

Abstract
Cerebral ischemia induces a complex transcriptional response with global changes in gene expression. It is essentially regulated by transcription factors as well as epigenetic players. While it is well known that the inhibition of transcriptionally repressive histone deacetylases leads to neuroprotection, the role of histone methyltransferases in the postischemic transcriptional response remains elusive. We investigated the effects of inhibition of the repressive H3K9 histone methyltransferases SUV39H1 and G9a on neuronal survival, H3K9 promoter signatures and gene expression. Their inhibition either with the specific blocker chaetocin or by use of RNA interference promoted neuronal survival in oxygen glucose deprivation (OGD). Brain-derived neurotrophic factor (BDNF) was upregulated and BDNF promoter regions showed an increase in histone marks characteristic for active transcription. The BDNF blockade with K252a abrogated the protective effect of chaetocin treatment. In conclusion, inhibition of histone methyltransferases SUV39H1 and G9a confers neuroprotection in a model of hypoxic metabolic stress, which is at least in part mediated by BDNF.
Keywords: BDNF, G9a, histone methylation, ischemia, SUV39H1
Introduction
Cerebral ischemia induces large changes in gene expression with a general tendency toward gene repression.1, 2, 3 Accordingly, repressive epigenetic marks accumulate: DNA methylation levels are reported to rise4 and vast histone deacetylation processes take place.5 Inhibiting epigenetic enzymes that function as transcriptional repressors, such as the DNA methyltransferase DNMT1, or the family of histone deacetylases (HDACs), leads to maintenance of activating epigenetic marks after ischemia. This, in turn, promotes the upregulation of genes, including key mediators of neuroprotection and regeneration and hence results in improved outcome after stroke.6, 7 Epigenetic drugs, such as pan-HDAC inhibitors, have been identified as promising treatment option for diverse neurologic diseases that involve epigenetic dysregulation including ischemia.8
In contrast to DNA methylation and histone acetylation, the role of histone methylation is poorly understood. Histone methylation is a posttranslational modification that can be found at lysine and arginine residues of histones (H). Methylation at lysine residues (K) occurs in three different states: mono-, di-, and tri-methylation (me 1/2/3). Different methylation states are associated with gene activation or repression. Tri- and di-methylation on histone 3, lysine 9 (H3K9) is linked to transcriptional repression. In contrast, mono-methylation and the unmethylated state of H3K9, that is often acetylated (ac), are found in transcriptionally active sites of the genome.9 Enzymes involved in the establishment of repressive H3K9 methylation marks are, among others, the methyltransferases G9a, catalyzing H3K9 me0 to me1 and me1 to me2 steps,10 as well as SUV39H1 that catalyzes H3K9 di- to tri-methylation.11 Both SUV39H1 and G9a are found in diverse repressive complexes and act in concert with other epigenetic players, such as DNA methyltransferases12 and diverse HDACs.13 As such they contribute to the induction and maintenance of transcriptional repression in the genome.
The aim of our study was to elucidate the role of histone methylation and selected associated enzymes SUV38H1 and G9a in the context of cerebral ischemia. We hypothesized that the inhibition of repressive histone methyltransferases could induce gene activation and confer neuroprotection in ischemia. We hence employed oxygen glucose deprivation (OGD), a widely used in vitro model of stroke, to investigate the effects of SUV39H1 and G9a inhibition on neuronal survival, H3K9 promoter signatures, and gene expression.
Materials and methods
Isolation of Rat Cortical Neurons and Neuronal Cell Culture
Primary rat cortical neurons were derived from embryos (E17) of Wistar rats (Bundesinstitut für gesundheitlichen Verbraucherschutz und Veterinärmedizin, Berlin, Germany). Cultivation took place in supplemented neurobasal medium in 24-well, or 6-well plates as described in detail in Meisel et al14 with a minor modification: cells were seeded at a density of 175 000 cells/cm2.
Chemicals and Drug Administration
Chaetocin (C9492), BIX-01294 (B9311), and K252a (K1639) were purchased from Sigma Aldrich (Taufkirchen, Germany). All were solved in dimethyl sulfoxide in 3 mmol/L stock solutions. After toxicity testing in neuronal cultures, chaetocin was used at a 30-nmol/L concentration, BIX-01294 at 100 nmol/L, and K252a at 50 nmol/L. In all cases, higher concentrations evoked signs of toxicity. Drug administration to neuronal cultures occurred as pretreatment on day in vitro (DIV) 8 by adding the respective amount of the inhibitor/dimethyl sulfoxide to the cultures. Twenty-four hours later, on DIV 9, OGD followed. The iron chelator desferrioxamine (DFO) was solved in phosphate-buffered saline (PBS) and applied in a 150-μmol/L concentration on DIV 7 for a duration of 48 hours before mRNA harvest on DIV 9. The PBS treatment served as a control.
