

Abstract
The transcription factor FoxG1 regulates neurogenesis in the embryonic telencephalon as well as a number of other neurodevelopmental processes. While FoxG1 continues to be expressed in neurons postnatally and through adulthood, its role in fully differentiated neurons is not known. The current study demonstrates that FoxG1 promotes the survival of postmitotic neurons. In cerebellar granule neurons primed to undergo apoptosis, FoxG1 expression is reduced. Ectopic expression of FoxG1 blocks neuronal death, whereas suppression of its expression induces death in otherwise healthy neurons. The first 36 residues of FoxG1 are necessary for its survival-promoting effect, while the C-terminal half of the protein is dispensable. Mutation of Asp219, a residue necessary for DNA binding, abrogates survival promotion by FoxG1. Survival promotion is also eliminated by mutation of Thr271, a residue phosphorylated by Akt. Pharmacological inhibition of Akt blocks the survival effects of wild-type FoxG1 but not forms in which Thr271 is replaced with phosphomimetic residues. Treatment of neurons with IGF-1, a neurotrophic factor that promotes neuronal survival by activating Akt, prevents the apoptosis-associated downregulation of FoxG1 expression. Moreover, the overexpression of dominant-negative forms of FoxG1 blocks the ability of IGF-1 to maintain neuronal survival suggesting that FoxG1 is a downstream mediator of IGF-1/Akt signaling. Our study identifies a new and important function for FoxG1 in differentiated neurons.
Introduction
FoxG1 (also referred to as BF-1) is a member of the winged-helix or forkhead family of transcription factors acting primarily as a transcriptional repressor through DNA binding (Murphy et al., 1994; Li et al., 1995; Bourguignon et al., 1998). During early brain development, FoxG1 is expressed selectively in rapidly proliferating cell populations comprising the telencephalon, where it functions to regulate the rate of neurogenesis by keeping cells in a proliferative state and by inhibiting their differentiation into neurons (Tao and Lai, 1992; Xuan et al., 1995; Hanashima et al., 2002, 2004). Neural progenitor cells in the telencephalon of mouse embryos lacking FoxG1 exit the cell cycle prematurely and differentiate into neurons. The depletion of the neural progenitor population leads to a marked reduction in the size of the FoxG1−/− telencephalon, culminating in a loss of ventral telencephalic structures and perinatal lethality (Xuan et al., 1995; Hanashima et al., 2002). FoxG1 continues to be expressed in neurogenic areas of the postnatal brain such as the subventricular zone and the hippocampal dentate gyrus. As in the telencephalon, FoxG1 functions as a regulator of neurogenesis in the postnatal hippocampus (Shen et al., 2006). Overexpression of FoxG1 in the developing chick neural tube caused a thickening of the neuroepithelium leading to large outgrowths in the telencephalon and mesencephalon (Ahlgren et al., 2003). The overgrowth was proposed to be due to a reduction in cell death within the neuroepithelium, rather than an increase in cell proliferation (Ahlgren et al., 2003). Likewise, an analysis of the postnatal hippocampus in FoxG1+/− mice showed reduction in the number of newly produced dentate gyrus neurons, which was suggested to be due to reduced survival of this postnatally generated cell population (Shen et al., 2006). However, another group analyzing FoxG1+/− embryos concluded that FoxG1 promotes cell death in the developing telencephalon rather than suppressing it (Martynoga et al., 2005).
In addition to regulating proliferation, differentiation, and possibly survival of neural progenitor cells, FoxG1 promotes axonal growth in the developing retina (Xuan et al., 1995; Trejo et al., 2004; Picker et al., 2009), regulates patterning of the developing forebrain (Xuan et al., 1995; Danesin et al., 2009), and is necessary for the proper formation of the inner ear (Pauley et al., 2006; Hwang et al., 2009), as well as the olfactory system (Duggan et al., 2008; Kawauchi et al., 2009a,b). Several recent studies have found that FoxG1 mutations are associated with the congenital form of Rett syndrome, a severe neurodevelopmental disorder (Jacob et al., 2009; Mencarelli et al., 2009, 2010; Philippe et al., 2010). Additionally, FoxG1 mutations have been reported to be associated with other neurodevelopmental disorders in humans, including epilepsy and microcephaly (Bahi-Buisson et al., 2010).
While being highly expressed in the fetal brain, FoxG1 is also expressed in the mammalian brain through adulthood (Shen et al., 2006; Obendorf et al., 2007). In contrast to the progress made in the understanding of its functions during nervous system development, nothing is known about what role FoxG1 plays in fully differentiated neurons. We find that FoxG1 expression in postmitotic and mature neurons is drastically reduced when these cells are induced to undergo apoptosis. Forced expression of FoxG1 completely inhibits apoptosis, whereas suppression of its expression induces cell death in otherwise healthy neurons. Based on these findings, we conclude that FoxG1 promotes survival of postmitotic neurons. We show that the survival-promoting activity of FoxG1 in neurons is mediated by the PI-3 kinase–Akt signaling pathway, and also that FoxG1 is a downstream target of IGF-1-mediated signal transduction.
Materials and Methods
Materials.
