

Abstract
Lactate has been shown to offer neuroprotection in several pathologic conditions. This beneficial effect has been attributed to its use as an alternative energy substrate. However, recent description of the expression of the HCA1 receptor for lactate in the central nervous system calls for reassessment of the mechanism by which lactate exerts its neuroprotective effects. Here, we show that HCA1 receptor expression is enhanced 24 hours after reperfusion in an middle cerebral artery occlusion stroke model, in the ischemic cortex. Interestingly, intravenous injection of L-lactate at reperfusion led to further enhancement of HCA1 receptor expression in the cortex and striatum. Using an in vitro oxygen-glucose deprivation model, we show that the HCA1 receptor agonist 3,5-dihydroxybenzoic acid reduces cell death. We also observed that D-lactate, a reputedly non-metabolizable substrate but partial HCA1 receptor agonist, also provided neuroprotection in both in vitro and in vivo ischemia models. Quite unexpectedly, we show D-lactate to be partly extracted and oxidized by the rodent brain. Finally, pyruvate offered neuroprotection in vitro whereas acetate was ineffective. Our data suggest that L- and D-lactate offer neuroprotection in ischemia most likely by acting as both an HCA1 receptor agonist for non-astrocytic (most likely neuronal) cells as well as an energy substrate.
Keywords: energy metabolism, focal ischemia, lactate, neuron-glia interactions, neuroprotection
Introduction
Stroke is a frequent and often disabling disease whose early identification and treatment within a few hours from symptom onset are essential. In ischemic stroke, the most common stroke subtype resulting from an arterial occlusion, energy failure because of lack of oxygen and glucose supply to the brain is the central mechanism leading to neural damage. Brain metabolism therefore has a critical role in the pathophysiologic mechanisms underlying neuronal damage in stroke and for recovery. Even though glucose is known as the brain energy substrate par excellence, other metabolic intermediates such as monocarboxylates including lactate, pyruvate, acetate, and ketone bodies have been shown to be oxidized for energy production1, 2, 3 under certain conditions. Brain energy metabolism is a compartmentalized process involving neurons as well as astrocytes and oligodendrocytes, with metabolic interactions between different cell types. Monocarboxylates can be transported to and from different cell types by monocarboxylate transporters (MCTs) with different degrees of affinity. Monocarboxylate transporters are a family of proton-dependent carriers where MCT1, MCT2, and MCT4 are the main MCTs in the central nervous system (CNS). Under physiologic conditions, MCT1 is expressed in endothelial cells, astrocytes, and oligodendrocytes, MCT4 in astrocytes, and MCT2 is the main transporter in neurons. Under hypoxic conditions, MCT1 and MCT2 are also expressed in activated microglia.4, 5, 6
Beside its role as an energy substrate, lactate also acts as a signaling molecule via the HCA1 receptor in adipose tissue and skeletal muscle.7 Interestingly, recent work revealed the presence of the HCA1 receptor in different brain structures, including cortex, hippocampus, and cerebellum,8 and it has been shown that lactate administration to cortical neurons induced specific modulations in calcium transients frequencies in the same manner as a specific HCA1 receptor agonist.9 HCA1 receptor involvement has also been suggested in neurologic disorders such as cerebral malaria.10
We have previously shown that L-lactate administration is neuroprotective in models of cerebral ischemia both in vitro and in vivo: it attenuated neuronal death in vitro in organotypic hippocampal slice cultures subjected to oxygen and glucose deprivation.11 Both, intracerebroventricular and intravenous administration of L-lactate after transient middle cerebral artery occlusion (tMCAO) lead to a reduction in lesion size and improved neurologic outcome.11, 12 Although it was assumed so far that lactate exerts its neuroprotective effect by acting as a metabolic substrate for energy-deprived neurons, the mode of action is so far not characterized and recent data on the presence and role of the lactate receptor HCA1 in the CNS challenges this assumption. In the present study, we have explored both possible modes of action, as a metabolic substrate and signaling molecule. Understanding the mechanisms by which lactate exerts its neuroprotective effect is essential to adequately translate this promising preclinical neuroprotection strategy to a clinical setting.
Materials and methods
All experiments were conducted in accordance with ordinance 455.163 of the Swiss Federal Veterinary Office and approved by the Service Cantonal des Affaires Vétérinaires (license number VD2017.4b to LH) and the Kanton Zürich Gesundheitsdirektion Veterinäramt (license number ZH53/2007 to BW) (cantonal veterinary authority). The animal studies are reported according to the ARRIVE guidelines.
Transient Middle Cerebral Artery Occlusion in the Mouse
A total of 55 male CD1 mice (body weight 26 to 35 g, Charles River, L'arbresle, France) were housed in the animal facility of the Department of Fundamental Neuroscience of the Université de Lausanne with 12:12 light/dark cycle with normal illumination, in groups of 5 per cage during 1 week for acclimatization before surgery. For experiments, mice were maintained anesthetized with isoflurane (1.5% to 2% in nitrous oxide/oxygen 70%/30%) using a face mask. Body temperature was maintained at 37.0±0.5 °C throughout surgery (FHC, Bowdoinham, ME, USA). Regional cerebral blood flow (rCBF) was measured and continuously recorded throughout the operation in all animals by laser-Doppler flowmetry (Perimed AB, Stockholm, Sweden) with a flexible probe fixed on the skull (1 mm posterior and 6 mm lateral from bregma). Transient focal cerebral ischemia (30 or 45 minutes depending on the set of experiments) was induced by occlusion of the left middle cerebral artery (MCA) with an intraarterial suture as described elsewhere.11 Briefly, the left common carotid artery and the left external carotid artery were exposed and ligated after a ventral midline neck incision. Ischemia was induced by inserting a silicon-coated nylon monofilament (0.17 mm diameter) through the left common carotid artery into the internal carotid artery until mild resistance was felt and a drop to less than 20% of initial rCBF was registered. Regional cerebral blood flow was monitored and maintained below 20% of the baseline level during ischemia. Reperfusion was considered successful if the rCBF rose above 50% of baseline. Only mice that had a drop of 80% of rCBF and accomplished a reperfusion of above 50% of baseline in the rCBF were included in the study. They received 5 μL per gram of body weight of either sodium D-lactate solution (200 mmol/L), pyruvate solution (200 mmol/L), or vehicle solution (phosphate-buffered saline, PBS) injected randomly in the tail vein at reperfusion using a 1- mL syringe with a 25-gauge needle and a mouse restrainer (Braintree Scientific, Braintree, MA, USA). All surgeries were performed during day-light, between 9 am and 5 pm. The surgeon was not masked. Each mouse was considered as an experimental unit.