Lentivirus Production, Titration, and Application
Interfering RNA target sequences were designed using the internet applications of Invitrogen (http://www5.invitrogen.com/custom-genomic-products/tools/mirna/). Selected sequences (SUV39H1: A GGA CAA GAA AGC TTG GCT AG and G9a: T AGA GCT TCG ACT TCA GAC TT) were BLAST-searched against rat and mouse genome sequences to ensure target match and to exclude unspecific targets. A nontargeting ‘scrambled' construct was used as a control (A AAT GTA CTG CGC GTG GAG AC). With the BLOCK-iT Pol II miR Expression Vector Kit (Invitrogen, Darmstadt, Germany) microRNA-embedded short hairpin RNA (miR-shRNA) constructs were generated containing the respective sequences. The miR-shRNA constructs were finally cloned into a third-generation lentiviral vector based on Addgene plasmid 27232 (ref 15) with the following modifications: microRNA delivery was driven by a ubiquitin promoter and the initial reporter protein was exchanged with a myc-tagged red fluorescent protein (RFP). HEK 293FT cells (Invitrogen, Darmstadt, Germany) were used as a producer cell line. Cotransfection with the packaging vectors psPAX (Addgene 12259) and 7.5pMD2G (Addgene 12260) was performed as described in Dull et al16 except that HEK medium was additionally supplemented with 1% sodium pyruvate and 1% nonessential amino acids. Viral harvest took place 48 and 72 hours after transfection. Viral concentration steps were performed with PEG-it Virus Precipitation Solution (System Biosciences, Heidelberg, Germany) and viral pellets were taken up in 100 μL PBS with magnesium and calcium (Gibco, Carlsbad, CA, USA) and subsequently stored at −80°C. Viral transduction efficiencies in neuronal cultures were determined and calculated from serial dilutions using RFP as a reporter for microscopic evaluation after 96 hours. For OGD experiments, neuronal cultures were transduced with miR-shRNA constructs on late DIV 1, fed fresh B27 on DIV 2 and 7 and in between a complete medium exchange was performed on DIV 4. Included in the final evaluation were experiments with transduction efficiencies above 80% as assessed by comparing cell counts from phase-contrast microscopy and RFP-positive neurons.
Oxygen Glucose Deprivation
Primary neuronal cultures were subjected to OGD on DIV 9. In experiments establishing the OGD model it was shown that 100 μmol/L glutamate or 100 μmol/L NMDA induces cell death on DIV 9 already (data not shown). In OGD experiments, medium was removed from cells and preserved. Cells were once rinsed in PBS and subjected to OGD for 135±5 minutes in a balanced salt solution at pO2<2 mm Hg. Upon reexposure to normoxia the preserved cell culture medium together with fresh cultivating medium (1:1) was added to the cultures and likewise exchanged in the control plates not subjected to OGD. Thereafter cells were kept under normal cell culturing conditions.
Evaluation of Cell Survival and Damage: Lactate Dehydrogenase Assay
Neuronal injury was assessed by measuring lactate dehydrogenase release (LDH) in the supernatant 24 hours after the injury paradigm on DIV 10. Further, a second measurement followed after total lysis of cells using 0.5% Triton-X for 30 minutes at 37°C. Data are presented as percentage of cell death calculated from the ratios between the unit of LDH activity per mL per well and the maximum LDH activity per well after total lysis.
Evaluation of Cell Survival and Damage: Cell Counts
Fluorescent microscopic images of neuronal cultures were taken on DIV 9 directly pre OGD, as well as on DIV 10, 24 hours after OGD, with a Leica DFC360 FX microscope combined with a Leica DFC360 FX camera (Leica, Wetzlar, Germany) and a computerized software program (Leica Application Suite V3.3.0, Heerbrugg, Switzerland). Ten regions of interest (ROIs) per well were preselected and repeatedly analyzed at × 400 magnification, maintaining identical settings for all experiments. Red fluorescent protein-positive cells were counted in a blinded manner (around 60 cells per ROI pre OGD). Mean values of the 10 ROIs were calculated per well and counted as N=1. All in all 9 wells were analyzed resulting in average cell counts of 10 800 cells per miRNA-shRNA construct (60 cells × 10 ROIs × 9 wells × 2 OGD/CTRL condition) leading to a total of approximately 32 400 cells counted before cell injury. To compare the effects of SUV39H1/G9a knockdown on survival after OGD, ratios of mean values per well post/pre OGD were calculated as previously described.17 For microscopic visualization of cell survival, emitted fluorescence was pseudo-colored in green (before OGD) and red (24 hours after OGD) and images were merged resulting in yellow surviving neurons. To ensure exact cell death measurement by cell counting, we took the following measures. First, cell counts were performed using both the magnified red fluorescent image and the phase-contrast image to assess the cellular morphology in case of doubt. The phase-contrast microscopic image at a × 400 magnification allowed the distinction of healthy neurons from dead cells. Second, all cell counts were performed in a blinded manner to prevent an investigator bias. Third, high numbers of neurons were counted to increase the statistical power.
Immunoblots
After whole cell protein harvest and electrophoretic separation, proteins were transferred onto cellulose membranes and probed with the following antibodies purchased from Cell Signaling (Frankfurt am Main, Germany): G9a: CS 3306; SUV39H1: CS 8729; Myc: CS 2272; ß-actin: CS 4967; H3K9me3: CS 9754; H3K9me2: CS 4658; and H3K9ac: CS 9671. Primary antibodies were applied in a 5% bovine serum albumin tris-buffered saline solution with 0.1% Tween-20 (TBST). Blocking and second antibody incubation were performed in 5% milk TBST.