Unless indicated otherwise, all materials were purchased from Sigma-Aldrich. Tissue culture reagents, including culture medium and fetal bovine serum, were purchased from Invitrogen. Antibodies used in this paper were as follows: Flag (catalog #F1804, Sigma-Aldrich), HA (Y-11 catalog# sc-805, F-7 catalog #sc-7392, Santa Cruz Biotechnology), α-Tubulin (TU-02 catalog #sc-8035, Santa Cruz Biotechnology), GFP (B-2 catalog #sc-9996, FL catalog #sc-8334, Santa Cruz Biotechnology), Phospho-Akt Substrate (RXRXXS/T, 110B7E, catalog #9614, Cell Signaling Technology), and Akt (pan, C67E7, catalog #4691, Cell Signaling Technology). In addition to these commercially available antibodies, a rabbit polyclonal FoxG1 antibody was generated using peptides corresponding to residues 163–179 of rat FoxG1 by 21st Century Biochemicals. Primary antibodies were used at concentrations ranging from 1:250 to 1:1000 in 5% bovine serum albumin. Secondary antibodies (from Pierce) were used at concentrations of either 1:10,000 or 1:20,000. For immunocytochemistry, primary antibodies were used at a dilution of 1:200. Texas Red- or fluorescein (FITC)-tagged secondary antibodies for immunocytochemistry were purchased from Jackson ImmunoResearch Laboratories and used at a dilution of 1:85.
Plasmids.
The FoxG1-Flag expression vector was a kind gift from Dr. Stefano Stifani of McGill University (Montreal, Quebec, Canada). The N-terminal Flag tag in this construct was replaced with a C-terminal hemagglutinin (HA) tag to generate FoxG1-HA. Also, the Flag-FoxG1 construct was used as a template to generate deletion mutants lacking residues 1–36, 1–171, 275–481, and 337–481 by PCR. These deletion constructs were Flag-tagged at the C terminus and cloned into the pTarget mammalian expression vector (Promega). All the constructs were sequenced to rule out the presence of mutations and then expressed in HEK293T cells to check for protein expression (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The HA-Akt CA plasmid generated in the laboratory of Dr. M. C. Hung (University of Texas M. D. Anderson Cancer Center, Houston, TX) (Zhou et al., 2000) and the pKLO.1-TRC shRNA control plasmid generated in the laboratory of Dr. D. E. Root (Broad Institute of MIT and Harvard, Cambridge, MA) (Moffat et al., 2006) were both purchased from Addgene.
Cerebellar granule neuron culture and transfection.
Cerebellar granule neurons (CGNs) were cultured from dissociated cerebella of 7- to 8-d-old Wistar rats and plated on dishes coated with poly-l-lysine in Basal Eagle's medium with Earle's salts (BME) supplemented with 10% fetal bovine serum, 25 mm KCl, 2 mm glutamine, and 100 μg/ml gentamicin as previously described (D'Mello et al., 1997; D'Mello et al., 1993). To prevent proliferation of non-neuronal cells, 10 μm cytosine arabinofuranoside was added to the culture medium ∼17 h after plating. Cells were maintained for 4–5 d before transient transfection using the calcium phosphate method as previously described (Koulich et al., 2001; Yalcin et al., 2003). Control cultures were transfected with CMV-GFP. The day following transfection, the cells were switched to either high-K+ (HK) medium (serum-free BME medium, supplemented with 20 mm KCl) or low-K+ (LK) medium (serum-free BME medium, 5 mm KCl) as indicated in the figure legends. When used, pharmacological inhibitors were added at the time of the switch. Transfected neurons were detected by GFP fluorescence or by immunostaining using Flag antibody as described previously (Koulich et al., 2001; Yalcin et al., 2003). Neuronal viability was quantified by staining cell nuclei with 4′, 6′-diamidino-2-phenylindole hydrochloride (DAPI) as previously described (Koulich et al., 2001; Yalcin et al., 2003). Cells with condensed or fragmented nuclei were scored as apoptotic.
For experiments involving pharmacological inhibitors, CGN cultures were switched to HK or LK medium 12 h after transfection. The inhibitors were added to the medium at the time of the switch at the following concentrations: PD98059 at 40 μm, U0126 at 10 μm, wortmannin at 200 nm, LY294002 at 20 μm, Akt inhibitor X at 5 μm, KN62 at 50 μm, trichostatin A (TSA) at 1 μm, IC261 at 10 μm, and D4476 at 10 μm. All pharmacological inhibitors were purchased from EMD Biosciences. The effectiveness of these inhibitors to inhibit their targets at the concentration at which they were used was confirmed by studying their effects on viability or on phosphorylation of target proteins (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Cortical neuron culture and transfection.
Embryonic cortical cultures were prepared from embryonic day 17 rat pups as described previously (Morrison et al., 2006; Chen et al., 2008). The cortical cultures were grown in DMEM + Glutamax-I medium (Invitrogen) supplemented with 10% FBS in the presence of penicillin-streptomycin (Invitrogen). About 26 h after plating, the cultures were treated with 15 μg/ml 5-fluoro-2′-deoxyuridine and 35 μg/ml uridine to prevent mitotic cell proliferation. Transfections were performed on 2-d-old cultures using the calcium phosphate method as described above for cerebellar granule neurons. When used, homocysteic acid (Sigma-Aldrich) was added at 1 mm concentration 24 h after transfection. Viability of successfully transfected cells was quantified following immunocytochemistry and DAPI staining (1:10,000 in PBS).
Site-directed mutagenesis.