At the beginning of the surgery, mice were administered 0.025 mg/kg of buprenorphine subcutaneously for post-surgery analgesia. Once the animals were awake, they were housed overnight in an incubator at 28 °C.
Behavioral Evaluation
The neurologic deficit was evaluated after reperfusion and before euthanasia, with a composite neuroscore graded for severity after surgery (0: no observable neurologic deficit; 1: failure to extend the right forepaw; 2: circling to the contralateral side, and 3: loss of walking or righting reflex) as previously described11 and motor performance assessed on the Rotarod treadmill (UgoBasile, Milan, Italy). In this test, mice were placed on the rotating cylinder, set to accelerate uniformly from 4 to 40 rpm, and their latency to fall was recorded before 600 seconds. The animals were trained on two different days before surgery, with two trials in each training session. The test was then performed 24 and 48 hours after MCAO, with two consecutive trials for each animal. The better of the two trials was selected. Points were attributed on the basis of performances expressed as a percentage of the best performance before ischemia (0: 90% to 100% and then 0.5 point for each decrease of 15% until a maximum of 5 points for the highest neurologic deficit). The behavior evaluation was assessed in a masked manner.
Determination of Ischemic Lesion Volumes
Animals were killed 48 hours after the onset of focal ischemia and 20-μm-thick, 720-μm-distant, coronal cryostat sections were stained with cresyl violet for histologic determination of lesion size. Digitalized images of the Nissl-stained tissue were acquired under a light stereomicroscope (Leica MZ16FA, Heerbrugg, Switzerland) and the lesion area was measured using ImageJ software (ImageJ 1.36b, National Institute of Health). Infarct volume was calculated by multiplying the sum of the lesion areas on each section by the distance between sections.13
Immunohistochemistry
Mice were injected intraperitoneally with a lethal dose of pentobarbital (10 mL/kg, Sigma, Buchs, Switzerland) and then perfused with 150 mL of 4% paraformaldehyde (Sigma-Aldrich, St Louis, MO, USA) dissolved in 1 × PBS at pH 7.4. Brains were dissected, postfixed overnight at 4 °C, cryoprotected 24 hours in 30% sucrose solution (Sigma-Aldrich), and rapidly frozen. Twenty micrometer thick coronal microtome-cryostat (Leica MC 3050S) sections were stored in cryoprotectant (30% ethylene glycol and 25% glycerin in 1 × PBS) at −20 °C. For immunostaining, sections were washed three times in 1 × PBS and blocking of non-specific binding was achieved by incubating in 1 × PBS containing 1% bovine serum albumin, 0.1% Triton X-100, and 10% normal goat serum during 1 hour. Double labeling was performed overnight at 4 °C in the 1 × PBS solution without normal goat serum and different antibodies: monoclonal mouse anti-neuronal nuclear antigen (NeuN) antibody (1:500 dilution, Sigma) or monoclonal mouse anti-microtubule associated protein (MAP2) antibody (1:300 dilution, Sigma) and polyclonal rabbit anti-hydroxycarboxylic acid receptor (HCA1) (1:500 dilution; Sigma). After washing three times with 1 × PBS, brain sections were incubated for 2 hours at room temperature with the following fluorescent secondary antibodies: donkey anti-rabbit Alexa-594 (1:250, Invitrogen, Eugene, OR, USA), donkey anti-mouse Alexa-488 (1:250, Invitrogen). After immunostaining, brain sections were incubated for 10 minutes with 4,6 diamidino-2-phenylindole (Sigma) dissolved in 1 × PBS (1/100,000) to reveal nuclei. Preparations were then maintained at 4 °C until observation with a Zeiss LSM 710 Quasar Confocal Microscope (Zeiss, Feldbach, Switzerland).
Western Blotting
For protein expression experiments, young adult male mice were subjected to 30 minutes MCAO as described above and killed either at 1, 3, 8, 24, or 48 hours after reperfusion. Striatum (caudate and putamen), primary motor, and somatosensory cortex as well as hippocampus were collected using a rodent brain matrix (adult mouse, coronal sections, ASI Warren, MI, USA).
Total proteins were extracted from brain tissues by cellular lysis in ice-cold RIPA buffer (#9806, Cell Signaling, Beverly, MA, USA) supplemented with a mixture of protease inhibitors (Complete 11257000; Roche, Basel, Switzerland). Protein quantification was performed with the Pierce BCA Protein Assay kit (#23227, Thermo Fisher Scientific, Pierce, Lausanne, Switzerland) and approximately 20 μg of proteins were denatured (95 °C) for 5 minutes in sodium dodecyl sufate-polyacrylamide gel electrophoresis sample buffer (60 mmol/L Tris-HCl pH 6.8, 5% SDS, 6.6% glycerol, 5 mmol/L EDTA, 5% β-mercaptoethanol, and 0.1% bromophenol blue). Samples were separated on a 10% acrylamide gel with a 4% stacking gel using an Electrophoresis Unit (Bio-Rad, Cressier, Switzerland). Proteins were then electroblotted onto nitrocellulose membranes (0.45 μm; #162-0115, Bio-Rad) using the Electrophoresis Unit. Nonspecific binding sites were blocked for 1 hour at room temperature with a solution of Tris-Buffered-Saline (50 mmol/L Tris-HCl pH 7.5, 150 mmol/L NaCl) supplemented with 0.1% Tween-20 and containing 10% (wt/vol) skimmed milk. Blots were then incubated overnight at 4 °C with specific primary antibodies in Tris-Buffered-Saline 0.1% containing 1% skimmed milk: rabbit anti-mouse GPR81-S296 (HCA1) (1:500 dilution; #SAB1300790, Sigma). Blots were washed three times in Tris-Buffered-Saline 0.1% and subsequently incubated 2 hours at room temperature with horseradish peroxidase-conjugated donkey anti-rabbit IgG (#NA9340V, 1:10,000 dilution; GE Healthcare, Glattbrugg, Switzerland). After being washed three times in Tris-Buffered-Saline 0.1%, blots were processed using Immun-StarTMWesternCTM Chemiluminescent Kit (#170-5070, Bio-Rad). Chemiluminescence detection was performed with the ChemiDoc XRS System (#170-8070, Bio-Rad). Total protein content assay was performed with the Pierce Reversible Protein Stain kit (#24580, Thermo Fisher Scientific, Pierce) and revealed with the ChemiDoc XRS System (#170-8070, Bio-Rad). Both types of labeling were quantified with the ImageLab 3.0 software (Bio-Rad) and the HCA1 (GPR81) protein expression was normalized to the total protein content.