Chromatin Immunoprecipitation and Sequencing
Around 50 million rat cortical neurons per condition (chaetocin versus vehicle-treated cells, both pre and post OGD) were cultivated, pretreated with 30 nmol/L chaetocin/vehicle on DIV 8 and harvested on DIV 9, 1 hour after OGD. Cells were crosslinked in 1% formaldehyde in growth medium for 10 minutes at 4°C, and the reaction stopped with 125 mmol/L glycine. Cultures were rinsed in cold PBS, scraped, centrifuged, and washed twice with PBS, and the pellet snap frozen in liquid nitrogen. Resuspension took place in a first lysis buffer (50 mmol/L HEPES-KOH, pH 7.5, 140 mmol/L NaCl, 1 mmol/L EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, freshly added protease inhibitors) for 10 minutes at 4°C followed by centrifugation and uptake in a second lysis buffer (10 mmol/L Tris-HCl, pH 8.0, 200 mmol/L NaCl, 1 mmol/L EDTA, 0.5 mmol/L EGTA) for 10 minutes, RT. For nuclear lysis, the pelleted chromatin was solubilized (10 mmol/L Tris-HCl, pH 8.0, 100 mmol/L NaCl, 1 mmol/L EDTA, 0.5 mmol/L EGTA, 0.1% Na-Deoxycholat, 0.5% N-Laroylsarcosine) and sheared by pulsed sonication for 40 minutes at 4°C in a bioruptor (Diagenode, Seraing, Belgium) to a bulk DNA reduction to less than 400 bp. To get rid of cell debris, the sheared chromatin was clarified by centrifugation at 20 000 g for 10 minutes. Per immune precipitation, 15 μg of DNA were used with 5 μg of H3K9ac antibody. Immune complexes were blocked with magnetic Dynabeads in RIPA complemented with 0.25% bovine serum albumin for 4 hours while rotating at 4°C, rinsed 5 times with cold RIPA. Precipitated complexes were finally separated from the beads with elution buffer (50 mmol/L Tris-HCl, pH 8.0, 10 mmol/L EDTA, 1.0% SDS) for 30 minutes at 65°C, followed by centrifugation and collection of the supernatant. Crosslink reversal took place by adding 5 mol/L NaCl at 65°C overnight. DNA was purified and precipitated with ethanol. Library preparation was conducted using the NEB-Next ChIP-seq Library Prep Master Mix for Illumina (New England Biolabs, Frankfurt am Main, Germany) according to the manufacturer's protocol. Sequencing of 50 bp single reads was performed on an Illumina HiSeq 1500 (San Diego, CA, USA) using TruSeq v3 chemistry. Read mapping against rat rn5 reference genome was performed using BWA (version 0.5.9-r16)18 allowing up to two mismatches per read. Nonuniquely mapped reads were discarded and redundant reads were removed using SAMtools rmdup.19 Read counts in ROI were determined using BEDtools. Within the brain-derived neurotrophic factor (BDNF) gene, three different transcription start sites (TSS) were chosen for analysis: BDNF1: accession number: NC_005102.3, TSS: 107371329 chr3:107371329-107421906; BDNF2: accession number: NM_001270631.1, TSS:107372531 chr3:107372531-107421906; BDNF3: accession number: NM_001270638.1, TSS: 107390141 chr3:107390141-107421906; H3K9ac enrichment within a smaller 2-kb and a larger 5-kb region around the respective TSS was analyzed (−1 kb to +1 kb and –3 kb to +2 kb around TSS, respectively). Absolute read counts per region were normalized to total reads × 1 000 000.
Expression Analysis and Polymerase Chain Reaction
For mRNA expression analysis, real-time quantitative reverse transcription PCR (qRT-PCR) was performed using LightCycler 2.0 (Roche, Mannheim, Germany). Briefly, whole-cell mRNA was harvested in Trizol (1 mL per 3 Mio cells). After chloroform extraction, RNA containing pellets were solved in diethylpyrocarbonate-treated water. After DNase digest, PCR inhibitors were removed with the help of NucleoSpin RNA clean-up KIT (Macherey Nagel, Düren, Germany). Equal amounts of RNA were reverse transcribed with M-MLV reverse transcriptase and random hexamers. For qRT-PCR, the LightCycler FastStart DNA Master SYBR Green I Kit (Roche) was used. All PCRs were performed in duplicate. The relative expression of each gene of interest (GOI) was calculated compared with a reference gene (ref) by the delta Cp (crossing point) method with efficiency correction using the equation E(GOI)–Cp(GOI)/E(ref)–Cp (ref). Mean values of duplicates were determined. The reference genes ß-actin and reep5 with the most stable expression patterns in OGD were chosen in a previously performed methodological study using NormFinder Software.20 In all experiments, PCRs for both reference genes were performed and similar results obtained with either reference. Amplification efficiencies (E) were determined for each primer pair by a serial dilution curve and calculated using LightCycler 2.0 software (Roche).
Thermal cycling conditions and amplification efficiencies for each primer pair are listed in Table 1.
Table 1. PCR primer pairs.
| Gene | Sequence 5'-3' fwd. and rev. | Annealing temperature (°C) | Melting curve temperature (°C) | Elongation time (°C/s) | Efficiency |
|---|---|---|---|---|---|
| ß-actin | Acccacactgtgcccatcta | 68 | 82 | 72/15 | 1.86 |
| Gccacaggattccataccca | |||||
| reep5 | Ctgataggtttcggatacccag | 68 | 84 | 72/15 | 1.97 |
| Gactcgtgcttcaggaagatgg | |||||
| SUV39H1 | Cgtgtagtccagaaaggcatcc | 68 | 84 | 72/15 | 1.94 |
| Ccacgtagtccaggtcaaagag | |||||
| G9a | Tagcaacggacagcctccaatc | 68 | 86 | 72/15 | 1.98 |
| Cttcagacttgctgtcagagtcc | |||||
| KDM3A | Cagactcccccagcccggaa | 59 | 81 | 72/15 | 1.94 |
| Gggcagctgtgccacgatgt | |||||
| BDNF | Cgtggggagctgagcgtgtg | 66 | 82 | 72/10 | 2.14 |
| Tgcccctgcagccttccttc |
Abbreviation: BDNF, brain-derived neurotrophic factor.
