

Abstract
A liquid-liquid extraction and liquid chromatography-electrospray ionization tandem mass spectrometry (LC-MS/MS) method to determine iodothyronines and thyronamines (TAM) from cell culture media was developed. Thyroid hormones (TH) are metabolized by sequential deiodination to eventually yield thyronine (T0), but can also be decarboxylated, resulting in TAM. The method presented here for extraction of DMEM/F12 cell culture media is a fundamental procedure for a precise determination of 9 TH and 6 TAM from a single LC run. Analytes and internal standards (IS) were extracted from DMEM/F12 (w/o phenol red) by liquid-liquid extraction using isopropanol-TBME (30:70 v/v). Measurement of TH and TAM was performed during a 10-min run time using 13C6-T4, 13C6-T3, 13C6-rT3, 13C6-3,3′T2 and 2H4-T1AM as IS. Calibration curves covered 11 calibrators measured as triplicates each for the analysis of the 9 TH and 6 TAM metabolites, and the 5 IS were linear and reproducible in the range of 0.12-120 nM (R2 0.991-0.999) for all calibrators. The lower limit of quantification was 0.078-0.234 nM. Method validation and robustness were demonstrated by the analysis of precision, accuracy, process efficiency, matrix effects and recoveries, as well as intra- and interassay stability. These parameters were investigated for high, middle and low concentrations of quality controls of all 9 TH and 6 TAM metabolites. This validated, sensitive and interaction-free LC-MS/MS method allows rapid analysis and accurate determination of TH and TAM from DMEM/F12 (w/o phenol red) conditioned media and seems to be easily transferable and applied to commonly used buffers and cell culture media.
Key Words: Thyroid hormone metabolite, Culture media, LC-MS/MS, Liquid-liquid extraction, Thyronamine, Iodothyronine
Introduction
The biologically active thyroid hormone (TH) 3,5,3′-triiodo-L-thyronine (T3) is the classic TH pivotal to a wide variety of developmental, growth and metabolic processes. T3 is only one of nine members of the thyronine family, which differ in the number and/or position of their iodine substituents. The reductive removal of iodide atoms from TH is catalyzed by three selenoproteins, deiodinases type 1-3 (Dio1, Dio2, Dio3), which are responsible for the phenolic ring deiodination (Dio1 and Dio2) and/or tyrosyl ring deiodination (Dio1 and Dio3) [1]. Alternative pathways for TH metabolism are based on conjugation of the 4′-phenolic group to functional residues, e.g. sulfoconjugation or glucuronidation, or modifications of the alanine side chain [2]. Side chain deamination results in thyroacetic acids, whereas side chain decarboxylation generates the novel class of biologically active metabolites called thyronamines (TAM; see table 1). So far, only two representatives of TAM, namely 3-iodothyronamine (3-T1AM) and thyronamine (T0AM), have been detected in vivo [3,4,5].
Table 1.
Analyte-specific relative retention times and tandem mass spectrometer working parameters for IS, TH and TAM
| No. | Analyte | IS reference | Relative RT ± SD | PI (m/z) | Pro 1 (m/z) | Pro 2 (m/z) | DP, eV | CE, eV | CXP, eV | |
|---|---|---|---|---|---|---|---|---|---|---|
| 13C6-3,5,3′,5′-T4 | 13C6-T4 | 1 | 783.6 | 738.0 | 611.0 | 106 | 33 | 20 | 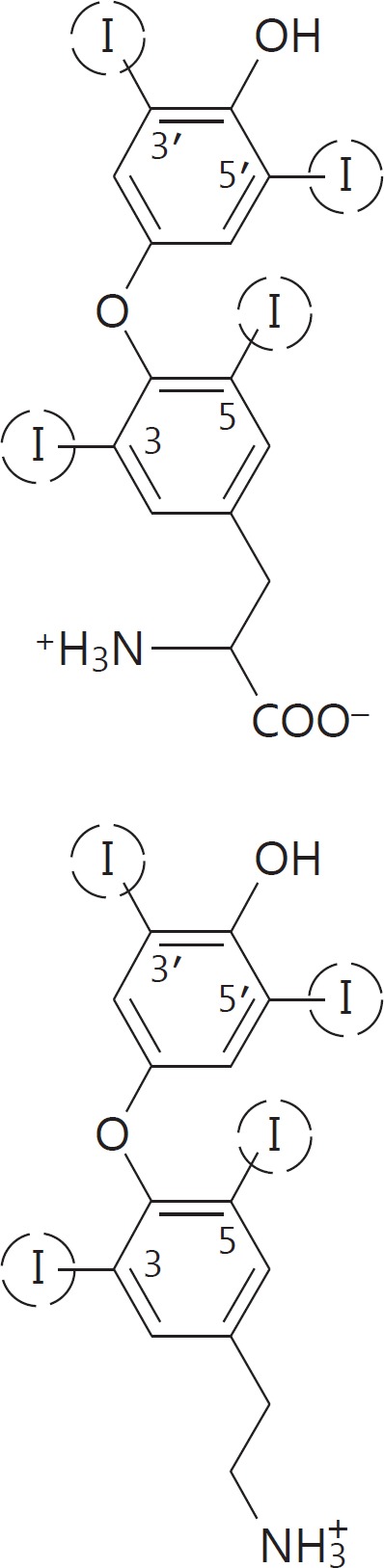 |
|
| 13C6-3,5,3′-T3 | 13C6-T4 | 0.930 ±0.001 | 657.7 | 611.8 | 203.1 | 80 | 30 | 20 | ||
| 13C6-3,3′,5′-rT3 | 13C6-T4 | 1.063 ±0.003 | 657.7 | 611.6 | 514.0 | 166 | 33 | 22 | ||
| 13C6-3,3′-T2 | 13C6-T4 | 0.967 ±0.002 | 531.8 | 485.7 | 359.0 | 116 | 29 | 18 | ||
| 2H4-3-T1AM | 2h4-t1am | 1 | 359.9 | 342.8 | 216.1 | 81 | 17 | 12 | ||
| 1 | T0 | 0.757 ±0.001 | 274.0 | 215.1 | 118.1 | 51 | 25 | 6 | ||
| 2 | 3-T1 | 0.840 ±0.002 | 399.9 | 353.9 | 341.0 | 96 | 23 | 14 | ||
| 3 | 3′-T1 | 0.895 ±0.001 | 399.9 | 341.0 | 353.9 | 131 | 29 | 14 | ||
| 4 | 3,5-T2 | 0.846 ±0.003 | 525.8 | 479.8 | 353.0 | 91 | 27 | 18 | ||
| 5 | 3,3′-T2 | 13C6-T4 | 0.967 ±0.002 | 525.8 | 479.9 | 381.9 | 116 | 27 | 18 | |
| 6 | 3′,5′-T2 | 1.007 ±0.002 | 525.8 | 508.8 | 466.8 | 146 | 23 | 18 | ||
| 7 | 3,5,3′-T3 | 0.929 ±0.001 | 651.7 | 605.6 | 478.9 | 131 | 33 | 24 | ||
| 8 | 3,3′,5′-rT3 | 1.062 ±0.003 | 651.7 | 605.8 | 478.9 | 130 | 47 | 9 | ||
| 9 | 3,5,3′,5′-T4 | 0.999 ±0.001 | 777.6 | 731.6 | 604.7 | 121 | 37 | 28 | ||
| 10 | 3,5,3′,5′-T4AM | 1.248 ±0.002 | 733.6 | 716.5 | 335.8 | 156 | 29 | 28 | ||
| 11 | 3,5,3′-T3AM | 1.130 ±0.002 | 607.7 | 590.7 | 209.9 | 141 | 25 | 24 | ||