Organotypic Hippocampal Slice Cultures
Hippocampal slice cultures were prepared as described elsewhere11 from P8 to P10 rats (OFA Sprague Dawley, Janvier, France). Coronal hippocampal sections (350 μm) were cultured on sterile porous membrane units (Millicell, Millipore, Billerica, MA, USA) in wells containing 1 mL of culture medium with D-glucose 36 mmol/L, 25% horse serum, 50% minimal essential medium (supplemented with HEPES and sodium bicarbonate; Gibco, Paisley, UK), 25% HBSS (Hank's balanced salt solution; Gibco), and L-glutamine 2 mmol/L (Sigma-Aldrich). Cultures were kept at 33 °C, 5% CO2, 100% humidity for 4 days. The medium was replaced by fresh identical medium at day 4, and then by culture medium with 15% horse serum, 60% minimal essential medium, 25% HBSS, and L-glutamine 2 mmol/L at day 7 and 10. Experiments were conducted after 10 days of culture.
Oxygen and Glucose Deprivation on Organotypic Hippocampal Slice Cultures
Oxygen and glucose deprivation (OGD) experiments were performed in serum-free low glucose medium, Dulbecco's modified Eagle's medium (D5030, Sigma-Aldrich) supplemented with D-glucose 1 mmol/L and L-glutamine 2 mmol/L and equilibrated for 1 hour at 37 °C, in a humidified hypoxic chamber (COY, Grass Lake, MI, USA) with an atmosphere of 5% O2, 5% CO2, completed by N2. Hippocampal slices were transferred into this medium and placed into the hypoxic chamber for 1 hour. Control cultures were kept in 60% minimal essential medium, 15% horse serum, 25% HBSS, and L-glutamine 2 mmol/L for 1 hour at 37 °C in a humid normoxic atmosphere.11, 14 For recovery, cultures where then transferred into fresh culture medium at 33 °C for 48 hours. Cultures were randomly treated with either D-lactate, pyruvate, acetate (Sigma-Aldrich; diluted in 1 × PBS, pH 7), 3,5-dihydroxybenzoic acid (Sigma-Aldrich; diluted in ethanol) or an equal volume of culture medium. Sodium D-lactate at a final concentration of 4 mmol/L, pyruvate at 10 mmol/L, sodium acetate at 0.13, 0.2, 4, or 8 mmol/L, or 3,5-dihydroxybenzoic acid at 4 mmol/L was administered immediately after OGD.
Assessment of Cell Death in Hippocampal Slices
Cell death was determined in the CA1 region using the fluorescent viability indicator propidium iodide. Propidium iodide was applied in each dish (50 μg/mL) 1 hour before measurement. Propidium iodide fluorescence emission (excitation wavelength 536 nm, emission wavelength 617 nm) was measured 48 hours after hypoxia using an epifluorescence microscope with a × 5 lens coupled to a camera (Leica). Propidium iodide images were acquired with standardized camera settings and signal intensity was measured with ImageJ software. After subtracting the background fluorescence on each slice, the results were expressed as a percentage of maximal cell death obtained by submerging slices in PBS for 24 hours at 4 °C. Cell death was averaged for the four slices of each culture well. The experimenter was not masked.
Experiments Using 11C-D-lactate in Sprague Dawley Rats
Radiotracer
The D- and L-enantiomers of 1-11C-lactic acid were produced by a previously described radiosynthetic method, which includes separation of racemic 1-11C-lactic acid by preparative chiral ligand exchange high-performance liquid chromatography (HPLC). Before use, the fractions corresponding to 1-11C-D- or 1-11C-L-lactic acid were further purified and finally formulated as sodium salts in physiologically acceptable sodium phosphate-buffered saline (pH 7). The chemical and enantiomeric purity of each isolated radiolabeled enantiomer was routinely monitored by analytical chiral ligand exchange HPLC as part of the radiosynthesis quality control and typically revealed more than 99% enantiomeric excess of the final product. The specific activity at the end of the synthesis was approximately 400 GBq/μmol. Both preparative and analytical chiral ligand exchange chromatography were performed on reversed-phase C18 columns (octadecyl-silica), coated with a D- or L-penicillamine-derived chiral selector. The preparation of the chiral selectors, the coating procedures for the HPLC columns, and the HPLC separation conditions have been described in detail earlier.15
Surgical preparation
Surgery was performed under isoflurane anesthesia (2% to 3% in air/oxygen 70%/30%) and involved the placement of an arteriovenous shunt from the right femoral artery to the right femoral vein, tracheotomy for mechanical ventilation, and craniotomy for the placement of the beta scintillator. The actual experiments were performed under α-chloralose anesthesia (44 mg/kg subcutaneously). The arteriovenous shunt was run through a coincidence scintillator (GE Medical Systems, Waukesha, WI, USA). The online arterial sampling procedure is described in detail elsewhere.15 In short, total radioactivity in arterial blood was continuously recorded and was then corrected for (1) a different tracer concentration in whole blood and plasma and (2) the build-up of labeled metabolites.