The site-directed mutagenesis was accomplished by using QuikChange II Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer's instructions. All the constructs were sequenced to check against mutations and expressed in HEK293T cells to check for protein expression (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). The following primers were used to create site-directed mutants: FoxG1 T271→A (top strand): 5′-gccgctccaccgcgtctcgggcc-3′; FoxG1 T271→A (bottom strand): 5′-ggcccgagacgcggtggagcggc-3′; FoxG1 N219→A (top strand): 5′-gcagggctggcaggcctccatccgccac-3′; FoxG1 N219→A (bottom strand): 5′-gtggcggatggaggcctgccagccctgc-3′; FoxG1 T271→D (top strand): 5′-cggcgccgctccaccgattctcgggccaagctg-3′; FoxG1 T271→D (bottom strand): 5′-cagcttggcccgagaatcggtggagcggcgccg-3′; FoxG1 T271→E (top strand): 5′-gcgccgctccaccgagtctcgggccaag-3′; FoxG1 T271→E (bottom strand): 5′-cttggcccgagactcggtggagcggcgc-3′.
Cell lines and transfection.
Mouse neuroblastoma HT22 cells were a kind gift from Dr. R. Ratan (Burke Medical Research Institute, White Plains, NY). Human embryonic kidney cells (HEK293T) were purchased from ATCC. Both cell lines were grown in DMEM plus 10% FBS and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
RT-PCR.
RNAs were extracted from cultured neurons or HT22 cell lines by using Trizol (Invitrogen) according to the manufacturer's instructions. Three micrograms of RNA were used to prepare cDNA using the Superscript First Strand Synthesis System for RT-PCR (Invitrogen) according to the manufacturer's instructions. PCR was performed with GoTaq Green Master Mix (Promega). The primers used for PCR amplification were as follows: FoxG1 forward: 5′-ACAAGAAGAACGGCAAGTACGAGA-3′; FoxG1 reverse: 5′-GTTGGCGGTGGAGAAGGAGT-3′; actin forward: 5′-AGGACTCCTATGTGGGTGACGA-3′; actin reverse: 5′-CGTTGCCAATAGTGATGACCTG-3′; cJun forward: 5′-GATGGAAACGACCTTCTACG-3′; cJun reverse: 5′-GTTGAAGTTGCTGAGGTTGG-3′; 18S forward: 5′-GCTACCACATCCAAGGAAGG-3′; 18S reverse: 5′-GGCCTCGAAAGAGTCCTGTA-3′.
shRNA-mediated suppression.
The pLKO.1-TRC control vector (indicated in the text and figure as pKLO.1), which contains a non-hairpin 18 bp insert, served as the transfection control. Four FoxG1 shRNAs were used in this study: TRCN0000081744, TRCN0000081745, TRCN0000081746, and TRCN0000081747 (purchased from Sigma-Aldrich), denoted in this study as FG1744, FG1745, FG1746, and FG1747, respectively. Ability to suppress expression was performed using HT22 cells. Briefly, HT22 cells were transfected with pKLO.1 or with FoxG1 shRNA's plasmids. RNA was harvested 24 h later and subjected to RT-PCR analysis as described above. To examine effect on neuronal viability, CGNs were cotransfected on day 4 with either pKLO.1 control plasmid or one of the FoxG1 shRNA-expressing plasmids along with CMV-GFP in a ratio of 6.5: 1. Two days later, the neurons were treated with HK/LK followed by immunocytochemistry after 24 h. Viability of the GFP-expressing cells was quantified by DAPI staining. HT22 cells were transfected with pKLO.1 or with FoxG1 shRNA's lentiviral plasmids using Lipofectamine 2000 as described above for 72 h. RNA was harvested using Trizol followed by cDNA preparation and PCR analysis as described above.
Western blot analysis.
Cell lysate was prepared by using 1× cell lysis buffer (Cell Signaling Technology). Equivalent amounts of protein from each sample (50–100 μg) were mixed with 6× SDS sample buffer [375 mm Tris-HCl (pH 6.8 at 25°C), 12% SDS, 60% glycerol, 300 mm dithiothreitol (DTT), 0.012% bromophenol blue]. This was followed by heating at 95°C for 5 min, subjected to SDS-PAGE, and transferred electrophoretically onto polyvinylidene difluoride membrane. After staining with Ponceau S to verify uniformity of protein loads/transfer, the membranes were blocked in 5% nonfat dry milk in PBS. Incubation with primary antibodies was performed overnight at 4°C or for 1 h at room temperature followed by secondary antibodies for 1 h at room temperature. Immunoreactivity was developed by enhanced chemiluminescence and visualized by autoradiography.
Immunoprecipitation.
Cell lysates were harvested with 300 μl of 1× cell lysis buffer. Lysates were centrifuged at 10,000 × g for 10 min at 4°C. Twenty-five microliters of the protein lysate were mixed with 6× SDS, boiled at 95°C for 5 min, and subjected to Western blot analysis to check for protein overexpression (in case of transfected cell lines) and to check for equal protein usage for immunoprecipitation. Thirty microliters of Protein A/G Plus-Agarose beads (Santa Cruz Biotechnology) were preincubated with 1 μg of pull-down antibody in 300 μl of cell lysis buffer for 1 h at 4°C with constant rocking. Beads bound with pull-down antibody were precipitated by centrifugation at 3300 × g for 2 min, and the supernatant was discarded. Two hundred fifty microliters of protein lysate were added to the beads and incubated overnight at 4°C with constant rocking. Immunoprecipitate was collected by centrifugation at 3300 × g for 2 min and washed thrice with 500 μl of cell lysis buffer. Thirty microliters of 3× SDS were added to the immunoprecipitate, boiled for 5 min at 95°C, and then subjected to Western blot analysis. The probing antibody was chosen so that it was raised in a different species than the antibody used for pull-down.