| 12 | 3,3′,5′-rT3AM | 2h4-t1am | 1.354 ±0.004 | 607.7 | 590.7 | 209.9 | 136 | 23 | 22 | |
| 13 | 3,5-T2AM | 1.000 ±0.002 | 481.8 | 464.9 | 337.8 | 121 | 21 | 18 | ||
| 14 | 3-T1AM | 1.000 ±0.000 | 355.8 | 338.9 | 212.1 | 91 | 15 | 10 | ||
| 15 | T0AM | 0.865 ±0.001 | 230.0 | 213.0 | 108.8 | 11 | 15 | 12 | ||
The circled iodine atoms can be replaced by hydrogen, giving rise to the various thyronines (upper figure) or TAM (lower figure) metabolites. m/z = Mass-to-charge ratio; PI = precursor ion; Pro = product ion; DP = declustering potential; CE = collision energy; CXP = collision cell exit potential.
In recent years, various TAM and TH metabolites have attracted increased attention because of their thyromimetic and TH antagonistic effects [6,7,8]. These observations raised interest in the quantitative detection of the wider spectrum of TH and TAM in serum, tissues and various in vitro cell culture conditions. Classic TH concentrations are typically measured using antibody-based immunoassays specific for an individual analyte, thus requiring significant sample volumes and multiple assays if the full spectrum of the TH and TAM family is of analytical interest. Technical advances in HPLC liquid chromatography and its subsequent combination with tandem mass spectrometry (LC-MS/MS) provided a highly sensitive method to measure part – most often the classic metabolites – of the TH spectrum in human serum or plasma and selected animal tissue specimens [4,9,10,11,12,13,14,15]. This analytical technique identifies the molecule of interest in a complex sample by its retention time (RT) and the mass-to-charge ratios of parent and fragmentation ions. In order to determine concentrations of the wider spectrum of TH and TAM metabolites in cell culture media, in the absence or presence of fetal bovine serum, we have developed and applied a liquid-liquid extraction method for commonly used cell culture media. Preanalytical extraction of samples enriched the analytes of interest and removed contaminants, resulting in better signal-to-noise ratios and simultaneously limiting memory effects of the preanalytical column. In a single chromatographic run, the extracted spectrum of TH and TAM metabolites can then be unequivocally and concurrently identified, and quantified by the new LC-MS/MS method presented.
Materials and Methods
Reagents, Internal Standards, Calibrators and Quality Control Samples
High-purity TH metabolites, T4AM, T3AM and rT3AM, were provided by Dr. R. Thoma (Formula GmbH, Berlin, Germany), who also purified 3-T1AM by preparative HPLC. Synthesis of high-purity 3-T1AM and T0AM was performed by Dr. R. Smits (ABX Advanced Biochemical Compounds, Radeberg, Germany). 3,5-T2AM and the internal standard (IS) 2H4-3-T1AM were kindly provided by T.S. Scanlan (OHSU, Portland, Oreg., USA) [16]. Further IS, 13C6-T4, 13C6-T3, 13C6-rT3 and 13C6-3,3′-T2, were obtained from Isosciences LLC (King of Prussia, Pa., USA). 125I-radiolabelled T4, T3 and rT3 were purchased from PerkinElmer (Hamburg, Germany). Water was purified by a Milli-Q water purification system from Millipore (Billerica, Mass., USA). Dimethyl sulfoxide, TBME (tert-butyl methyl ether; CHROMASOLV® Plus), methanol, formic acid, 2-propanol, acetonitrile (LC-MS CHROMASOLV) and HCl (37%) were purchased from Sigma-Aldrich Chemie Gmbh (Munich, Germany). DMEM/F-12 (1:1 mixture of DMEM and Ham's F-12, containing 15 mM HEPES and 2.5 mM L-glutamine) without phenol red was purchased from Life Technologies GmbH (Darmstadt, Germany).
Preparation of Quality Controls in Dulbecco's Modified Eagle's Medium
IS mixture stock solution was prepared in methanol, using 13C6-T4, 13C6-T3, 13C6-rT3, 13C6-3,3′-T2 and 2H4-3-T1AM, each at a concentration of 1 µM. Two stock solutions containing either TH (T4, T3, rT3, 3,3′-T2, 3,5-T2, 3′,5′-T2, 3-T1, 3′-T1 and T0) or TAM (T4AM, T3AM, rT3AM, 3,5-T2AM, 3-T1AM and T0AM), each at a concentration of 20 µM, were separately prepared in methanol and stored at −20°C in brown glass vials with screw tops. Using pure DMEM/F12 without phenol red, we prepared several calibrators (0.015, 0.030, 0.039, 0.059, 0.078, 0.117, 0.234, 0.469, 0.937, 1.875, 3.75, 7.5, 15, 30, 60, 120, 240 and 480 nM of both TH and TAM) and in-house high- (80 nM), medium- (10 nM) and low- (0.75 nM) concentration quality controls (HQAL, MQAL and LQAL, respectively) of both TH and TAM for the assay.