D-Lactate kinetics in the somatosensory cortex
For the measurement of the time-course of 1-11C-D-lactate in the brain, an intracortical beta probe was used.16, 17 Twenty minutes of data were acquired at baseline condition after injection of 200 to 300 MBq of radiotracer in 5 animals. In one of these animals, baseline cerebral blood flow was determined before 1-11C-D-lactate acquisition using 15O-H2O and the methodology described previously.16 The first pass extraction fraction of 1-11C-D-lactate was then calculated using the relationship EF=K1/CBF where K1 is the transport parameter describing (1-11C-D-lactate) transport from blood to tissue (Figure 4A).
Kinetic modeling
Acquired radioactivity data were analyzed using the software package PMOD (PMOD Technologies GmbH, Zürich, Switzerland). The investigated methods consisted of standard compartmental modeling using the arterial input function and the one-tissue compartment model. The parameters are as follows: K1 describes the transport of the tracer across the blood brain barrier and k2 represents the back-diffusion of label from tissue to the blood system. Carbon is labeled as ‘C.' Label exchange between compartments is described by the following differential equation:
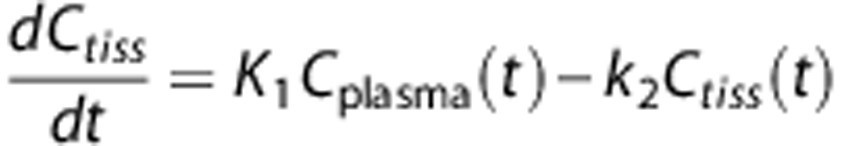 |
The basis of the calculation of the CBF measurement was also the one-tissue compartment model including a partition coefficient for 15O-H2O.16
Metabolite analysis in the blood
Samples (approximately 400 μL) were collected at different time points after tracer injection, with a maximum of 4 to 5 blood samples per animal, to determine the time-course of the ratio of the 11C activity in plasma to whole blood and for analysis of authentic tracer and metabolites. These samples were first centrifuged for 3 minutes at 270 g. Proteins were then precipitated with 75 μL acetonitrile in 50 μL plasma. After centrifugation for 3 minutes at 270 g, the composition of the 11C-derived radioactivity in the supernatant (80 μL) was analyzed by HPLC on a polymeric column (PRP-1, 5-μm, 250 × 4.1 mmol/L intradermally, Hamilton, Franklin, MA, USA) with 3 mmol/L phosphoric acid in water (pH 2.67) as the mobile phase (1 mL/minute). The retention times of 11C-HCO3− (3.3 minutes) and lactic acid (5.1 minutes) were determined by using aqueous solution of NaHCO3 and DL-lactic acid as reference compounds, detected by ultraviolet absorption at 220 nm. The amount of authentic tracer was expressed as a fraction of total plasma counts.
Metabolite analysis in brain
At the end of the experiment, the rats were perfused with PBS, brains were removed and prepared for measurements with the HPLC system. Each brain was first homogenized before adding acetonitrile (150%, i.e., 1.5 × volume/weight of brain sample). The subsequent procedure was the same as with the blood samples described above, except that the amount of supernatant injected into the HPLC system was 200 μL.
Statistical Analysis
All data are presented as mean±s.d. Statistical analyses were performed using non-parametric tests; one-way analysis of variance (Kruskal–Wallis followed by Dunn's multiple comparison test) was used for analysis of in vitro experiments and the Mann–Whitney test was used for comparison between two groups in the in vivo experiments and for the behavioral experiments. Paired t-test was used to compare protein expression. P<0.05 was considered statistically significant.
Results
HCA1 Receptor Expression and a Possible Neuroprotective Role in Ischemia
Recent studies revealed the presence and mechanism of action of the HCA1 receptor (formerly known as GPR81) in the CNS.8, 9, 10 Because L-lactate acts as an endogenous ligand for this receptor, it was of interest to describe the cellular expression of this receptor in several brain regions affected by ischemic damage in the mouse tMCAO model. We confirmed a strong neuronal expression of the HCA1 receptor in the hippocampus (CA1 and dentate gyrus), the cortex, and the striatum as revealed by double labeling immunofluorescence for the HCA1 receptor and the neuronal markers NeuN or MAP2 (Figure 1: NeuN (green), MAP2 (green), HCA1 (red), 4,6 diamidino-2-phenylindole (blue)). The next step was to evaluate whether such HCA1 receptor expression was altered at different time points after 30 minutes tMCAO. For this purpose, the level of HCA1 receptor expression was evaluated on Western blots in protein extracts from hippocampus, cerebral cortex and striatum at 1, 3, 8, 24, and 48 hours after 30 minutes of tMCAO. Results at 1, 3, and 8 hours did not reveal significant differences between the three brain structures investigated (see Supplementary Table 1). However, a significant increase in HCA1 receptor expression was seen in the ischemic cortex after 24 hours (1.2±0.2, P<0.05 vs. contralateral cortex; Figure 2A). Even though there is neuronal loss in the lesion core (ischemic striatum), we did not observe a significant decrease in HCA1 receptor expression in this structure (0.8±0.3, n.s. vs. contralateral cortex) which may reflect a compensatory increase in HCA1 receptor expression in remaining neurons (Figure 2A). At 48 hours after ischemia, no difference could be observed between ischemic and contralateral tissues (see Supplementary Table 1). On the basis of this result and the possibility that lactate regulates HCA1 receptor expression, we injected L-lactate intravenously at the beginning of the reperfusion period and monitored any changes in HCA1 receptor expression at 24 hours after the ischemic episode. We found an increase in HCA1 receptor expression in both the ischemic cortex (1.57±0.34, P<0.04 vs. contralateral cortex) and ischemic striatum (1.7±0.4, P<0.05 vs. contralateral striatum) while no significant difference was observed for the hippocampus (Figure 2B). These observations suggest that the expression of the HCA1 receptor may be modulated by ischemia and potentially by lactate injected at reperfusion.
Figure 1.