Statistical Analysis
Data are presented as box/scatter plots and mean±95% confidence interval as well as bars with mean±s.d. t-tests as well as one-way and two-way ANOVA (analysis of variance) were performed with Tukey's post hoc analysis21 using SPSS (IBM) and GraphPad Prism (GraphPad, San Diego, CA, USA). P-values<0.05 were considered as statistically significant.
To calculate significant differences between sequence reads in the ChIP-seq experiments the following assumptions were made: Given are two datasets for two conditions C1 and C2 consisting of N1 and N2 uniquely mapped reads. In a given genomic region for C1 and C2 we observe k1 and k2 mapped reads, respectively. The probability is modelled as a series of n=k1+k2 Bernoulli experiments with probability of success of p=N1/(N1+N2). The probabilities follow the Binomial distribution:
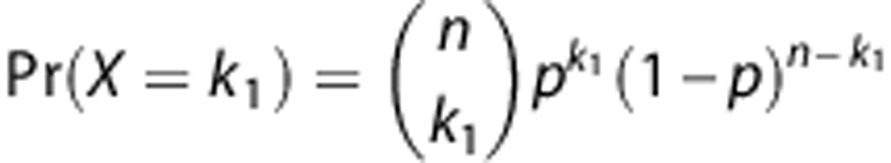 |
For the calculation of the P-values the probabilities were summed up:
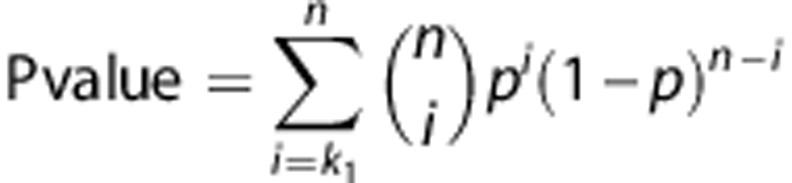 |
Results
Pharmacological Inhibition of the Histone Methyltransferases SUV39H1 and G9a with Chaetocin Promotes Neuronal Survival after Oxygen Glucose Deprivation
To test the hypothesis whether inhibition of the transcriptional repressors SUV39H1 and G9a induced neuroprotection in OGD, two pharmacological inhibitors were employed: chaetocin, an inhibitor of members of the SUV39 family of histone methyltransferases with inhibitory effects against SUV39H1 and G9a22, 23 and second, BIX-01294, an inhibitor of G9a.24 Primary rat cortical neuron cultures were pretreated with chaetocin or BIX-01294 24 hours before OGD. Cell death was assessed by LDH measurements 24 hours after OGD. Cell death was significantly reduced in the chaetocin-treated group compared with the vehicle-treated group in OGD (Figure 1A). In contrast, BIX-01294 pretreatment had no significant effect on neuronal viability post OGD (Figure 1B).
Figure 1.

Chaetocin but not BIX-01294 protects neurons subjected to oxygen glucose deprivation (OGD). Percentage of cell death upon inhibitor/vehicle pretreatment in neurons subjected to OGD or control (CTRL) conditions as assessed by lactate dehydrogenase (LDH) measurement. A two-way analysis of variance (ANOVA) was conducted that examined the effect of OGD and treatment on cell survival followed by a Tukey post hoc analysis. Significant effects are indicated in the figure *P<0.005. Included are 10 independent experiments each consisting of 3 wells/condition. (A) Treatment with 30 nmol/L chaetocin. Significant interaction between effects of OGD and treatment, F(1,36)=4.714 with P=0.037 for interaction. (B) Treatment with 100 nmol/L BIX-01294. No significant interaction between effects of OGD and treatment, F(1,36)=0.204 with P=0.654 for interaction.
Knockdown of the Histone Methyltransferases SUV39H1 or G9a Protects Rat Cortical Neurons in Oxygen Glucose Deprivation
The miR-shRNA constructs targeting SUV39H1 or G9a were delivered to cortical neurons via lentiviral transduction resulting in a successful knockdown of SUV39H1/G9a compared with a nontargeting scrambled miR-shRNA (Figure 2A). G9a knockdown increased global H3K9 acetylation levels whereas global H3K9me3/me2 levels remained unchanged upon knockdown of either G9a or SUV39H1 (Figure 2A). To determine the influence of SUV39H1 and G9a knockdown on neuronal survival after OGD, LDH measurements and a cell survival assay based on the ratio of neuronal cell counts immediately before and 24 h after OGD were performed. The knockdown of either histone methyltransferase did not impact cell viability under control conditions (CTRL) as indicated by a stable cell count ratio around 1 (Figure 2C) and low LDH release (Figure 2D). However, 24 hours after OGD cell survival was increased upon SUV39H1 or G9a knockdown compared with scrambled control treatment. This is demonstrated by representative images of fluorescent and phase-contrast microscopy (examples in Figure 2B, surviving cells in yellow) as well as significantly higher survival rates compared with scrambled control according to cell count ratios (Figure 2C). In scrambled control cultures, the number of red labelled neurons decreased significantly after OGD suggesting that dead cells have neither been counted and nor did they interfere with the cell viability assay.
Figure 2.