Sample Preparation of TH and TAM for LC-MS/MS
Sample preparation was performed in 2.0-ml Eppendorf tubes. Four hundred microliters of DMEM/F12 (w/o phenol red) was spiked with ice-cold 10-µl IS solution, acidified with 5 µl of 30% HCl, vortex mixed for 15 s and incubated for 30 min at 37°C in the dark. Liquid-liquid extraction was performed by two subsequent extractions, using 1 ml of freshly mixed 30/70 2-propanol/TBME (v/v). The upper organic phases were combined in a 1.5-ml Eppendorf tube, evaporated to dryness (Eppendorf concentrator 5301 at 45°C), and reconstituted in 100 μl of 50/50 methanol/water (v/v; containing 0.1% formic acid) by vortex-mixing for 20 s. Samples were centrifuged at 14,000 rpm for 5 min and stored at −20°C until LC-MSMS investigation.
LC-MS/MS Analysis
For the measurement of TH, TAM and IS analytes, a HSS PFP 2.5 µm 3.0 × 100-mm column (Waters, Milford, Mass., USA) maintained at 40°C was used for separation by an 1260 quaternary HPLC system (Agilent Technologies GmbH, Waldbronn, Germany) directly coupled to a mass spectrometer QTrap 6500 (AB SCIEX Germany GmbH, Darmstadt, Germany) fitted with a Turbo Spray IonDrive. The PAL HTC-xt auto-sampler (CTC Analytics AG, Zwingen, Switzerland) used wash 1 solution of 25/25/25/25 water/acetonitrile/2-propanol/methanol (v/v/v/v) and wash 2 solution of 50/50 water/methanol (v/v). A 20-µl sample volume was injected at an HPLC flow rate of 0.9 ml/min. The HPLC solvents consisted of 0.1% (v/v) formic acid in H2O (eluent A) and 0.1% (v/v) formic acid in methanol (eluent B). The total run time of the gradient program was 10 min and is described in online supplementary figure 1 (for all online suppl. material, see www.karger.com/doi/10.1159/000430840).
The MS was operated in electrospray positive ionization low-mass mode, system control and data acquisition were conducted by Analyst 1.6.2, and data were processed using the Multiquant 2.1.1 software provided with the instrument. Nitrogen was used as the nebulizing and collision gas. The instrument settings were as follows: source temperature, 600°C; curtain gas, 45; IonSpray gas 1, 55; IonSpray gas 2, 70, and IonSpray voltage, 4,500 V. Sample analysis was recorded in the multiple reaction monitoring (MRM) mode of the instrument, with a dwell time of 10-15 ms. MRM transitions were individually optimized for each single analyte (table 1). The entrance potential was set to 10 V for all transitions.
Method Validation
The assay was validated according to the US Food and Drug Administration (FDA) guidelines, determining the most important test characteristics, such as selectivity, accuracy, precision, recovery, matrix effect (ME), linearity, lower limit of detection (LLOD) and lower limit of quantification (LLOQ) [17].
System Suitability and Chromatographic Stability
System suitability experiments were performed by injecting an aqueous standard mixture of TH and TAM (100 nM) at the start of each batch during the method validation. The carryover effect of the auto-sampler was evaluated by injecting a sequence of solutions of blank (water), aqueous standard, mobile phase, blank (water) and extracted standard equivalent to the highest standard in the calibration range. As per the acceptance criteria, the response in the blank should not be greater than 20% of the LLOQ response [18].
To verify the robustness of the proposed HPLC method, a column method validation kit, consisting of 3 analytical columns, each derived from a different batch, was tested under various calibrator concentrations to confirm the full separation of TH and TAM metabolites. Each column was individually tested and the relative RT and peak resolution investigated.
Linearity Studies
Matrix-based calibration curves for both TH and TAM metabolites were constructed using pure DMEM/F12 w/o phenol red. One hundred microliters of the TAM (20 µM) and 100 µl of the TH (20 µM) stock solutions were mixed; this solution was used as the starting calibrator solution and when used at 19.2 µl in 400 µl DMEM/F12 w/o phenol red gave a concentration of 480 nM. The starting calibrator solution was serial diluted 1:1 or 1:2 with ice-cold methanol. The linearity was assessed by analysis of four linear curves containing eleven calibrators in triplicate and ranging from 0.117 to 120 nM (n = 2) or 0.078 to 80 nM (n = 2). The ratio of area under the curve (AUC) response for analyte and IS was used for regression analysis. For each calibration curve a coefficient of determination was calculated by using least square weighted (1/x2) linear regression.
Lower Limit of Detection or Quantification
The LLOQ was calculated as the minimum concentration at which each TH and TAM can be reliably quantified with a precision ≤20%, signal-to-noise ratio (S/N) >10 and an accuracy within the range of 80-120%. The LLOD was calculated as the smallest detectable peak above baseline noise (here conservatively set as S/N >6:1). The deviation of standards other than LLOQ from nominal concentration should not to be more than ±15.0%.
Selectivity
The selectivity of the method towards endogenous DMEM/F12 w/o phenol red matrix components was assessed in 5 measurements of blank extracted DMEM/F12 w/o phenol red. This was done to estimate the extent to which endogenous components contribute towards interference at the RT of the analytes and IS.
Precision and Accuracy
Interassay precision and accuracy were assessed on HQAL, MQAL and LQAL samples, in each performed in triplicate for 10 different days (n = 10). To determine intra-assay accuracy and precision, ten samples of HQAL, MQAL and LQAL were extracted and analyzed on the same day. The precision of the method was determined by calculating the percent coefficient of variation (CV) for each TH and TAM concentration from HQAL, MQAL and LQAL samples. The deviation at each concentration level from the nominal concentration was expected to be within ±15.0% except LLOQ, for which it should be within ±20.0%.