Confocal visualization of HCA1 receptor expression. Using two different antibodies against neuronal cells, we confirmed with immunohistochemistry the localization of the HCA1 receptor is in neurons. The brain structures analyzed were hippocampus (CA1 (1a–1f) and Dentate Gyrus (DG) (2a–2f) regions) primary cortex (3a–3f) and Striatum (4a–4f). Color code: NeuN (green, a and c), MAP2 (green, d and f), HCA1 (red, b and c and e and f), 4,6 diamidino-2-phenylindole (blue). Magnification: 40 μm.
Figure 2.

The HCA1 receptor expression is promoted by ischemia and has a role in the neuroprotective effect. (A) Western blot analysis showed an increase in the HCA1 receptor expression at 24 hours after transient middle cerebral artery occlusion (tMCAO) in the region surrounding the lesion site (primary motor and somatosensory cortex), in comparison with the contralateral hemisphere. (B) Intravenous L-lactate injection prompted an increase also in the lesion site (striatum) at the same time point (*P<0.05; paired t-test ischemic vs. contralateral hemisphere). (C) Western blot analysis showed an increase in the HCA1 receptor expression at 48 hours after oxygen and glucose deprivation (OGD) (**P<0.01 paired t-test). (D) Administration of 4 mmol/L 3,5-dihydroxybenzoic acid (3,5-DHBA), an HCA1 receptor agonist, attenuated cell death when administered directly to the medium after OGD (*P<0.05, ***P<0.001 one-way analysis of variance (ANOVA) Kruskal–Wallis test followed by Dunn's multiple comparison test).
We went on to further investigate the role of HCA1 receptors under ischemic conditions in an in vitro model. For this purpose, we used rat organotypic hippocampal slices exposed to an OGD protocol. We observed a significant increase in HCA1 receptor expression 48 hours after OGD (1.45±0.16, P<0.01 vs. control slices; Figure 2C). To determine whether the enhanced HCA1 receptor expression could have a role in the neuroprotective effect of L-lactate in this model, organotypic hippocampal slices were exposed to the HCA1 receptor agonist 3,5-dihydroxybenzoic acid after OGD. Results show that 3,5-dihydroxybenzoic acid produced significant protection as it reduced neuronal cell death in the CA1 region of the hippocampus 48 hours after insult from 13.5±7.2% to 7.4±5.2% (P<0.05; Figure 2D).
D-Lactate Neuroprotection in Ischemia Models
The D enantiomer of lactate has been described to act at least as a partial agonist of the HCA1 receptor.9, 18 For this reason, we tested the neuroprotective effect of D-lactate both in vitro and in vivo. The administration of 4 mmol/L D-lactate to rat organotypic hippocampal slices directly after OGD also resulted in significantly reduced neuronal cell death in the CA1 region of the hippocampus 48 hours after OGD, from 29.2±14.3% to 11.3±7.3% (P<0.05; Figure 3A). It is noteworthy that the neuroprotective effect approaches that previously reported for L-lactate in this model.11 We then tested D-lactate in vivo by intravenous administration of 1 μmol/g D-lactate after 45 minutes tMCAO. Mice with satisfactory ischemia (rCBF<20% of baseline) and reperfusion (rCBF above 50% of baseline) received a single intravenous administration of D-lactate or vehicle, 10 to 15 minutes after the silicon-coated monofilament was removed allowing reperfusion. A total of nine mice were not included in the study as they did not fulfill the rCBF inclusion criteria. Single administration of D-lactate significantly decreased the infarct volume from 81.8±40.1 mm3 (control group) to 45.5±32 mm3 (D-lactate group) measured 48 hours after ischemia (P<0.05; Figure 3B). Again, the magnitude of neuroprotection was comparable with that reported previously for L-lactate in vivo.12 Behavioral performances on motor task assessment showed a clearly milder neurological deficit at 48 hours (P<0.05) in the D-Lactate group, from a median of 2.5, with scores ranging from 1 to 5 (control group), to a median of 1, with scores ranging from 0 to 1 (D-lactate group, P<0.05, Figure 3C). One mouse from the control group and two mice from the D-lactate group died during the first night after surgery. Three mice per group were killed within the first 6 hours after surgery because of the presence of repeated seizures as required by the veterinary authority.
Figure 3.

Neuroprotection by D-lactate. (A) In vitro, administration of 4 mmol/L D-lactate induced a significant reduction in cell death, assessed by propidium iodide (PI) staining, 48 hours after treatment (*P<0.05, one-way analysis of variance (ANOVA) Kruskal–Wallis followed by Dunn's multiple comparison test). In vivo, intravenous injection of 1 μmol/g D-lactate after 45 minutes transient middle cerebral artery occlusion (tMCAO) decreased total infarct volumes measured 48 hours after ischemia (B) and improved the neurologic outcome (C), neurologic deficit scores from 0, no deficit—white, to 5 worst outcome—black; *P<0.05 Mann–Whitney test, two-tailed P-value.
Cerebral D-Lactate Metabolism
Different in vivo and in vitro studies have suggested that D-lactate is not metabolized in cerebral tissue6, 19, 20 based on the apparent lack of expression of the specific enzyme D-lactate dehydrogenase in mammals. To clarify this issue in the rodent brain, we set up a series of experiments using radiolabeled D-lactate to trace its metabolism and kinetics (Figure 4A). To evaluate the brain uptake of D-lactate, we determined the first pass extraction fraction, which was 16%. The tissue kinetics of 1-11C-D-lactate displayed an increase of more than 5 minutes and slow washout thereafter on tracer injection (Figure 4B). The average respective first-order kinetic rate constants obtained by kinetic modeling were 0.059±0.01 mL/minute/mL tissue (K1) and 0.063±0.01 per minute (k2). Average lactate levels in the blood were 2.2±0.93 mmol/L.
Figure 4.

D-lactate metabolism. (A) Schematic of biochemical pathways involved in the degradation of lactate and the proposed interpretation of the rate constants K1 and k2 which are mathematically defined by the one-tissue compartment model used for 1-11C-D-lactate data analysis. (B) Measured radioactivity concentration in the brain (open circles), model fit (black line), and arterial input curve (gray line). The inset displays the residuals of the fitting to the one-tissue compartment model. Equal distribution around the neutral line supports the adequacy of the applied model. (C) Fraction of native radiolabeled lactate more than 40 minutes after intravenous injection of 1-11C-D-lactate in the blood. The filled circles represent data points from individual measurements and the solid line is the corresponding fit. (D) Percentage of identified metabolites in brain tissue after 40 minutes.