Specific miRNA-based knockdown of histone methyltransferases SUV39H1 or G9a protects neurons subjected to oxygen glucose deprivation (OGD). (A) Representative immunoblots from day in vitro (DIV) 9, 8 days after lentiviral delivery. Left blot: knockdown of SUV39H1, same samples (scrambled ctrl and mi-SUV39H1) run twice on the same gel, incubated with anti-LSD1 (100 kb protein) and anti-ß-actin as loading controls, anti-Myc indicating transduction efficiency and anti-H3K9me3 detecting the product of SUV39H1 activity; right blot: knockdown of G9a, same samples (scrambled ctrl and mi-G9a) run twice, incubated with anti-ß-actin as a loading control, anti-Myc indicating transduction efficiency, anti-H3K9me2 and anti-H3K9ac as indicators of G9a activity. (B) Representative microscopic images of neuronal cultures transduced with microRNA-embedded short hairpin RNA (miR-shRNA) constructs (scrambled ctrl, mi-SUV39H1, mi-G9a); scale: 100 μm. Column 1: RFP fluorescence of cortical neurons before OGD (DIV 9) pseudo-colored in green; column 2: same region of interest (ROI) 24 hours after OGD (DIV 10), red fluorescent protein (RFP) fluorescence in red; column 3: merged images pre and post OGD, surviving neurons shown in yellow; column 4: magnified detail from the overlay image showing surviving neurons in yellow; column 5: phase-contrast image post OGD corresponding to the magnified detail image of column 4. (C) Ratio of mean cell counts per well post/pre OGD; RFP-positive cells were counted in a blinded manner. Per construct (scrambled ctrl, mi-SUV39H1, mi-G9a) and condition (CTRL/OGD) cells from 9 independent wells were counted and mean values of 10 ROIs calculated per well. Survival ratios of 1 represent a 100% survival rate. A two-way analysis of variance (ANOVA) was conducted that examined the effect of OGD and microRNA application on cell survival. There was a significant interaction between effects of OGD and microRNA, F(2,48)=8.7 with P<0.001 for interaction. A Tukey post hoc analysis was performed. Significant effects are indicated in the figure *P<0.001. (D) Lactate dehydrogenase (LDH) measurements 24 hours after OGD; N=9. A two-way ANOVA was conducted that examined the effect of OGD and microRNA treatment on cell survival. There was a significant interaction between effects of OGD and microRNA, F(2,48)=7.7 with P=0.001 for interaction. A Tukey post hoc analysis was performed. Significant effects are indicated in the figure *P<0.001. (E) Correlation of cell count data (survival) and the corresponding LDH measurement (% cell death) per well. Each dot represents one well (N=54). Included are all wells from all constructs (scrambled ctrl, mi-SUV39H1, mi-G9a) from OGD and control conditions. Person's r=−0.87, P<0.001.
The neuroprotective effect of SUV39H1 and G9a knockdown in OGD assessed by cell counts was confirmed by LDH measurement of cell death (Figure 2D). Lactate dehydrogenase measurements and cell counts correlated significantly — as previously shown in a similar experimental setting17, 25 — which corroborated the robust neuroprotective effect upon SUV39H1 and G9a knockdown (Figure 2E).
SUV39H1 and G9a Expression Is Not Induced by Desferrioxamine
As the transcription factor hypoxia inducible factor 1 (HIF-1) regulates a large number of genes in response to hypoxia, we tested, whether the mRNA expression of either SUV39H1 or G9a was induced by 48 hours treatment with the iron chelator DFO, a known activator of HIF. The histone demethylase KDM3A, shown to be HIF-1 induced in hypoxic cancer cells26 was upregulated in primary rat cortical neurons upon DFO treatment and served as a positive control. However, neither SUV39H1 nor G9a mRNA expression was affected by DFO (Figure 3).
Figure 3.

KDM3A but not SUV39H1 or G9a mRNA expression is upregulated by desferrioxamine (DFO). Expression of KDM3a, SUV39H1 and G9a mRNA after 48 hours DFO or phosphate-buffered saline (PBS) (=CTRL) treatment was determined by quantitative reverse transcription PCR (qRT-PCR) and normalized to reference gene reep5. A one-way analysis of variance (ANOVA) was used to determine mRNA expression difference upon DFO treatment followed by Tukey's multiple comparison test to isolate differences among groups F(3,8)=34.77, P<0.0001.
Chaetocin Treatment Increases Activating H3K9 Marks at Brain-Derived Neurotrophic Factor Promoter
To capture alterations in epigenetic profiles upon SUV39H1 and G9a inhibition by chaetocin, promoter regions of genes involved in neuroprotection and regeneration were analyzed by chromatin immunoprecipitation followed by sequencing (ChIP-seq) 1 hour after OGD. Chromatin immunoprecipitation was performed using an antibody against unmethylated, but acetylated H3K9, assuming that the inhibition of SUV39H1 and G9a would induce a shift from repressive (methylated) to activating (unmethylated) H3K9 posttranslational modifications around target genes. Brain-derived neurotrophic factor, which is known to have a crucial role in promoting postischemic survival,27 was chosen for closer analysis. Several TSS were analyzed (BDNF1, BDNF2, BDNF3). Per TSS two regions of different size were selected for the assessment of H3K9ac enrichment: a smaller locus ranging from −1 kb to +1 kb around the respective TSS and a larger region spanning −3 kb to +2 kb. Chaetocin-treated neurons showed increased H3K9ac levels 1 hour after OGD in all analyzed BDNF promoter regions independent of the selected region size (Figures 4A and 4B).
Figure 4.