Recovery, Matrix Effect and Process Efficiency
The process efficiency (PE), relative recovery (RE) and ME of DMEM/F12 extraction were assessed at 3 different TH and TAM concentrations (0.75, 10 and 80 nM) in triplicate over 10 different days. On each day further triplicates were made of the standards spiked into the neat resuspension buffer (hence without DMEM/F12) and were not extracted. The PE was calculated by the ratio of the mean area response of extracted samples spiked before extraction (SB) to that with mean area of neat standard solutions (SN) at each TH and TAM concentration [(SB/SN) × 100] [19]. ME was calculated using the postextraction addition technique, the ratio of the mean area response of extracted samples spiked after extraction SA and that of SN [(SA/SN) × 100] [20,21]. The RE was calculated as the mean area response of [(SB/SA) × 100] [19].
Method Comparison and Application
The recovery values for T4, T3 and rT3, derived from our proposed LC-MS/MS method, were compared with the extraction yield of the 125I-labelled T4, T3 or rT3, respectively, from DMEM/F12 w/o phenol red following our presented liquid-liquid extraction protocol. The 125I was counted using a γ-counter (1277 Gammamaster, LKB Wallac, Turku, Finland). The recovery was calculated as the counts per minute (cpm) after extraction/cpm total counts.
The proposed LC-MS/MS method was applied to medium (DMEM; Biochrom GmbH, Berlin, Germany) containing 1.5 g/l glucose, 1% glutamine, 0.5% DMSO, 100 IU/ml penicillin G, 100 IU/ml streptomycin, 100 nM Na2SeO3 with or without 100 nM T3 for 24 h (n = 3). The primary hepatocytes were isolated from 3-month-old adult C57BL/6 N male mice. Animal care and experiments were performed in accordance with the institutional guidelines of the German Federal Law and local authorities of Berlin. The mice were sacrificed and livers were initially perfused with a calcium-free Earle's Balanced Salt Solution (Gibco, Life Technologies, Carlsbad, Calif., USA) containing 50 mM EGTA, and subsequently digested with 0.3 mg/ml type II collagenase (Worthington Biochemical Co., Lakewood N.J., USA) in Hanks' balanced salt solution (Biochrom GmbH) via the inferior vena cava to isolate hepatocytes. A total of 4 × 105 cells/well were seeded on collagen-coated 6-well plates. Cells were cultured in medium as described above but with 10% fetal calf serum (FCS) and 4.5 g/l glucose. Before the start of the experiment, the cells were FCS starved/1% bovine serum albumin (BSA) supplemented for 24 h.
Results
System Suitability and General Characterization of the LC-MS/MS Method
Carryover from a methanol-based 100-nM TH and TAM calibrator mixture (10 µl injected) to subsequently measured neat water blank was not observed. The injection served additionally as a quality control for chromatographic separation of analytes (online suppl. fig. 1) for the following analytical run. To calculate the relative RT, the absolute RT of the analyte is divided by the RT of the corresponding IS reference (n = 123). For thyronines the IS reference RT is 13C6-T4 (RT 5.48-5.67 min), whereas 2H4-T1AM is the reference for TAM (RT 5.71-5.87 min). The relative RT of all IS, TH and TAM remained very consistent, with CV <0.4% for any analyte (table 1). 3-T1AM and 3,5-T2AM have a similar RT, but specific ion fragment selectivity of the mass spectrometer is sufficient for an individual and precise quantification of the peaks. No crosstalk for 3-T1AM, which might occur due to deiodination, was observed after injecting high 3,5-T2AM concentrations. Maximum sensitivity for the calibrators and the IS was achieved by monitoring the fragmentation of single-charged molecule ions (analyte + H+) with m/z transitions as described in table 1 (see also online suppl. fig. 2). The most intense peak was used for quantification, except for 13C6-3,3′-T2, 3,3′-T2 and 3-T1AM. The slightly less intense peak was chosen for reliable quantification of the latter analytes to overcome the background signals at the LLOQ. The IS were used to minimize any analytical variation due to solvent evaporation, column integrity and ionization efficiency of analytes. The related IS for the TH and TAM used to establish the calibration curves are listed in table 2. The structurally identical IS for the corresponding analytes elute at the same RT; therefore, quantitative errors resulting from potential ion suppression are compensated via IS for T4, T3, rT3, 3,3′-T2 and 3-T1AM. For all analytes without a structural identical IS, we have chosen the most similar IS in terms of structure and functional groups.
Table 2.