11C-CO2 turned out to be the only metabolite in blood and brain. The time course of the fraction of authentic tracer in arterial plasma is shown in Figure 4C. The pooled fraction data of all animals were approximated by a fit of a quadratic polynomial. This function was subsequently used to convert the total plasma activity to the time-course of authentic 11C-lactate (= input curve). At 40 minutes, the fraction of true tracer in plasma dropped to approximately 30%. Measurements in brain tissue at the end of the experiment revealed approximately one-third CO2 and two-thirds lactate (Figure 4D).
Effect of Pyruvate and Acetate in Ischemia Models
Results obtained on D-lactate metabolism suggest that in addition to a possible activation of the HCA1 receptor, the use of D-lactate as an energy substrate may also contribute to its beneficial effect in ischemia. Therefore, we explored the specificity of its metabolic mode of action by testing the neuroprotective effect of two other monocarboxylates. Pyruvate is the immediate downstream product of either L- or D-lactate and is the main substrate entering the Krebs cycle to provide energy. Other groups have previously described the neuroprotective effect of pyruvate in different models of cerebral ischemia.2, 21 In agreement with these observations, administration of 10 mmol/L pyruvate directly after OGD significantly decreased cell death in the hippocampal CA1 region at 48 hours from 25.4±14.5% to 13.9±10.3% (P<0.05; Figures 5A and 5B). In contrast, we did not observe a significant benefit in vivo after tMCAO. In our hands, pyruvate administration at reperfusion after 30 minutes tMCAO induced only a slight reduction in lesion size (P=0.4), from 91.9±53.6 mm3 (control group, n= 7) to 64.6±53.4 mm3 (pyruvate, n=5) 48 hours after ischemia. A total of four mice were not included because they did not fulfill rCBF inclusion criteria. There was no mortality in this experiment. Because of the inherent variability of the model, our experiment was, however, not powered to detect a significant neuroprotective effect (the sample size calculation using an alpha error of 5% and a beta error of 50% resulted in a sample size of 21 per group, www.dssresearch.com).
Figure 5.

Administration of pyruvate and acetate after OGD. (A) Administration of 10 mmol/L pyruvate directly to the medium after 1 hour oxygen-glucose deprivation (OGD) in rat organotypic hippocampal slices was able to protect against ischemic damage. (B) Administration of various doses of acetate did not protect against ischemic damage. Cell death was assessed by propidium iodide (PI) staining 48 hours after treatment. (C) Representative images of PI staining among each group can be observed in panel (*P<0.05; one-way analysis of variance (ANOVA) Kruskal–Wallis test followed by Dunn's multiple comparison test).
As both L- and D-lactate could be taken up and metabolized in principle by either neurons or astrocytes, we wanted to determine whether the beneficial metabolic effect of lactate in vitro could be exerted via its use by astrocytes. For this purpose, we made use of acetate which is known to be specifically taken up and used as energy substrate by astrocytes.22 However, the addition of different concentrations of acetate (0.13, 0.2, 4, and 8 mmol/L) to organotypic hippocampal slices 1 hour after OGD did not provide significant neuroprotection (Figures 5A and 5C).
Discussion
After being considered for decades as a waste-product of metabolism, lactate has been shown to be able to support synaptic activity1, 23 and is nowadays recognized as a prominent energy substrate in the CNS.24 Lactate is produced under physiologic conditions,25, 26 and it is suggested to have an important role in the metabolic support of long axons6, 27 as well as in long-term memory formation.28 Lack of adequate lactate supply has also been implicated in some neurodegenerative diseases such as amyotrophic lateral sclerosis.29 Moreover, exogenous lactate supplementation seems beneficial after traumatic brain injury.30, 31, 32 In ischemic conditions, our group and others have shown neuroprotection after L-lactate administration at reperfusion after tMCAO.11, 12, 33 Similarly, brain lactate was found to be essential to improve post-ischemic outcome after cardiac arrest.34 Lactate can be transported from one cell type to another by MCTs that are selectively expressed in different cell types and which can transport monocarboxylates with different affinities.35 More recently, lactate has been described to act as a signaling molecule in the CNS, either through the HCA1 receptor8, 9 or through an unknown receptor yet to be specified.36
Our initial characterization of the distribution of the HCA1 receptor in three brain regions affected after tMCAO confirmed a prominent neuronal expression in the CNS.8, 9 Interestingly, our data also suggested that the cerebral expression of HCA1 can be modulated under certain conditions. Lactate itself seems to be a signal for the induction of its own receptor. However, whether lactate accumulation after ischemia is responsible for the enhancement of the HCA1 receptor observed after 24 hours of reperfusion remains to be directly shown. Nonetheless, our results provide the first evidence that activation of HCA1 receptors alone is sufficient to provide neuroprotection, at least in an in vitro ischemic model. Our demonstration that neuroprotection was also achieved with D-Lactate confirms this observation in that D-lactate was shown to exhibit at least partial agonist activity on HCA1/GPR81 receptor18, 37 and that D-lactate was considered as a non-metabolizable lactate enantiomer and often used as a negative control to show the necessity of L-lactate use as an alternative energy substrate. However, unexpectedly, we found that D-lactate is extracted by the rodent brain and oxidized. In fact, one third of D-lactate was metabolized to CO2, which is only marginally less than the previously reported fraction of CO2 produced from L-lactate (42±8% CO2 after 40 minutes1). This observation suggests that the rodent brain may possess a D-lactate dehydrogenase activity. In accordance, the isolation of human and mouse transcripts encoding a homolog of the yeast D-lactate dehydrogenase was reported and its expression was found in various tissues, including the brain.38 However, it remains to be determined whether the presence of a cerebral D-Lactate dehydrogenase enzyme is sufficient to explain our results or whether the activity is performed by another a priori unrelated enzyme.