Chaetocin induces H3K9 promoter acetylation and transcription of brain-derived neurotrophic factor (BDNF) post oxygen glucose deprivation (OGD). (A and B) Comparison of chromatin immunoprecipitation followed by sequencing (ChIP-seq) sequence reads chaetocin/vehicle, immunoprecipitated with anti-H3K9ac. Significant effects are indicated in the figure, *P<0.05. H3K9ac enrichment either 2 kb (A) or 5 kb (B) around three selected transcription start sites (TSS) of the rat BDNF gene, indicated as BDNF 1, 2, and 3. (C) mRNA of neurons harvested at time of reoxygenation (0 hour) and 1, 4, and 24 hours after OGD, analyzed using quantitative reverse transcription PCR (qRT-PCR) normalized to the reference gene reep5; values of chaetocin-treated cells were normalized to values of the respective vehicle-treated cells (0, 1, 4, and 24 hours) and subsequently related to the onset of reoxygenation (0 hour). Included are four independent experiments. A two-way analysis of variance (ANOVA) was conducted to evaluate transcriptional changes of candidate genes over time. A Tukey post hoc analysis was further employed to evaluate group differences. Significant effects are indicated in the figure, *P<0.05. There is no significant interaction of OGD and time, P=0.610. However, the factor OGD versus CTRL differs significantly, P<0.001.
Chaetocin Inhibition of SUV39H1 and G9a during Oxygen Glucose Deprivation Increases Brain-Derived Neurotrophic Factor mRNA
After SUV39H1 and G9a inhibition via chaetocin, the mRNA of neuronal cultures subjected to OGD or CTRL was harvested at the time of reoxygenation (0 hour) and subsequently at 1, 4, and 24 hours after OGD to assess changes in gene expression. Compared with CTRL cultures, chaetocin treatment led to increased BDNF mRNA levels already 1 hour after OGD (Figure 4E).
Brain-Derived Neurotrophic Factor Upregulation Is Essential for Chaetocin-Induced Neuroprotection in Oxygen Glucose Deprivation
To investigate whether chaetocin exerted its neuroprotective effect by inducing BDNF, BDNF-TrkB signaling was blocked by applying K252a, a selective inhibitor of tyrosine kinases (TrK). Rat cortical neurons were pretreated with chaetocin or vehicle or chaetocin+K252a or K252a alone on DIV 8 and subjected to OGD on DIV 9 followed by cell death assessment on DIV 10. While chaetocin treatment again significantly protected neurons against OGD, the additional administration of K252a attenuated the protective effect of chaetocin. BDNF-TrkB blockade by K252a administration alone did not significantly alter cellular survival upon OGD (Figure 5).
Figure 5.

BDNF-TrkB blockade attenuates chaetocin-induced protection in oxygen glucose deprivation (OGD). Blockade of BDNF-TrkB with K252a and SUV39H1 and G9a inhibition with chaetocin. Evaluation of cell death by lactate dehydrogenase (LDH) measurement. Included are 10 independent experiments, consisting of 3 wells/condition each. A two-way analysis of variance (ANOVA) was conducted that examined the effect of OGD and treatment on cell survival. There was a significant interaction between effects of OGD and treatment, F(3,72)=3.546 with P=0.019 for interaction. A Tukey post hoc analysis was performed. Selected significant effects are indicated in the figure *P<0.05. BDNF, brain-derived neurotrophic factor.
Discussion
We identified the histone methyltransferases SUV39H1 and G9a as crucial players in OGD-induced cell damage. Inhibition of SUV39H1 and G9a, by knockdown or pharmacological inhibition, conferred significant protection to neurons subjected to OGD. SUV39H1 and G9a inhibition increased H3K9ac in promoter regions of BDNF post OGD — a sign for increased transcriptional activity — and resulted in elevated BDNF mRNA levels after experimental ischemia. Blockade of BDNF-TrkB signaling attenuated chaetocin-induced protection of rat cortical neurons subjected to OGD.
From DNA methylation and histone acetylation studies in stroke models, it is known that postischemic gene repression can be attenuated through epigenetic remodeling and that a subsequent neuroprotective effect is based on transcriptional activation of cytoprotective genes.5, 6, 7 With the current study we could for the first time demonstrate that inhibition of transcriptional repressors on the level of histone methylation can effectively promote neuronal survival in an in vitro model of cerebral ischemia. The employed OGD model had been validated regarding its capacity to elicit essential signaling cascades of ischemic brain damage, e.g., excitotoxicity.