Interday validation outcome for TH and TAM extraction
| Interday (n = 10)/hormone | Linear regression |
HQAL (80 nM) |
MQAL (10 nM) |
LQAL (0.75 nM) |
LLOQ, pM | LLOD, pM | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| related IS | R2 range (n = 4) | mean conc., nM % | CV, % | accuracy, % | mean conc., nM | CV, % | accuracy, % | mean conc., nM | CV, % | accuracy, % | |||
| T0 | 13C6-3,3′-T2 | 0.996–0.999 | 82.97 | 7.9 | 103.7 | 11.41 | 9.8 | 114.1 | 2.29 | 33.0 | 296.2 | – | – |
| 3-T1 | 13C6-3,3′-T2 | 0.991 – 0.997 | 80.17 | 5.0 | 100.2 | 10.20 | 5.5 | 102.0 | 0.79 | 13.7 | 102.8 | 78 | 30 |
| 3′-T1 | 13C6-3,3 ′-T2 | 0.994 – 0.998 | 80.47 | 5.3 | 100.6 | 10.36 | 4.8 | 103.6 | 0.78 | 8.7 | 101.1 | 39 | 15 |
| 3,5-T2 | 13C6-3,3 ′-T2 | 0.997 – 0.999 | 77.36 | 4.9 | 96.7 | 10.33 | 5.4 | 103.3 | 0.77 | 8.5 | 100.4 | 78 | 30 |
| 3,3′ -T2 | 13C6-3,3 ′-T2 | 0.997 – 0.998 | 79.76 | 5.7 | 99.7 | 10.34 | 6.6 | 103.4 | 0.77 | 8.9 | 100.5 | 78 | 15 |
| 3 ′, 5 ′-T2 | 13C6-3,3 ′-T2 | 0.994 – 0.998 | 80.70 | 6.7 | 100.9 | 10.43 | 7.7 | 104.3 | 0.77 | 9.5 | 100.5 | 78 | 30 |
| T3 | 13C6-T3 | 0.998 – 0.999 | 78.91 | 8.1 | 98.6 | 10.24 | 7.6 | 102.4 | 0.77 | 7.7 | 100.1 | 78 | 15 |
| rT3 | 13C6-rT3 | 0.994 – 0.999 | 83.08 | 5.2 | 103.9 | 10.36 | 6.6 | 103.6 | 0.79 | 8.6 | 103.0 | 78 | 15 |
| T4 | 13C6-T4 | 0.995 – 0.998 | 75.28 | 12.2 | 94.1 | 9.99 | 11.0 | 99.9 | 0.75 | 10.8 | 97.7 | 78 | 15 |
| T4AM | 2H4-3-T1AM | 0.994 – 0.997 | 90.56 | 12.0 | 113.2 | 11.08 | 11.2 | 110.8 | 0.82 | 10.9 | 106.7 | 234 | 78 |
| t3am | 2H4-3-T1AM | 0.998 – 0.999 | 85.68 | 7.2 | 107.1 | 10.90 | 8.9 | 109.0 | 0.82 | 10.1 | 106.0 | 39 | 15 |
| rT3AM | 2H4-3-T1AM | 0.998 – 0.998 | 88.25 | 6.4 | 110.3 | 11.41 | 7.2 | 114.1 | 0.85 | 9.6 | 110.3 | 78 | 15 |
| 3,5-T2AM | 2H4-3-T1AM | 0.997 – 0.998 | 85.23 | 5.1 | 106.5 | 10.98 | 7.3 | 109.8 | 0.82 | 7.9 | 106.9 | 78 | 15 |
| 3-T1AM | 2H4-3-T1AM | 0.997 – 0.999 | 84.28 | 4.6 | 105.4 | 10.92 | 7.1 | 109.2 | 0.82 | 8.5 | 105.8 | 78 | 15 |
| T0AM | 2H4-3-T1AM | 0.994 – 0.998 | 85.11 | 4.8 | 106.4 | 10.96 | 6.6 | 109.6 | 0.87 | 10.1 | 112.3 | 78 | 59 |
R2 = Coefficient of determination; mean conc. = mean determined concentration. DMEM/F12 contains approximately 1.54 nM T0 as endogenous background concentration.
Linearity and LLOQ
The chosen related IS were used to establish calibration curves for each analyte, resulting in linear regression curves over the working range of 0.117-120 nM with a coefficient of determination of at least R2 ≥0.991 (table 2). The LLOD of DMEM/F12 w/o phenol red extracted analytes was identified in the range of 15-78 pM, whereas the LLOQ ranged from 39 to 234 pM (table 2).
Accuracy and Precision
The intra- and interbatch precision and accuracy were measured for several concentrations of TH and TAM analytes (HQAL, MQAL and LQAL). The within-run CVs were all <6.4% (table 3), whereas the between-run CVs were <13.7%, except for T0 with 33.0% (table 2). The interday batch analysis of the LQAL revealed an endogenous concentration of 1.54 nM for T0 that is present in the matrix (DMEM/F12 w/o phenol red; table 2). The accuracy values for the interday batch analysis were within 96.7-113.2% for all analytes except T0 (table 2), whereas intraday accuracy ranged from 89.0 to 119.2% for all analytes except T0 (table 3).
Table 3.
Intraday validation outcome for TH and TAM extraction
| Intraday (n = 10)/ analytes | HQAL (80 nM) |
MQAL (10 nM) |
LQAL (0.75 nM) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| mean conc., nM | CV, % | accuracy, % | mean conc., nM | CV, % | accuracy, % | mean conc., nM | CV, % | accuracy, % | |
| T0 | 82.61 | 3.7 | 103.3 | 12.62 | 2.1 | 126.2 | 2.93 | 1.9 | 391.1 |
| 3-T1 | 77.08 | 2.7 | 96.4 | 10.62 | 1.9 | 106.2 | 0.82 | 4.1 | 108.7 |
| 3′-T1 | 73.07 | 2.6 | 91.3 | 10.23 | 2.9 | 102.3 | 0.82 | 3.9 | 109.7 |
| 3,5-T2 | 71.71 | 1.6 | 89.6 | 10.16 | 3.1 | 101.6 | 0.80 | 2.7 | 106.6 |
| 3,3′-T2 | 73.67 | 2.7 | 92.1 | 9.94 | 2.9 | 99.4 | 0.79 | 3.6 | 104.7 |
| 3′,5′-T2 | 75.24 | 2.3 | 94.0 | 10.51 | 3.3 | 105.1 | 0.81 | 3.9 | 107.8 |
| T3 | 71.21 | 2.4 | 89.0 | 9.70 | 3.0 | 97.0 | 0.76 | 3.1 | 100.7 |
| rT3 | 78.45 | 2.3 | 98.1 | 10.36 | 2.6 | 103.6 | 0.77 | 3.1 | 102.4 |
| T4 | 72.20 | 2.0 | 90.2 | 9.62 | 4.1 | 96.2 | 0.72 | 6.4 | 96.5 |
| t4am | 90.07 | 4.6 | 112.6 | 11.60 | 2.5 | 116.0 | 0.89 | 5.1 | 119.2 |
| t3am | 84.32 | 1.8 | 105.4 | 11.34 | 2.5 | 113.4 | 0.88 | 3.5 | 116.7 |
| rT3AM | 83.66 | 2.1 | 104.6 | 11.35 | 2.9 | 113.5 | 0.86 | 4.5 | 114.0 |
| 3,5-T2AM | 83.55 | 2.0 | 104.4 | 11.25 | 2.2 | 112.5 | 0.86 | 3.5 | 115.2 |
| 3-T1AM | 82.72 | 2.6 | 103.4 | 11.31 | 2.8 | 113.1 | 0.88 | 3.1 | 117.3 |
| T0AM | 88.76 | 1.2 | 111.0 | 11.50 | 1.5 | 115.0 | 1.02 | 3.9 | 111.2 |
R2 = Coefficient of determination; mean conc. = mean determined concentration. DMEM/F12 contains approximately 1.54 nM T0 as endogenous background concentration.