In vitro kinase assay.
HEK293T cells were transfected with Flag-FoxG1 plasmid and kept overnight. Cell lysate was harvested as discussed above and subjected to immunoprecipitation with flag antibody. The immunoprecipitate was washed twice with cell lysis buffer and twice with kinase buffer (40 mm Tris-HCl, pH 7.5; 8 mm MgCl2; 50 mm β-glycerol phosphate; 1 mm DTT). The immunoprecipitate was resuspended in 10 μl of kinase buffer by gently tapping the sides of the tube. Whenever used, 10 μl of Akt inhibitor X (5 μm, diluted in kinase buffer) was preincubated with the immunoprecipitate for 15 min at 30°C in a water bath. One hundred nanograms of Active Akt 1 (catalog #A16-10G, SignalChem) were then added to the immunoprecipitate. Five microliters of 250 μm ATP kinase assay cocktail [15 μl of 10 μm ATP and 10 μl of [32P]ATP (1 mCi/100 μl, BLU002Z, PerkinElmer] was added in 575 μl of kinase buffer. The total volume of the reaction mixture was adjusted to 25 μl by adding the required amount of kinase buffer, and the reaction pot was incubated at 30°C for 30 min. The reaction was stopped by adding 25 μl of 3× SDS and boiled for 5 min at 95°C. The beads were pulled down by centrifugation at 3300 × g for 2 min, and the supernatant was run in SDS polyacrylamide gel. The gel was dried and then subjected to autoradiography.
Statistical analysis.
All the graphs in this study were generated using GraphPad Prism 5 software. Unless otherwise mentioned in the figure legends, statistical analysis was done using unpaired two-tailed t test (Student's t test), and the results are shown as mean ± SEM. p values of <0.05 were deemed statistically significant. Asterisks indicate the following p values: *p < 0.05, **p < 0.01, ***p < 0.001. Unless mentioned otherwise, all viability experiments were done in duplicate and repeated three times. For each experiment, ≥200 transfected cells were counted.
Results
Expression of FoxG1 is reduced in neurons primed to undergo apoptosis
CGNs undergo apoptosis when switched from HK medium (containing depolarizing levels of potassium) to LK medium (containing non-depolarizing concentrations of potassium) (D'Mello et al., 1993). Previous work has shown that in this paradigm, commitment to death occurs at 4–6 h after LK treatment (Galli et al., 1995; Nardi et al., 1997; Borodezt and D'Mello, 1998). As shown in Figure 1A, FoxG1 mRNA expression is substantially reduced within 6 h of the switch to LK. At the same time, the expression of c-jun mRNA, a transcription factor whose activation is involved in promoting apoptosis in most paradigms of neuronal death (Ham et al., 2000), is increased. To confirm the findings observed at the mRNA level, we performed Western blot analyses. A 58 kDa FoxG1-immunoreactive band is observed in CGN lysates (Fig. 1B), which is sharply downregulated in neurons primed to die. Ectopic expression of FoxG1 in HEK293T cells also produces a 58 kDa band (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Further analysis showed that the downregulation of FoxG1 mRNA and protein was an early event, being discernible within 1 h of LK treatment (Fig. 1C,D). By 9 h of LK treatment, FoxG1 protein levels were reduced to ∼10% of that seen in healthy neuronal cultures.
Figure 1.

Expression of FoxG1 in postmitotic neurons. A, RNA isolated from CGNs treated with HK or LK for 6 h were subjected to RT-PCR analysis of FoxG1 expression. FoxG1 mRNA expression is reduced at 6 h of LK treatment. In contrast, the expression of the proapoptotic c-Jun gene is induced in neurons primed to die by LK treatment. Actin serves as the loading control. B, Whole-cell lysates were prepared from CGNs treated with HK or LK for 6 h and subjected to Western blot analysis using a FoxG1 antibody. FoxG1 is downregulated in LK. Tubulin serves as the loading control. C, RNA isolated from CGNs treated with LK for 0, 1, 3, 6, and 9 h was subjected to RT-PCR analysis of FoxG1 expression. Actin served as loading control. Densitometric analysis indicates reduction of FoxG1 mRNA level with time. Results were obtained from three separate experiments. **p < 0.01. For statistical analysis, one-way ANOVA was performed using Bonferroni's multiple-comparison test. D, Whole-cell lysates prepared from CGNs treated with LK for 0, 1, 3, 6, and 9 h were subjected to Western blot analysis using a FoxG1 antibody. Tubulin serves as the loading control. Densitometric analysis indicated reduction of FoxG1 protein level with time. Results were obtained from three separate experiments. ***p < 0.001, **p < 0.01. For statistical analysis, one-way ANOVA was performed using Bonferroni's multiple-comparison test.
FoxG1 expression is required for neuronal survival
The sharp reduction of FoxG1 expression suggests that an elevated level of this protein might be necessary for neuronal survival. To investigate this issue, we transfected neurons with an expression plasmid encoding Flag-tagged FoxG1. Ectopically expressed FoxG1 localizes exclusively to the nucleus in both HK and LK (Fig. 2A). Forced expression of FoxG1 completely prevents death induced by LK (Fig. 2B). No effect on neuronal viability in HK was observed. To rule out the possibility that the N-terminal Flag-tag contributes to the effectiveness of the tagged FoxG1 protein to promote neuronal survival, we used a FoxG1 construct with a HA-tag at the C terminus. Expression of this FoxG1 construct also prevented LK-induced neuronal death completely (Fig. 2C). To examine whether FoxG1 could prevent apoptosis in other neuronal types, we used embryonic cortical neurons. Treatment of these cultures with HCA induces oxidative stress leading to cell death (Chen et al., 2008; Majdzadeh et al., 2008). As shown in Figure 2D, HCA-mediated neuronal death is inhibited by ectopic expression of FoxG1.
