kg; a proteasome inhibitor) 30 minutes postinjury prevented proteasomal degradation of the glucocorticoid receptor after TBI, which critically enabled glucocorticoid-mediated recovery of BBB function.19 After stabilizing glucocorticoid receptor function, DEX treatment increased occludin expression, and decreased neurologic impairment, brain edema, and lesion size after controlled cortical impact in mice.19 Such recent insights and new mechanistic understanding may stimulate renewed clinical interest in the development of glucocorticoid therapies for TBI and disorders of the central nervous system.
In summary, this study has elucidated important cellular mechanisms underlying potentiated barrier recovery with DEX treatment after primary blast exposure, showing that glucocorticoid-mediated upregulation of ZO-1 tight junction protein was associated with functional restoration of an in vitro BBB model. These findings hold important implications for future development of glucocorticoid therapies and for potentially reducing mandatory rest periods for personnel exposed to blast injury.
Acknowledgments
The authors thank Gwen B Effgen and Edward W Vogel III for providing scientific insight and helpful suggestions pertaining to experimental design and development of the blast injury model. They also thank Michael R Lamprecht for skilled technical guidance in the development of the western blotting protocol.
Author Contributions
CDH designed and performed all experiments, analyzed and interpreted all experimental data, and wrote the paper; FSC performed shock-tube experiments, immunostaining, and western blotting assays; SC performed shock-tube experiments, hydraulic conductivity, and western blotting assays; CRB and DFM interpreted experimental data and edited the paper; and BM designed experiments, interpreted all experimental data, wrote and edited the paper, and provided funding.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This work was supported by a Multidisciplinary University Research Initiative from the Army Research Office (W911MF–10–1–0526), and a National Science Foundation Graduate Research Fellowship (CDH; DGE–07–07425).
Supplementary Material
References
- 1Warden D. Military TBI during the Iraq and Afghanistan wars. J Head Trauma Rehabil 2006; 21: 398–402. [DOI] [PubMed] [Google Scholar]
- 2Hoge CW, McGurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med 2008; 358: 453–463. [DOI] [PubMed] [Google Scholar]
- 3Garman RH, Jenkins LW, Switzer RC, Bauman RA, Tong LC, Swauger PV et al. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma 2011; 28: 947–959. [DOI] [PubMed] [Google Scholar]
- 4Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR, McCarron RM et al. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J Neurosci Res 2010; 88: 3530–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med 2013; 60: 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6Yeoh S, Bell ED, Monson KL. Distribution of blood-brain barrier disruption in primary blast injury. Ann Biomed Eng 2013; 41: 2206–2214. [DOI] [PubMed] [Google Scholar]
- 7Hue CD, Cao S, Dale Bass CR, Meaney DF, Morrison B, 3rd. Repeated primary blast injury causes delayed recovery, but not additive disruption, in an in vitro blood-brain barrier model. J Neurotrauma 2014; 31: 951–960. [DOI] [PubMed] [Google Scholar]
- 8Hue CD, Cao S, Haider SF, Vo KV, Effgen GB, Vogel E, 3rd et al. Blood-brain barrier dysfunction after primary blast injury in vitro. J Neurotrauma 2013; 30: 1652–1663. [DOI] [PubMed] [Google Scholar]
- 9Skotak M, Wang F, Alai A, Holmberg A, Harris S, Switzer RC et al. Rat injury model under controlled field-relevant primary blast conditions: acute response to a wide range of peak overpressures. J Neurotrauma 2013; 30: 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10Cucullo L, Hallene K, Dini G, Dal Toso R, Janigro D. Glycerophosphoinositol and dexamethasone improve transendothelial electrical resistance in an in vitro study of the blood-brain barrier. Brain Res 2004; 997: 147–151. [DOI] [PubMed] [Google Scholar]
- 11Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis 2010; 37: 13–25. [DOI] [PubMed] [Google Scholar]
- 12Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 2010; 6: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13Forster C, Waschke J, Burek M, Leers J, Drenckhahn D. Glucocorticoid effects on mouse microvascular endothelial barrier permeability are brain specific. J Physiol 2006; 573: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14Kaal EC, Vecht CJ. The management of brain edema in brain tumors. Curr Opin Oncol 2004; 16: 593–600. [DOI] [PubMed] [Google Scholar]
- 15Miller DH, Thompson AJ, Morrissey SP, MacManus DG, Moore SG, Kendall BE et al. High dose steroids in acute relapses of multiple sclerosis: MRI evidence for a possible mechanism of therapeutic effect. J Neurol Neurosurg Psychiatry 1992; 55: 450–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16Campolo M, Ahmad A, Crupi R, Impellizzeri D, Morabito R, Esposito E et al. Combination therapy with melatonin and dexamethasone in a mouse model of traumatic brain injury. J Endocrinol 2013; 217: 291–301. [DOI] [PubMed] [Google Scholar]