Recovery, Matrix Effect and Process Efficiency
The PE, ME and recovery data at HQAL, MQAL and LQAL concentrations are presented in table 4. The recoveries for all TAM, TH and IS were in the range of 80-101%. Matching these findings were the recoveries of the 125I-labelled T4, T3 and rT3, which ranged between 91 and 94%. The ME were between 77 and 105%, resulting in solid PE from 70 to 101%, except for T0 which is present in the matrix at a nanomolar concentration.
Table 4.
PE, ME and RE for the IS, TH and TAM extraction
| HQAL (80 nM) |
MQAL (10 nM) |
LQAL (0.75 nM) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| PE | ME | RE | PE | ME | RE | PE | ME | RE | |
| n = 9 | |||||||||
| 13C6-T4 | 85± 6 | 91 ± 8 | 88 ± 7 | 82 ±6 | 96 ± 7 | 87 ± 8 | 81 ± 4 | 97 ± 6 | 84 ±5 |
| 13c6-t3 | 86± 9 | 90 ± 18 | 88± 9 | 85 ±7 | 99 ± 12 | 88 ± 10 | 83 ± 4 | 99 ± 10 | 84 ±5 |
| 13C6-rT3 | 86± 6 | 91 ± 13 | 90± 9 | 83 ±6 | 93 ± 10 | 90 ± 10 | 79 ± 5 | 93 ± 9 | 86 ±6 |
| 13C6-3,3′-T2 | 89± 7 | 92 ± 12 | 91± 9 | 87 ±4 | 98 ± 9 | 91 ± 9 | 85 ± 3 | 96 ± 5 | 88 ±5 |
| 2H4-3-T1AM | 90± 10 | 93 ± 18 | 93± 8 | 88 ±6 | 97 ± 13 | 93 ± 9 | 85 ± 5 | 95 ± 10 | 90 ±6 |
| n = 10 | |||||||||
| T0 | 70± 16 | 77 ± 19 | 80± 9 | 80 ±16 | 97 ± 23 | 85 ± 10 | 209 ± 97 | 226 ± 96 | 92 ±9 |
| 3-T1 | 84± 15 | 87 ± 14 | 88± 7 | 86 ±13 | 97 ± 16 | 89 ± 9 | 84 ± 11 | 97 ± 14 | 87 ±9 |
| 3′-T1 | 87± 13 | 89 ± 13 | 89± 6 | 90 ±15 | 98 ± 15 | 93 ± 9 | 84 ± 10 | 93 ± 12 | 90 ±5 |
| 3,5-T2 | 84± 14 | 88 ± 13 | 88± 6 | 87 ±14 | 98 ± 14 | 90 ± 9 | 83 ± 11 | 94 ± 14 | 88 ±6 |
| 3,3′-T2 | 90± 12 | 91 ± 8 | 90 ± 6 | 94 ±16 | 101 ± 14 | 95 ± 10 | 88 ± 9 | 96 ± 12 | 92 ±7 |
| 3′,5′-T2 | 90± 11 | 92 ± 9 | 92 ± 5 | 91 ±15 | 96 ± 13 | 96 ± 10 | 85 ± 10 | 92 ± 11 | 93 ±7 |
| T3 | 91± 13 | 91 ± 13 | 90± 6 | 93 ±17 | 101 ± 15 | 93 ± 11 | 89 ± 11 | 99 ± 12 | 90 ±7 |
| rT3 | 92± 12 | 91 ± 11 | 90± 6 | 94 ±15 | 101 ± 15 | 94 ± 10 | 89 ± 11 | 100 ± 15 | 90 ±7 |
| T4 | 91± 11 | 93 ± 6 | 92 ± 6 | 94 ±17 | 99 ± 14 | 96 ± 11 | 89 ± 12 | 94 ± 13 | 95 ±9 |
| T4AM | 96± 14 | 96 ± 15 | 97± 8 | 97 ±16 | 97 ± 16 | 101 ± 10 | 96 ± 18 | 99 ± 22 | 98 ±13 |
| T3AM | 95± 13 | 94 ± 12 | 93± 6 | 100±18 | 104 ± 16 | 98 ± 11 | 96 ± 12 | 100 ± 14 | 97 ±8 |
| rT3AM | 95± 13 | 93 ± 13 | 92± 7 | 101±20 | 105 ± 17 | 96 ± 13 | 94 ± 12 | 100 ± 12 | 94 ±10 |
| 3,5-T2AM | 89± 13 | 90 ± 13 | 89± 5 | 91 ±16 | 99 ± 16 | 94 ± 9 | 88 ± 12 | 97 ± 13 | 91 ±6 |
| 3-T1AM | 89± 13 | 90 ± 13 | 90± 5 | 91 ±14 | 99 ± 14 | 94 ± 9 | 86 ± 10 | 95 ± 13 | 91 ±6 |
| T0AM | 83± 14 | 85 ± 15 | 84± 6 | 85 ±14 | 98 ± 16 | 87 ± 9 | 85 ± 19 | 101 ± 16 | 84 ±8 |
All values are percentages (±SD). DMEM/F12 contains approximately 1.54 nM T0 as endogenous background concentration, the findings for the PE and ME of this analyte are confounded.
Method Applied to Cell Culture Media Containing Stripped FCS
The transferability of our presented LC-MS/MS method was investigated via extracting 400 µl of 10% stripped (hormone reduced) FCS in DMEM/F12 w/o phenol red. The established calibration curves were linear (R2 0.991-0.998) over the concentration range of 0.117 to 120 nM for all TH and TAM (T4AM ranges from 0.23 to 120 nM; online suppl. table 1). The accuracy of determination of QALs were 88.4-111.7% with an interbatch precision from 1.7-10.0% for all TH and TAM, except T0 due to the endogenous matrix content. The PE, ME and RE were in the same range as those for the DMEM/F12 w/o FCS (online suppl. table 2).