Figure 2.
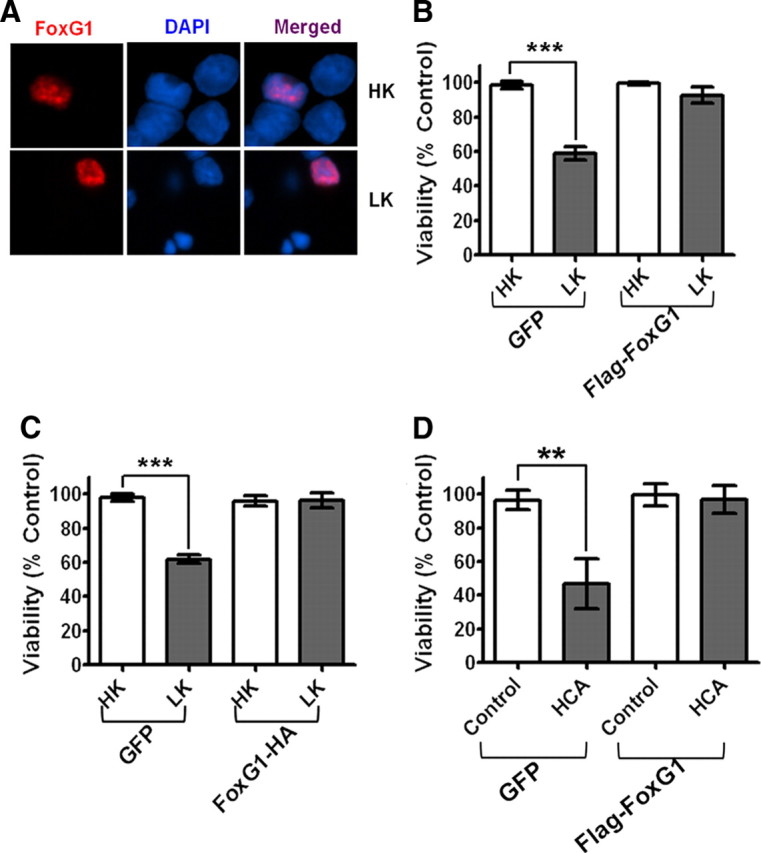
Effect of FoxG1 overexpression in postmitotic neuronal survival: CGN cultures were transfected with GFP and Flag-FoxG1 or FoxG1-HA for 12 h and then switched to HK or LK medium for 24 h followed by immunocytochemistry. The viability of transfected neurons was quantified by DAPI staining and normalized to GFP-transfected cells in HK. FoxG1 protects neurons independent of its tag. A, Localization of ectopically expressed FoxG1 in CGNs. Transfected neurons were identified by immunocytochemistry using Flag antibody. FoxG1 localizes exclusively in the nucleus in HK- and LK-treated neurons. B, FoxG1 protects CGNs from LK-induced apoptosis. C, CGN cultures transfected with either GFP or FoxG1-HA. FoxG1-HA protects neurons from LK-induced apoptosis. Results were obtained from three separate experiments done in duplicate. ***p < 0.001 as compared to control (GFP-transfected neurons in HK). D, Two-day-old cultures of primary cortical neurons transfected with GFP or Flag FoxG1 were treated with either medium having no additives (NA) or medium containing 1 mm HCA for 20 h. Viability of transfected neurons was quantified after immunocytochemistry using GFP or Flag antibody and normalized to the viability of control cultures (GFP-transfected cells receiving no additives). **p < 0.01 as compared to GFP-transfected cortical neurons with NA.
Although ectopic expression of FoxG1 protects CGNs from death, it was not clear whether its expression is normally necessary for neuronal survival. To address this issue, we suppressed endogenous FoxG1 expression in healthy cultures of neurons (maintained in HK medium) by expressing shRNA against FoxG1 mRNA. Two shRNA constructs FG1745 and FG1746 were initially tested for their effectiveness in the HT22 neuroblastoma cell line. While FG1745 reduced FoxG1 mRNA expression cells by ∼ 30%, transfection of FG1746 lowered expression by ∼70% (Fig. 3A, upper panel). The level of actin mRNA or 18S rRNA was not affected by either of these shRNAs. When expressed in CGNs, FG1746 reduced survival by >60% in HK medium and increased the extent of cell death in LK (Fig. 3B). In comparison, FG1745 had an insignificant effect on cell viability. These results were confirmed with two other FoxG1 shRNA constructs, FG1744 and FG1747. Expression studies in HT22 cells showed that FG1744 reduced FoxG1 mRNA expression substantially, whereas FG1747 had only a modest effect (Fig. 3A, lower panel). As shown in Figure 3, B and C, expressing FG1744 reduced neuronal survival in HK, while FG1747 did not have a statistically significant effect.
Figure 3.