In the CNS, pyruvate is transported mainly by MCT1 and MCT2 in the direction of the concentration gradient, and these MCTs are located in astrocytes and neurons, respectively.4, 39 Possible explanations for the neuroprotection against ischemic damage induced by either L- or D-lactate are the fact that lactate is converted to pyruvate by the mitochondrial lactate dehydrogenase to fuel the Krebs cycle and therefore used as energetic substrate and that this conversion produces reduced nicotinamide adenine dinucleotide that will in turn act as a reactive oxygen species scavenger.40 Evidence pointing towards this metabolic explanation is the protection exerted by the administration of 10 mmol/L pyruvate in vitro. Protection is not seen in vivo though. However, in support of a metabolism-mediated neuroprotective effect, others2, 21 have shown protection in vivo using an extended intravenous mode of administration of pyruvate.21 This difference in the mode of administration may explain the different in vivo results, although we cannot exclude that lactate may prove to be a more efficient neuroprotective agent, notably in vivo, in particular because of the ability of both L- and D-lactate to activate the HCA1 receptor.
It has been shown that acetate is preferentially transported by MCT1 and metabolized in astrocytes.17, 22 During ischemia, it was observed that 14C-acetate uptake is decreased as early as 3 minutes after the onset of ischemia. The degree of reduction in 14C-acetate uptake correlates with the severity of ischemia which, according to Hosoi et al,41 relates to the depression of glial metabolism. Even though the reduction in 14C-acetate uptake was reversible and reached full recovery 3 hours after 30 minutes MCAO, Hosoi et al did not evaluate the functional outcome of acetate administration. We showed here that the administration of acetate did not exert protection against neuronal death in hippocampal slices subjected to oxygen and glucose deprivation. Even though there is an increased energy demand in neurons and astrocytes subjected to ischemic insult, this result suggests that energy supply to astrocytes is not enough to exert neuroprotection. It should be noted that using an in vitro assay of ligand-induced 35S-GTPγS binding with membranes from CHO cells expressing human HCA1 receptor, Cai et al18 described that neither sodium pyruvate nor sodium acetate showed binding activity. As neither acetate nor pyruvate binds to the HCA-1 receptor, the results shown here cannot be related to HCA1 signaling pointing to the use of pyruvate and acetate as metabolic substrates.
Some limitations to consider in the present study are that (1) our in vivo stroke model involves a transient occlusion with removal of the silicon filament, therefore our results are limited to be translated to patients with either a spontaneous reperfusion or successful intravenous thrombolysis or intrarterial recanalisation. Evaluating the effect of lactate administration in permanent ischemia is an important step before aiming for translation in clinical practice. (2) Ischemia is induced under isoflurane anesthesia and this compound has been described as neuroprotective by itself. Therefore, we may not be observing the neurologic damage to its fullness. In organotypic hippocampal slice cultures the tissue architecture is only partially preserved and while some neuronal connections are maintained many are interrupted. Furthermore, there is no blood circulation. Despite these limitations, both models complement each other and strengthen our observations. (3) It should be considered as well that lactate was shown to increase regional CBF in physiologically activated but not resting human brain.42 Although we cannot rule out a contribution from CBF changes, the effect observed in the in vitro model, independently from blood flow, shows that lactate exerts a neuroprotective effect independently from possible effects on cerebral blood flow.
Taken together, these data point to the possibility that lactate, independently of the enantiomer type, may confer neuroprotection after ischemia by two distinct mechanisms: One, through the classic metabolic pathway providing an alternative energy supply to deprived neurons and the other through a receptor-mediated signal transduction mechanism in neurons. In both cases, the exact targets within the cell still need to be identified.
Acknowledgments
The authors thank Maité Willaredt for her help with behavioral evaluation, Carole Berthet for her preliminary observation on the role of D-lactate in ischemia and Melanie Price for critically reading the manuscript.
Author Contributions
XC, KR, MW, KD, and AB designed and performed experiments; LP, BW, and LH designed experiments. XC, MW, LP, and LH wrote the paper.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This work was supported by a CONACYT fellowship to XC and a grant from the Biaggi Foundation to LH.
Supplementary Material
References
- 1Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci 2011; 31: 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2Lee JY, Kim YH, Koh JY. Protection by pyruvate against transient forebrain ischemia in rats. J Neurosci 2001; 21: RC171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Muller-Fielitz H et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat Commun 2014; 5: 3944. [DOI] [PubMed] [Google Scholar]
- 4Pellerin L, Bergersen LH, Halestrap AP, Pierre K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res 2005; 79: 55–64. [DOI] [PubMed] [Google Scholar]
- 5Moreira TJ, Pierre K, Maekawa F, Repond C, Cebere A, Liljequist S et al. Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J Cereb Blood Flow Metab 2009; 29: 1273–1283. [DOI] [PubMed] [Google Scholar]
- 6Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci 2011; 31: 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7Rooney K, Trayhurn P. Lactate and the GPR81 receptor in metabolic regulation: implications for adipose tissue function and fatty acid utilisation by muscle during exercise. Br J Nutr 2011; 106: 1310–1316. [DOI] [PubMed] [Google Scholar]
- 8Lauritzen KH, Morland C, Puchades M, Holm-Hansen S, Hagelin EM, Lauritzen F et al. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb Cortex 2014; 24: 2784–2795. [DOI] [PubMed] [Google Scholar]