Method Applied to Cell Culture Media after Incubation with Primary Hepatocytes
The LC-MS/MS method was applied to investigate the appearance of TH metabolites in culture media after incubation of T3 with primary hepatocytes as a model for cellular deiodinase activity. A 24-hour incubation of 100 nM T3 resulted in deiodination of T3 into 3,3′-T2 (1.59 ± 0.20 nM) and its further metabolites 3-T1 (8.16 ± 0.55 nM) and T0 (2.04 ± 0.06 nM). The initial 100 nM T3 concentration was reduced to 36.25 (± 1.82) nM. The preliminary findings of extracted hepatocyte homogenates indicate that 44% of the initial T3 added was bound to or taken up by the hepatocytes. The endogenous TH metabolite concentrations in the control group treated with DMSO were below the LLOD. These findings indicate that the primary hepatocytes exhibit an effective phenolic and tyrosyl ring deiodination capacity.
Discussion
Liquid chromatography mass spectrometry is becoming increasingly important as a major tool for the precise and simultaneous analysis of a broad range of biological molecules. The use of MS/MS and stable isotope IS permits the development of highly sensitive and specific assays [22,23,24]. Here, we report the validation of the first high-performance stable isotope dilution LC-MS/MS method to measure 9 TH and 6 TAM from a single cell culture media sample using a simple and fast liquid-liquid extraction. Compared to the cell homogenate LC-MS/MS method from Piehl et al. [9] (LLOQ 250-5,000 pM) we were able to increase the LLOD and LLOQ, and therefore the sensitivity of all TH and TAM up to 64-fold. In addition, for the first time the full separation of all TH was achieved within a significantly reduced chromatographic run time from 29 min down to 10 min (online suppl. fig. 1) [9]. Our experience underlines that the herein performed usage of structurally identical or, if not available, at least similar IS for TH or TAM is very beneficial. The appropriate IS can compensate for the alteration of analytes in complex matrices during sample preparation, adhesion effects or ion suppression during electrospray ionization during the MS measurement by coelution from the LC [12].
Recent LC-MS/MS investigations of TH or TAM were based on direct injection of diluted cell lysates and/or cell culture media (w/o FCS) without analyte extraction [25,26,27]. Sample preparation (solid-phase extraction, SPE, liquid-liquid extraction or online SPE) prior to LC-MS/MS analysis can improve solid and high RE with a minor ME, while preventing accelerated deterioration of analytical columns and contamination of the LC-MS/MS system. Here we have shown a validated LC-MS/MS method, following the FDA guidelines [17], and successfully transferred the method to extract all TH, except T0, and TAM from DMEM/F12 containing 10% TH-depleted FCS. The residual TH metabolites in culture media or TH-depleted FCS influences the ME and PE. The existence and role of T0 in cell culture media (T0 is also described in humans [28]) has yet to be studied in any detail. We could also show that proteins are fully separated from the TH/TAM/IS by the liquid-liquid extraction (online suppl. fig. 3) since RE of TH and TAM remain high (online suppl. table 1S). In addition, we could prove a good correlation between our LC-MS/MS assay recovery (T3, rT3 and T4) and the radiolabeled iodine tracer recovery of T3, rT3 and T4 from DMEM/F12 in the absence or presence of 10% FCS, resulting in 91-94% return of tracers. Preliminary tracer experiments using RPMI 1640 and Coons F12 both with or without 10% FCS revealed >90% return of the tracer after extraction. The incubation of 100 nM T3 in DMEM on primary hepatocytes shows that the preanalytical extraction method and the LC-MS/MS procedure can be applied to identify the appearance of TH metabolites. This supports our idea that the presented method can very easily be transferable to any other cell culture media/buffer system, since DMEM/F12 + 10% stripped FCS most likely represents one of the more complex matrices, but of course this needs to be tested before application.
Disclosure Statement
The authors declare that there are no competing interests.
Supplementary Material
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgements
The authors gratefully acknowledge Raymond Monk and Anja Fischbach for their expert technical assistance, and Dr. R. Thoma (Formula GmbH, Berlin, Germany), for generous provision of some of the rare TH metabolites. Dr. T.S. Scanlan (Portland, Oreg., USA) kindly supplied isotope-labeled TAM standards and some of the TAM metabolites, and is highly supportive of this project with continuing advice.
This work is supported by grants from the Deutsche Forschungsgemeinschaft within the DFG-SPP 1629 Thyroid Trans Act KO 922/17-1 to J.K.