Recently, it was also shown in an in vivo model of stroke that enhanced cell survival correlates with higher levels of epigenetic histone methylation marks of active gene expression. Aged female rats, that are more severely affected by ischemic injury compared with younger ones, show less activating H3K4me3 peaks at transcriptional start sites and more repressive H3K9me3 marks in their astrocytes. The more active chromatin in younger rats is associated with higher expression levels of cytoprotective genes.28
For actual changes in gene transcription, an orchestrated interplay between different epigenetic levels, such as histone methylation and acetylation, is necessary.9 Both SUV39H1 and G9a act in multiple repressive complexes and interact with regulatory elements to silence transcription.12, 13, 29 The inhibition of these two histone methyltransferases can induce a shift from repressive to activating posttranslational histone modifications beyond H3 demethylation. Our observation that the knockdown of the transcriptional repressor G9a alone increases global H3K9 acetylation levels (Figure 2A) — a sign for transcriptional derepression — is consistent with findings of others. A reduction of global H3K9me3 or H3K9me2 levels is not visible in the immunoblots upon SUV39H1 or G9a knockdown. Other enzymes known to be involved in the establishment or eradication of H3K9 methylation such as ESET or members of the KDM4 family that are not blocked by SUV39H1 or G9a knockdown might account for this finding. Moreover, the lack of alterations of the H3K9 methylation status on the global level does not exclude changes at single gene promoters. Discrepancies between histone lysine modifications on the global level and locally at promoters of single genes have been described. For example, ischemic preconditioning induced a global decrease of activating H3K4me2 and H3K4me1 compared with controls, while H3K4me3 levels remained unchanged. Repressive global H3K9me2 levels increased by almost 60%.30 However, subsequent analysis of histone methylation at promoters of induced neuroprotective genes showed methylation changes contrary to those assessed upon global analysis, e.g., locally increased H3K4me2 and decreased H3K9me2 levels. The authors explained this discrepancy by the fact that preconditioning led to decreased H3K4me2 and increased H3K9me2 in noncoding regions of frequent long interspersed nuclear element sequences, correlating with the globally found histone methylation pattern.31
Histone deacetylases 1 and 2 interact with G9a in a repressor complex assembled around the REST (repressor-element-1-transcription-factor), which is induced upon experimental stroke. The REST depletion was shown to prevent epigenetic silencing and rescue neurons.29 Thus, the neuroprotective effect by G9a knockdown in our paradigm might be mediated by inhibiting the REST complex, which links histone deacetylation to histone methylation, subsequently followed by transcriptional derepression of key cytoprotective players.
Interestingly, blocking G9a with the inhibitor BIX-01294 did not yield neuroprotection in OGD in contrast to both pharmacological blockade with chaetocin and G9a knockdown. This discrepancy might be due to the concentrations of BIX-01294 we used in our primary neuronal culture. To avoid neurotoxicity we used concentrations that might be too low for induction of neuroprotection. Initially, BIX-01294 was tested in much less sensitive cells (murine fibroblasts and stem cells)24 and later found to display a poor separation of toxicity and functional potency.32 A number of new G9a inhibitors are currently being designed and constantly improved.32 Cellular application, however, still seems to be a challenge with the compound BIX-01294 as well as with more recent products.33
The exclusive inhibitory specificity toward SUV39 family members as well as the exact mechanism of action of chaetocin are a matter of debate; its inhibitory potency against SUV39H1 and G9a, however, has been confirmed by several research groups.22, 23, 34, 35 In the present study, chaetocin was for the first time employed in neurons. We could hence identify it as a novel neuroprotective agent, which might promote survival not only in models of cerebral ischemia but also in other models of neurodegeneration.
As observed with HDAC inhibition in experimental ischemia36, 37 the neuroprotective effect of chaetocin seems to rely on the induction of important neuroprotective and regenerative players, such as BDNF. The neurotrophin BDNF has an important role in neuronal plasticity and survival. It exerts its function through binding to its receptor tyrosine kinase B (TrkB) and subsequent induction of downstream signaling cascades. In the ischemic brain, BDNF induction promotes protective and neurogenic developments. The crucial role of BDNF-TrkB signaling in neuroprotection was observed in diverse models of ischemia. In a model of glutamate excitotoxicity, BDNF-TrkB blockade partially inhibited the protective effect of lithium.38 Neuroprotection provided by PBI-05204, a plant extract from Nerium oleander, is blocked by BDNF-TrkB inhibition with K252a in ischemic brain slices.39 Furthermore, neurogenic effects of HDAC inhibitors were attenuated upon BDNF blockade with BDNF antibodies in neurons40 as well as with K252a in a rodent model of ischemia: sodium butyrate-induced neurogenesis in various brain regions41 as well as oligodendrogenesis and reduction of white-matter injury were demonstrated to depend on BDNF-TrkB signaling.42 Here, we showed that chaetocin-induced neuroprotection in OGD was diminished by BDNF-TrkB blockade with K252a (Figure 5). This finding identifies BDNF as one crucial mediator of chaetocin-induced regeneration and protection in experimental ischemia.
Conclusion
Pharmacological as well as genetic inhibition of the histone methyltransferases SUV39H1 and G9a significantly promoted neuronal survival post OGD. The protective effect was based on a shift in promoter signatures toward transcriptional activity and mediated by upregulation of protective genes such as BDNF.
The authors declare no conflict of interest.
Footnotes
Author Contributions
SS contributed to experimental design, data acquisition and analysis, writing article and approval of current version; CH contributed to RNA interference experiments: miRNA design, data acquisition and analysis, revising article and approval of current version; HL contributed to BDNF experiments, PCR data acquisition and analysis, revising article and approval of current version; JF contributed to experimental design of miRNAs, revising article and approval of current version; JH contributed to ChIP-data acquisition and analysis, revising article and approval of current version; FY and AM contributed to study design, revising article and approval of current version. SM contributed to experimental design and analysis, revising article and approval of current version.
This work was supported by German Research Foundation (EXC 257 NeuroCure), the Federal Ministry of Education and Research (01 EO 08 01), the Helmholtz Association (SO-022NG) and has received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no 201024.