Suppression of FoxG1 expression promotes neuronal death. CGN cultures were cotransfected with plasmids expressing GFP and either pKLO.1 or one of the four different FoxG1 shRNAs (FG1744, FG1745, FG1746, and FG1747). The ability of the shRNAs to suppress FoxG1 expression was evaluated in HT22 cells, and their effect on neuronal survival was analyzed in CGNs. A, RNA was extracted from HT22 cells 72 h after transfection with shRNA constructs and subjected to RT-PCR analysis using primers against FoxG1, 18S ribosomal RNA, and actin. Results for the four FoxG1 shRNAs [FG1745 and FG1746 (upper panel) and FG1747 and FG1744 (lower panel)] along with the control (pKLO.1) are shown. Densitometric analysis of the FoxG1 band from three separate experiments was performed using Kodak 1D software, and the results are shown in the graph. ***p < 0.001, *p < 0.05. For statistical analysis, one-way ANOVA was performed using Bonferroni's multiple-comparison test. B, CGNs were cotransfected with plasmids expressing GFP and pKLO.1 or one of the four FoxG1 shRNAs and treated with HK or LK medium. Results come from three separate experiments performed in duplicate. ***p < 0.001 as compared to neurons cotransfected with GFP and pKLO.1 in HK. **p < 0.01 as compared to neurons cotransfected with GFP and pKLO.1 in LK. C, Appearance of neurons transfected with pKLO.1 plasmid or FoxG1 shRNA (FG1747, FG1746, FG1744). Transfected neurons were identified by GFP immunocytochemistry and nuclei stained with DAPI.
The survival-promoting effect of FoxG1 is mediated by the PI-3 kinase–Akt signaling pathway
It has been reported that the ability of FoxG1 to maintain neural progenitor cells in a proliferative state depends on its subcellular localization, which is regulated by casein kinase-I (CKI) and Akt (Regad et al., 2007). Phosphorylation of FoxG1 by CKI induces the import of FoxG1 into the nucleus, promoting the differentiation of these progenitor cells into neurons (Regad et al., 2007). In contrast, the phosphorylation of FoxG1 by Akt at Thr271 causes its export to the cytoplasm (Regad et al., 2007). Pharmacological inhibition of Akt or its upstream activator, PI-3 kinase traps FoxG1 in the nucleus, keeping the cortical progenitors in a proliferative state, thus blocking their differentiation (Regad et al., 2007). To examine whether the survival-promoting activity of FoxG1 is regulated by mechanisms similar to its neural progenitor-specifying activity, we treated CGNs with IC261 and D4476, two widely used and highly selective CK1 inhibitors that were also used in the study by Regad et al. (2007). In contrast to what was observed by Regad et al. (2007), however, the inhibition of CKI had no effect on the nuclear localization of FoxG1 in postmitotic neurons (Fig. 4A). Additionally, neither IC261 nor D4476 had any effect on the ability of FoxG1 to promote CGN survival (Fig. 4B). Both compounds inhibited HK-mediated neuronal survival, however (supplemental Table 1, available at www.jneurosci.org as supplemental material). As observed in CGNs, the CK1 inhibitors did not alter the nuclear localization pattern of FoxG1 in HT22 cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 4.
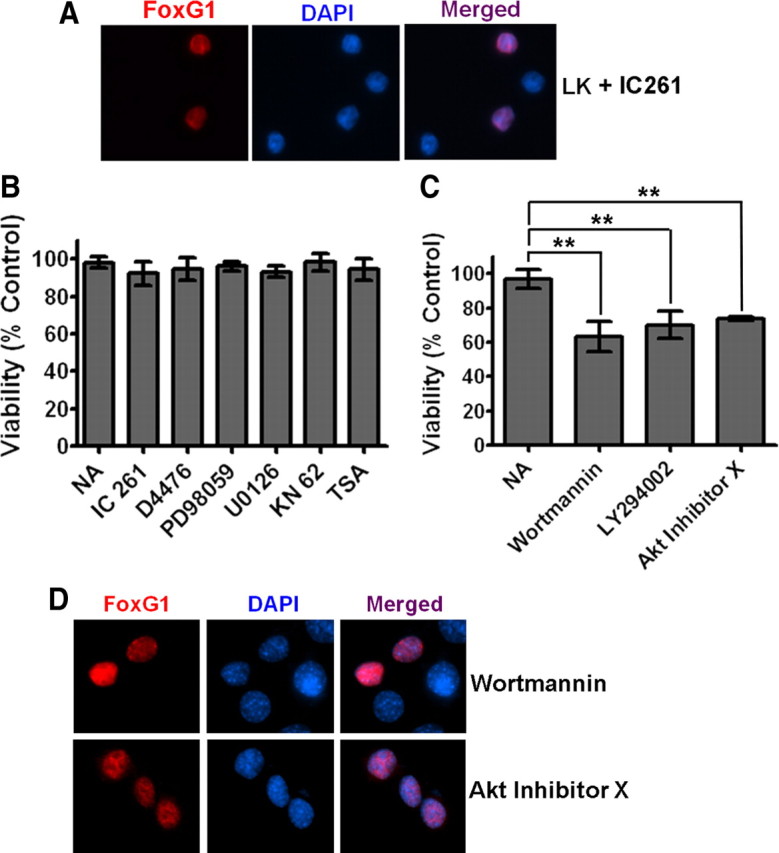
The PI-3 kinase/Akt pathway mediates the anti-apoptotic effect of FoxG1. A, Ectopically expressed FoxG1 was detected immunocytochemically with a Flag antibody and nuclei were stained with DAPI. The figure shows nuclear localization of FoxG1 in neurons treated with LK in presence of IC261. CKI inhibition does not alter the nuclear localization of FoxG1. B, CGN cultures transfected with Flag-FoxG1 were switched to LK medium having no additives (NA), LK medium containing 10 μm IC261, 10 μm D4476, 40 μm PD98059, 10 μm U0126, 50 μm KN62, or 1 μm trichostatin A. The survival status of transfected neurons was quantified 24 h later by Flag immunocytochemistry and DAPI nuclear staining. Inhibitors against CKI, CaMK, HDACs, and the RAf-MEK-ERK pathway did not have any effect on FoxG1-mediated survival. Results were obtained from three independent experiments performed in duplicate. Control experiments (supplemental Table 1, available at www.jneurosci.org as supplemental material) confirmed that all the pharmacological agents used in this analysis inhibit the activities of their targets. C, CGN cultures transfected with Flag-FoxG1 were switched to LK medium with NA or LK medium containing 200 nm wortmannin, 20 μm LY294002, and 5 μm Akt inhibitor X. Inhibitors of the PI-3 kinase/Akt pathway blocked FoxG1 from helping CGNs to survive in LK conditions. **p < 0.01 as compared to FoxG1-transfected CGNs treated with LK. Results were obtained from three independent experiments performed in duplicates. D, HT22 cells were transfected with Flag-FoxG1. Six hours after transfection, the medium was changed to fresh medium supplemented with 200 nm wortmannin or 5 μm Akt inhibitor X for 12 h. Immunocytochemistry was performed with Flag antibody and nuclei were stained with DAPI. Blocking PI3K/Akt does not block nuclear translocation of FoxG1.