References
- 1.Köhrle J, Jakob F, Contempre B, Dumont JE. Selenium, the thyroid, and the endocrine system. Endocr Rev. 2005;26:944–984. doi: 10.1210/er.2001-0034. [DOI] [PubMed] [Google Scholar]
- 2.Wu SY, Green WL, Huang WS, Hays MT, Chopra IJ. Alternate pathways of thyroid hormone metabolism. Thyroid. 2005;15:943–958. doi: 10.1089/thy.2005.15.943. [DOI] [PubMed] [Google Scholar]
- 3.Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, Lin ET, Hatton D, Zucchi R, Grandy DK. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat Med. 2004;10:638–642. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- 4.DeBarber AE, Geraci T, Colasurdo VP, Hackenmueller SA, Scanlan TS. Validation of a liquid chromatography-tandem mass spectrometry method to enable quantification of 3-iodothyronamine from serum. J Chromatogr A. 2008;1210:55–59. doi: 10.1016/j.chroma.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hillmann G, Keil B, Taslimi P. Determination of thyroxamine in the thyroid gland and plasma (in German) Z Naturforsch B. 1958;13B:820–821. [PubMed] [Google Scholar]
- 6.Orozco A, Navarrete-Ramirez P, Olvera A, Garcia GC. 3,5-Diiodothyronine (T2) is on a role: a new hormone in search of recognition. Gen Comp Endocrinol. 2014;203:174–180. doi: 10.1016/j.ygcen.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 7.Piehl S, Hoefig CS, Scanlan TS, Köhrle J. Thyronamines – past, present, and future. Endocr Rev. 2011;32:64–80. doi: 10.1210/er.2009-0040. [DOI] [PubMed] [Google Scholar]
- 8.Vergani L. Lipid lowering effects of iodothyronines: in vivo and in vitro studies on rat liver. World J Hepatol. 2014;6:169–177. doi: 10.4254/wjh.v6.i4.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piehl S, Heberer T, Balizs G, Scanlan TS, Köhrle J. Development of a validated liquid chromatography/tandem mass spectrometry method for the distinction of thyronine and thyronamine constitutional isomers and for the identification of new deiodinase substrates. Rapid Commun Mass Spectrom. 2008;22:3286–3296. doi: 10.1002/rcm.3732. [DOI] [PubMed] [Google Scholar]
- 10.Wang D, Stapleton HM. Analysis of thyroid hormones in serum by liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2010;397:1831–1839. doi: 10.1007/s00216-010-3705-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soldin OP, Soldin SJ. Thyroid hormone testing by tandem mass spectrometry. Clin Biochem. 2011;44:89–94. doi: 10.1016/j.clinbiochem.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunisue T, Fisher JW, Kannan K. Determination of six thyroid hormones in the brain and thyroid gland using isotope-dilution liquid chromatography/tandem mass spectrometry. Anal Chem. 2011;83:417–424. doi: 10.1021/ac1026995. [DOI] [PubMed] [Google Scholar]
- 13.Kunisue T, Eguchi A, Iwata H, Tanabe S, Kannan K. Analysis of thyroid hormones in serum of Baikal seals and humans by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and immunoassay methods: application of the LC-MS/MS method to wildlife tissues. Environ Sci Technol. 2011;45:10140–10147. doi: 10.1021/es203002a. [DOI] [PubMed] [Google Scholar]
- 14.Hackenmueller SA, Scanlan TS. Identification and quantification of 3-iodothyronamine metabolites in mouse serum using liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2012;1256:89–97. doi: 10.1016/j.chroma.2012.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiebooms JA, Wauters J, Vanden Bussche J, Vanhaecke L. Validated ultra high performance liquid chromatography-tandem mass spectrometry method for quantitative analysis of total and free thyroid hormones in bovine serum. J Chromatogr A. 2014;1345:164–173. doi: 10.1016/j.chroma.2014.04.032. [DOI] [PubMed] [Google Scholar]
- 16.Miyakawa M, Scanlan TS. Synthesis of [125I]-, [2H]-, and [3H]-labeled 3-iodothyronamine (T1AM) Synthetic Commun. 2006;36:891–902. [Google Scholar]
- 17.US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research Center for Veterinary Medicine : Guidance for industry: bioanalytical method validation. 2001. http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf.
- 18.Viswanathan CT, Bansal S, Booth B, DeStefano AJ, Rose MJ, Sailstad J, Shah VP, Skelly JP, Swann PG, Weiner R. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharm Res. 2007;24:1962–1973. doi: 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- 19.Buhrman DL, Price PI, Rudewiczcor PJ. Quantitation of SR 27417 in human plasma using electrospray liquid chromatography-tandem mass spectrometry: a study of ion suppression. J Am Soc Mass Spectrom. 1996;7:1099–1105. doi: 10.1016/S1044-0305(96)00072-4. [DOI] [PubMed] [Google Scholar]
- 20.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 21.Taylor PJ. Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin Biochem. 2005;38:328–334. doi: 10.1016/j.clinbiochem.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 22.De Brabandere VI, Hou P, Stockl D, Thienpont LM, De Leenheer AP. Isotope dilution-liquid chromatography/electrospray ionization-tandem mass spectrometry for the determination of serum thyroxine as a potential reference method. Rapid Commun Mass Spectrom. 1998;12:1099–1103. doi: 10.1002/(SICI)1097-0231(19980831)12:16<1099::AID-RCM290>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 23.Tai SS, Bunk DM, White E, Welch MJ. Development and evaluation of a reference measurement procedure for the determination of total 3,3′,5-triiodothyronine in human serum using isotope-dilution liquid chromatography-tandem mass spectrometry. Anal Chem. 2004;76:5092–5096. doi: 10.1021/ac049516h. [DOI] [PubMed] [Google Scholar]
- 24.Soukhova N, Soldin OP, Soldin SJ. Isotope dilution tandem mass spectrometric method for T4/T3. Clin Chim Acta. 2004;343:185–190. doi: 10.1016/j.cccn.2004.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agretti P, De Marco G, Russo L, Saba A, Raffaelli A, Marchini M, Chiellini G, Grasso L, Pinchera A, Vitti P, Scanlan TS, Zucchi R, Tonacchera M. 3-Iodothyronamine metabolism and functional effects in FRTL5 thyroid cells. J Mol Endocrinol. 2011;47:23–32. doi: 10.1530/JME-10-0168. [DOI] [PubMed] [Google Scholar]
- 26.Ghelardoni S, Chiellini G, Frascarelli S, Saba A, Zucchi R. Uptake and metabolic effects of 3-iodothyronamine in hepatocytes. J Endocrinol. 2014;221:101–110. doi: 10.1530/JOE-13-0311. [DOI] [PubMed] [Google Scholar]
- 27.Saba A, Chiellini G, Frascarelli S, Marchini M, Ghelardoni S, Raffaelli A, Tonacchera M, Vitti P, Scanlan TS, Zucchi R. Tissue distribution and cardiac metabolism of 3-iodothyronamine. Endocrinology. 2010;151:5063–5073. doi: 10.1210/en.2010-0491. [DOI] [PubMed] [Google Scholar]
- 28.Willetts P, Crossley DN, Ramsden DB, Hoffenberg R. The role of thyronine in thyroid hormone metabolism. J Clin Endocrinol Metabol. 1979;49:658–660. doi: 10.1210/jcem-49-4-658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data
Supplementary data
Supplementary data