- 17Forster C, Silwedel C, Golenhofen N, Burek M, Kietz S, Mankertz J et al. Occludin as direct target for glucocorticoid-induced improvement of blood-brain barrier properties in a murine in vitro system. J Physiol 2005; 565: 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18Romero IA, Radewicz K, Jubin E, Michel CC, Greenwood J, Couraud PO et al. Changes in cytoskeletal and tight junctional proteins correlate with decreased permeability induced by dexamethasone in cultured rat brain endothelial cells. Neurosci Lett 2003; 344: 112–116. [DOI] [PubMed] [Google Scholar]
- 19Thal SC, Schaible EV, Neuhaus W, Scheffer D, Brandstetter M, Engelhard K et al. Inhibition of proteasomal glucocorticoid receptor degradation restores dexamethasone-mediated stabilization of the blood-brain barrier after traumatic brain injury. Crit Care Med 2013; 41: 1305–1315. [DOI] [PubMed] [Google Scholar]
- 20Singer KL, Stevenson BR, Woo PL, Firestone GL. Relationship of serine/threonine phosphorylation/dephosphorylation signaling to glucocorticoid regulation of tight junction permeability and ZO-1 distribution in nontransformed mammary epithelial cells. J Biol Chem 1994; 269: 16108–16115. [PubMed] [Google Scholar]
- 21Blecharz KG, Haghikia A, Stasiolek M, Kruse N, Drenckhahn D, Gold R et al. Glucocorticoid effects on endothelial barrier function in the murine brain endothelial cell line cEND incubated with sera from patients with multiple sclerosis. Mult Scler 2010; 16: 293–302. [DOI] [PubMed] [Google Scholar]
- 22Underwood JL, Murphy CG, Chen J, Franse-Carman L, Wood I, Epstein DL et al. Glucocorticoids regulate transendothelial fluid flow resistance and formation of intercellular junctions. Am J Physiol 1999; 277: C330–C342. [DOI] [PubMed] [Google Scholar]
- 23Balda MS, Anderson JM. Two classes of tight junctions are revealed by ZO-1 isoforms. Am J Physiol 1993; 264: C918–C924. [DOI] [PubMed] [Google Scholar]
- 24Willott E, Balda MS, Heintzelman M, Jameson B, Anderson JM. Localization and differential expression of two isoforms of the tight junction protein ZO-1. Am J Physiol 1992; 262: C1119–C1124. [DOI] [PubMed] [Google Scholar]
- 25Alvarado JA, Betanzos A, Franse-Carman L, Chen J, Gonzalez-Mariscal L. Endothelia of Schlemm's canal and trabecular meshwork: distinct molecular, functional, and anatomic features. Am J Physiol Cell Physiol 2004; 286: C621–C634. [DOI] [PubMed] [Google Scholar]
- 26Poungvarin N. Steroids have no role in stroke therapy. Stroke 2004; 35: 229–230. [DOI] [PubMed] [Google Scholar]
- 27Chen X, Lin YP, Wang D, Zhang JN. Dexamethasone exacerbates spatial acquisition deficits after traumatic brain injury in rats. Neurol Res 2010; 32: 1097–1102. [DOI] [PubMed] [Google Scholar]
- 28Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet 2004; 364: 1321–1328. [DOI] [PubMed] [Google Scholar]
- 29Effgen GB, Hue CD, Vogel E, 3rd, Panzer MB, Meaney DF, Bass CR et al. A multiscale approach to blast neurotrauma modeling: Part II: methodology for inducing blast injury to in vitro models. Front Neurol 2012; 3: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30Li G, Simon MJ, Cancel LM, Shi ZD, Ji X, Tarbell JM et al. Permeability of endothelial and astrocyte cocultures: in vitro blood-brain barrier models for drug delivery studies. Ann Biomed Eng 2010; 38: 2499–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31Lamprecht MR, Morrison B, 3rd. GPR30 activation is neither necessary nor sufficient for acute neuroprotection by 17beta-estradiol after an ischemic injury in organotypic hippocampal slice cultures. Brain Res 2014; 1563: 131–137. [DOI] [PubMed] [Google Scholar]
- 32Sheth B, Fesenko I, Collins JE, Moran B, Wild AE, Anderson JM et al. Tight junction assembly during mouse blastocyst formation is regulated by late expression of ZO-1 alpha+ isoform. Development 1997; 124: 2027–2037. [DOI] [PubMed] [Google Scholar]
- 33Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005; 353: 1711–1723. [DOI] [PubMed] [Google Scholar]
- 34Forster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol 2008; 586: 1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35Eigenmann DE, Xue G, Kim KS, Moses AV, Hamburger M, Oufir M. Comparative study of four immortalized human brain capillary endothelial cell lines, hCMEC/D3, hBMEC, TY10, and BB19, and optimization of culture conditions, for an in vitro blood-brain barrier model for drug permeability studies. Fluids Barriers CNS 2013; 10: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36Walsh D, Avashia J. Glucocorticoids in clinical oncology. Cleve Clin J Med 1992; 59: 505–515. [DOI] [PubMed] [Google Scholar]
- 37Zhuo YH, He Y, Leung KW, Hou F, Li YQ, Chai F et al. Dexamethasone disrupts intercellular junction formation and cytoskeleton organization in human trabecular meshwork cells. Mol Vis 2010; 16: 61–71. [PMC free article] [PubMed] [Google Scholar]
- 38Kimberly RP. Mechanisms of action, dosage schedules, and side effects of steroid therapy. Curr Opin Rheumatol 1991; 3: 373–379. [DOI] [PubMed] [Google Scholar]
- 39Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 2002; 96: 23–43. [DOI] [PubMed] [Google Scholar]
- 40Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z et al. Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol 2014, e-pub ahead of print. doi:10.1007/s12035-014-8902-7. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.