- 9Bozzo L, Puyal J, Chatton JY. Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PloS ONE 2013; 8: e71721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10Mariga ST, Kolko M, Gjedde A, Bergersen LH. Lactate transport and receptor actions in cerebral malaria. Front Neurosci 2014; 8: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11Berthet C, Lei H, Thevenet J, Gruetter R, Magistretti PJ, Hirt L. Neuroprotective role of lactate after cerebral ischemia. J Cereb Blood Flow Metab 2009; 29: 1780–1789. [DOI] [PubMed] [Google Scholar]
- 12Berthet C, Castillo Tovar X, Magistretti PJ, Hirt L. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc Dis 2012; 34: 329–335. [DOI] [PubMed] [Google Scholar]
- 13Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab 1990; 10: 290–293. [DOI] [PubMed] [Google Scholar]
- 14Badaut J, Hirt L, Price M, de Castro Ribeiro M, Magistretti PJ, Regli L. Hypoxia/hypoglycemia preconditioning prevents the loss of functional electrical activity in organotypic slice cultures. Brain Res 2005; 1051: 117–122. [DOI] [PubMed] [Google Scholar]
- 15Weber B, Burger C, Biro P, Buck A. A femoral arteriovenous shunt facilitates arterial whole blood sampling in animals. Eur J Nucl Med Mol Imaging 2002; 29: 319–323. [DOI] [PubMed] [Google Scholar]
- 16Weber B, Spath N, Wyss M, Wild D, Burger C, Stanley R et al. Quantitative cerebral blood flow measurements in the rat using a beta-probe and H2 15O. J Cereb Blood Flow Metab 2003; 23: 1455–1460. [DOI] [PubMed] [Google Scholar]
- 17Wyss MT, Weber B, Treyer V, Heer S, Pellerin L, Magistretti PJ et al. Stimulation-induced increases of astrocytic oxidative metabolism in rats and humans investigated with 1-11C-acetate. J Cereb Blood Flow Metab 2009; 29: 44–56. [DOI] [PubMed] [Google Scholar]
- 18Cai TQ, Ren N, Jin L, Cheng K, Kash S, Chen R et al. Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem Biophys Res Commun 2008; 377: 987–991. [DOI] [PubMed] [Google Scholar]
- 19Borg MA, Tamborlane WV, Shulman GI, Sherwin RS. Local lactate perfusion of the ventromedial hypothalamus suppresses hypoglycemic counterregulation. Diabetes 2003; 52: 663–666. [DOI] [PubMed] [Google Scholar]
- 20Tekkok SB, Brown AM, Westenbroek R, Pellerin L, Ransom BR. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J Neurosci Res 2005; 81: 644–652. [DOI] [PubMed] [Google Scholar]
- 21Ryou MG, Liu R, Ren M, Sun J, Mallet RT, Yang SH. Pyruvate protects the brain against ischemia-reperfusion injury by activating the erythropoietin signaling pathway. Stroke 2012; 43: 1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci 1998; 18: 5225–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 1988; 240: 1326–1328. [DOI] [PubMed] [Google Scholar]
- 24Bergersen LH. Lactate transport and signaling in the brain: potential therapeutic targets and roles in body-brain interaction. J Cereb Blood Flow Metab 2015; 35: 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25Fellows LK, Boutelle MG, Fillenz M. Physiological stimulation increases nonoxidative glucose metabolism in the brain of the freely moving rat. J Neurochem 1993; 60: 1258–1263. [DOI] [PubMed] [Google Scholar]
- 26Hu Y, Wilson GS. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem 1997; 69: 1484–1490. [DOI] [PubMed] [Google Scholar]
- 27Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012; 485: 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011; 144: 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012; 487: 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30Ichai C, Armando G, Orban JC, Berthier F, Rami L, Samat-Long C et al. Sodium lactate versus mannitol in the treatment of intracranial hypertensive episodes in severe traumatic brain-injured patients. Intensive Care Med 2009; 35: 471–479. [DOI] [PubMed] [Google Scholar]
- 31Ichai C, Payen JF, Orban JC, Quintard H, Roth H, Legrand R et al. Half-molar sodium lactate infusion to prevent intracranial hypertensive episodes in severe traumatic brain injured patients: a randomized controlled trial. Intensive Care Med 2013; 39: 1413–1422. [DOI] [PubMed] [Google Scholar]
- 32Bouzat P, Sala N, Suys T, Zerlauth JB, Marques-Vidal P, Feihl F et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med 2014; 40: 412–421. [DOI] [PubMed] [Google Scholar]
- 33Horn T, Klein J. Neuroprotective effects of lactate in brain ischemia: dependence on anesthetic drugs. Neurochem Int 2013; 62: 251–257. [DOI] [PubMed] [Google Scholar]
- 34Schurr A, Payne RS, Miller JJ, Tseng MT. Preischemic hyperglycemia-aggravated damage: evidence that lactate utilization is beneficial and glucose-induced corticosterone release is detrimental. J Neurosci Res 2001; 66: 782–789. [DOI] [PubMed] [Google Scholar]
- 35Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem 2005; 94: 1–14. [DOI] [PubMed] [Google Scholar]
- 36Tang F, Lane S, Korsak A, Paton JF, Gourine AV, Kasparov S et al. Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat Commun 2014; 5: 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37Liu C, Kuei C, Zhu J, Yu J, Zhang L, Shih A et al. 3,5-Dihydroxybenzoic acid, a specific agonist for hydroxycarboxylic acid 1, inhibits lipolysis in adipocytes. J Pharmacol Exp Ther 2012; 341: 794–801. [DOI] [PubMed] [Google Scholar]
- 38Flick MJ, Konieczny SF. Identification of putative mammalian D-lactate dehydrogenase enzymes. Biochem Biophys Res Commun 2002; 295: 910–916. [DOI] [PubMed] [Google Scholar]
- 39Pierre K, Magistretti PJ, Pellerin L. MCT2 is a major neuronal monocarboxylate transporter in the adult mouse brain. J Cereb Blood Flow Metab 2002; 22: 586–595. [DOI] [PubMed] [Google Scholar]
- 40Schurr A, Gozal E. Aerobic production and utilization of lactate satisfy increased energy demands upon neuronal activation in hippocampal slices and provide neuroprotection against oxidative stress. Front Pharmacol 2011; 2: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41Hosoi R, Kashiwagi Y, Tokumura M, Abe K, Hatazawa J, Inoue O. Sensitive reduction in 14C-acetate uptake in a short-term ischemic rat brain. J Stroke Cerebrovasc Dis 2007; 16: 77–81. [DOI] [PubMed] [Google Scholar]
- 42Mintun MA, Vlassenko AG, Rundle MM, Raichle ME. Increased lactate/pyruvate ratio augments blood flow in physiologically activated human brain. Proc Natl Acad Sci USA 2004; 101: 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.