We examined whether the inhibition of the PI-3 kinase–Akt pathway has any effect on the anti-apoptotic activity using LY294002 and wortmannin, two potent and specific inhibitors of PI-3 kinase, and an Akt inhibitor (Thimmaiah et al., 2005) [10-(4′-(N-diethylamino)butyl)-2-chlorophenoxazine] designated as Akt inhibitor-X. All three inhibitors inhibited the ability of FoxG1 to prevent LK-mediated apoptosis in CGNs (Fig. 4C). As seen in Figure 4D, these inhibitors had no effect on the nuclear localization of FoxG1.
To confirm that phosphorylation of FoxG1 by Akt was necessary for its survival-promoting effect, we mutated Thr271 in FoxG1 to a nonphosphorylatable alanine. The ectopic expression of Flag-FoxG1T271A fails to protect against LK-induced death (Fig. 5C). Furthermore, this mutant construct promoted death of the neurons even in HK (Fig. 5C). Like the wild-type protein, Flag-FoxG1T271A localized to the nucleus, arguing against the possibility that its failure to protect was due to a non-nuclear localization. (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Nuclear localization of Flag-FoxG1T271A was also confirmed in HT22 cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To confirm further that Akt-mediated phosphorylation of FoxG1 at Thr271 was necessary for survival promotion, we generated two mutants in which the threonine residue was substituted with aspartic acid or glutamic acid (Flag-FoxG1T271D and Flag-FoxG1T271E, respectively). Not unexpectedly, these “phosphomimetic” forms of FoxG1 promoted survival in HK (Fig. 6). In contrast to wild-type FoxG1, however, treatment with wortmannin or LY294002 had no effect on the ability of these mutant FoxG1 proteins to promote survival (Fig. 6).
Figure 5.

Sites or domains responsible for FoxG1 mediated neuronal survival. A, Schematic diagram of the wild-type FoxG1 protein from Xenopus [which was used by Regad et al. (2007)] and the mouse FoxG1 protein used in this study. Position of relevant amino acids as well as the conserved sequence region (CS) and the DBD is indicated. Also shown are the various FoxG1 deletion constructs used in this study. B, Immunocytochemistry result of CGN in HK transfected with Flag-FoxG1N219A and Flag-FoxG1T271A. C, CGNs were transfected with plasmids expressing wild-type FoxG1-Flag, the Akt phosphorylation site mutant Flag-FoxG1T271A, and Flag-FoxG1N219A, a mutant construct in which an aspartate residue essential for DNA binding is substituted with alanine. The neurons were then switched to HK or LK medium for 24 h, after which viability of transfected neurons was quantified. Both Flag-FoxG1N219A and Flag-FoxG1T271A failed to protect neurons from LK-induced death. Results are from three different experiments performed in duplicate. **p < 0.01 as compared to FoxG1-transfected CGNs in HK. *p < 0.05 as compared to FoxG1-transfected neurons in LK. D, Quantification of neuronal survival with the various FoxG1-Flag deletion constructs. Deletion of N-terminus regions destroys the ability of FoxG1 to prevent neuronal death in LK. Moreover, these constructs inhibit survival in HK, possibly through a dominant-negative mechanism. In contrast, C-terminus deletion constructs had no effect on HK-mediated survival. Results come from three different experiments performed in duplicate. **p < 0.01 as compared to FoxG1-transfected CGNs in HK or LK as indicated in the graph by bars.
Figure 6.

Phosphorylation at Thr271 by Akt is necessary for survival-promoting activity of FoxG1. CGNs were transfected with plasmids expressing two separate phosphomimetic forms of FoxG1, Flag-FoxG1T271D, and Flag-FoxG1T271E. The cultures were then switched to HK or LK media or LK media supplemented with 200 nm wortmannin (Wort) or 20 μm LY294002 (LY). Viability of transfected neurons was quantified after immunocytochemistry and normalized to the viability of GFP-transfected neurons in HK. Survival-promoting effect of Flag-FoxG1T271D and Flag-FoxG1T271E in LK medium cannot be inhibited by inhibition of the PI-3 kinase–Akt pathway. Columns compared are indicated with bars. ***p < 0.001.