References
- 1Trendelenburg G, Prass K, Priller J, Kapinya K, Polley A, Muselmann C et al. Serial analysis of gene expression identifies metallothionein-II as major neuroprotective gene in mouse focal cerebral ischemia. J Neurosci 2002; 22: 5879–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2VanGilder RL, Huber JD, Rosen CL, Barr TL. The transcriptome of cerebral ischemia. Brain Res Bull 2012; 88: 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3Hori M, Nakamachi T, Rakwal R, Shibato J, Nakamura K, Wada Y et al. Unraveling the ischemic brain transcriptome in a permanent middle cerebral artery occlusion mouse model by DNA microarray analysis. Dis Model Mech 2012; 5: 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4Endres M, Fan G, Meisel A, Dirnagl U, Jaenisch R. Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport 2001; 12: 3763–3766. [DOI] [PubMed] [Google Scholar]
- 5Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A et al. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol 2008; 210: 531–542. [DOI] [PubMed] [Google Scholar]
- 6Schweizer S, Meisel A, Marschenz S. Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab 2013; 33: 1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke 2009; 40: 2899–2905. [DOI] [PubMed] [Google Scholar]
- 8Gibson CL, Murphy SP. Benefits of histone deacetylase inhibitors for acute brain injury: a systematic review of animal studies. J Neurochem 2010; 115: 806–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40: 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 2002; 16: 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001; 410: 116–120. [DOI] [PubMed] [Google Scholar]
- 12Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 2006; 20: 3089–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13Vaute O, Nicolas E, Vandel L, Trouche D. Functional and physical interaction between the histone methyl transferase Suv39H1 and histone deacetylases. Nucleic Acids Res 2002; 30: 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14Meisel A, Harms C, Yildirim F, Bosel J, Kronenberg G, Harms U et al. Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem 2006; 98: 1019–1031. [DOI] [PubMed] [Google Scholar]
- 15Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW et al. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci USA 2004; 101: 18206–18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D et al. A third-generation lentivirus vector with a conditional packaging system. J Virol 1998; 72: 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17Datwyler AL, Lattig-Tunnemann G, Yang W, Paschen W, Lee SL, Dirnagl U et al. SUMO2/3 conjugation is an endogenous neuroprotective mechanism. J Cereb Blood Flow Metab 2011; 31: 2152–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004; 64: 5245–5250. [DOI] [PubMed] [Google Scholar]
- 21Schlattmann P, Dirnagl U. Statistics in experimental cerebrovascular research: comparison of more than two groups with a continuous outcome variable. J Cereb Blood Flow Metab 2010; 30: 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol 2005; 1: 143–145. [DOI] [PubMed] [Google Scholar]
- 23Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M et al. Total synthesis of (+)-chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity. J Am Chem Soc 2010; 132: 4078–4079. [DOI] [PubMed] [Google Scholar]
- 24Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 2007; 25: 473–481. [DOI] [PubMed] [Google Scholar]
- 25Yildirim F, Ji S, Kronenberg G, Barco A, Olivares R, Benito E et al. Histone acetylation and CREB binding protein are required for neuronal resistance against ischemic injury. PLoS ONE 2014; 9: e95465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem 2008; 283: 36542–36552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27Greenberg DA, Jin K. Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci 2013; 70: 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28Chisholm N, Henderson M, Selvamani A, Park MJ, Dindot S, Miranda R et al. Histone methylation patterns in astrocytes are influenced by age following ischemia. Epigenetics 2015; 10: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM et al. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci USA 2012; 109: E962–E971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30Passaro D, Rana G, Piscopo M, Viggiano E, De Luca B, Fucci L. Epigenetic chromatin modifications in the cortical spreading depression. Brain Res 2010; 1329: 1–9. [DOI] [PubMed] [Google Scholar]
- 31Rana G, Donizetti A, Virelli G, Piscopo M, Viggiano E, De Luca B et al. Cortical spreading depression differentially affects lysine methylation of H3 histone at neuroprotective genes and retrotransposon sequences. Brain Res 2012; 1467: 113–119. [DOI] [PubMed] [Google Scholar]
- 32Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol 2011; 7: 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33Liu F, Barsyte-Lovejoy D, Li F, Xiong Y, Korboukh V, Huang XP et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem 2013; 56: 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34Cherblanc FL, Chapman KL, Brown R, Fuchter MJ. Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat Chem Biol 2013; 9: 136–137. [DOI] [PubMed] [Google Scholar]
- 35Cherblanc FL, Chapman KL, Reid J, Borg AJ, Sundriyal S, Alcazar-Fuoli L et al. On the histone lysine methyltransferase activity of fungal metabolite chaetocin. J Med Chem 2013; 56: 8616–8625. [DOI] [PubMed] [Google Scholar]
- 36Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S et al. Valproic acid induces functional heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem 2009; 111: 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B et al. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci 2008; 28: 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM. Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 2002; 43: 1173–1179. [DOI] [PubMed] [Google Scholar]
- 39Van Kanegan MJ, He DN, Dunn DE, Yang P, Newman RA, West AE et al. BDNF mediates neuroprotection against oxygen-glucose deprivation by the cardiac glycoside oleandrin. J Neurosci 2014; 34: 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40Hasan MR, Kim JH, Kim YJ, Kwon KJ, Shin CY, Kim HY et al. Effect of HDAC inhibitors on neuroprotection and neurite outgrowth in primary rat cortical neurons following ischemic insult. Neurochem Res 2013; 38: 1921–1934. [DOI] [PubMed] [Google Scholar]
- 41Kim HJ, Leeds P, Chuang DM. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem 2009; 110: 1226–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42Kim HJ, Chuang DM. HDAC inhibitors mitigate ischemia-induced oligodendrocyte damage: potential roles of oligodendrogenesis, VEGF, and anti-inflammation. Am J Transl Res 2014; 6: 206–223. [PMC free article] [PubMed] [Google Scholar]