Another well characterized anti-apoptotic pathway is the Raf-MEK-ERK signaling pathway (Hetman and Gozdz, 2004). We investigated the contribution of this pathway in the survival-promoting effect of FoxG1 using U0126 and PD98059, two potent inhibitors of MEK. Neither of these inhibitors reduced the survival-promoting effect of FoxG1, indicating that the Raf-MEK-ERK signaling pathway was not involved (Fig. 4B). Control experiments (supplemental Table 1, available at www.jneurosci.org as supplemental material) as well as previously performed analyses (Chin et al., 2004; Chen et al., 2008) confirmed that ERK phosphorylation in CGNs is completely inhibited by these compounds when used at the doses shown in Figure 4B.
HK treatment of CGNs activates calcium-calmodulin kinase (CaMK), and pharmacological inhibition of CaMK blocks the survival-promoting effect of HK (Boutillier et al., 1999; Linseman et al., 2003; Morrison et al., 2006). Likewise, inhibition of histone deacetylases using pharmacological agents such as TSA blocks the ability of HK to promote CGN survival (Salminen et al., 1998; Boutillier et al., 2002; Morrison et al., 2006). As shown in Figure 4B, treatment with KN62 and TSA had no effect on the survival-promoting activity of FoxG1, suggesting that the involvement of CaMK and HDACs was not required.
DNA-binding activity and the conserved N-terminal region of FoxG1 are necessary for its anti-apoptotic effect
The transcriptional repressor activity of FoxG1 requires DNA binding to the high-affinity FoxG1-binding site (Li et al., 1995). However, FoxG1 can also function independently of DNA binding (Hanashima et al., 2002). Structural analyses of FoxG1 has identified two residues within its DNA-binding domain, N219 and H223, that make critical base contacts with double-stranded DNA containing the FoxG1-binding site (Clark et al., 1993; Dou et al., 2000; Hanashima et al., 2002). Mutation of these two residues abolishes the ability of FoxG1 to bind DNA and repress transcription (Hanashima et al., 2002). To examine whether the anti-apoptotic activity of FoxG1 requires its DNA-binding activity, we generated a point mutant in which residue N219 was changed to an alanine. Expression of Flag-FoxG1N219A was incapable of preventing neuronal apoptosis, suggesting that DNA binding is necessary for the survival-promoting activity of FoxG1 (Fig. 5C). As observed with the Akt phosphorylation-deficient mutant, expression of FoxG1N219A also reduced neuronal survival in HK. Both point-mutant forms of FoxG1 localized to the nucleus (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), indicating that the inability to promote survival was not due to defective localization.
To identify other regions necessary for the anti-apoptotic activity, we generated a construct that lacked the first 171 residues of FoxG1 (Fig. 5A). This construct, FoxG1172–481-Flag, which contains the DNA-binding domain (DBD) and the C-terminus transcriptional-regulatory region, could not prevent LK-induced death and, as observed with the two point-mutant constructs, inhibited survival in HK (Fig. 5D). Within the first 171 aa is a highly conserved 36 aa stretch at the N-terminal end of FoxG1 (designated as CS in Fig. 5A). The amino acid sequence of the CS region is identical in mammals, and the first 33 residues are identical in vertebrates ranging from fish and frogs to humans (Bredenkamp et al., 2007). To examine the significance of the CS region, we generated and tested a construct, FoxG137–481-Flag, lacking this domain. FoxG137–481-Flag fails to prevent LK-induced death (Fig. 5D). Moreover, and as observed with FoxG1172–481-Flag, the expression of FoxG137–481-Flag inhibits survival in HK (Fig. 5D). We also evaluated the significance of the C-terminal end of FoxG1 to survival promotion by expressing a construct that lacked the last 206 and 145 residues of the protein (Fig. 5A), designated as FoxG11–275-Flag and FoxG11–336-Flag. In contrast to the N-terminus deletion constructs, both C-terminus deletion constructs, FoxG11–275-Flag and FoxG11–336-Flag, were fully capable of preventing LK-induced death (Fig. 5D).
Together, our results indicate that the first 275 residues of FoxG1 are sufficient for its survival-promoting effect. Within this half of the protein, the CS region along with an intact Akt phosphorylation site and DNA-binding residues are essential for this function of FoxG1.
FoxG1 is a downstream effector of IGF-1 signaling in neurons
IGF-1 plays an important role in the development and function of the CNS (Bondy, 1991; Bondy and Lee, 1993; D'Ercole et al., 1996). Within the cerebellar cortex, IGF-1 is produced and released by Purkinje neurons and is believed to be the primary trophic factor for the survival of CGNs in vivo. The loss of Purkinje cells leads to a subsequent degeneration of CGNs because of the unavailability of IGF-1 (Bartlett et al., 1991; Zhang et al., 1997; Selimi et al., 2000). Recapitulating the situation in vivo, the death of cultured CGNs induced by LK-treatment can be fully prevented by addition of IGF-1 to the culture medium (D'Mello et al., 1997; D'Mello et al., 1993; Brunet et al., 1999). It is well established that IGF-1 acts by activating the PI-3 kinase–Akt signaling pathway (D'Mello et al., 1997; Dudek et al., 1997). Since the survival-promoting effect of FoxG1 is also dependent on PI-3 kinase–Akt signaling, we tested the possibility that FoxG1 participates in the signaling pathway activated by IGF-1. As shown in Figure 7A, neuroprotective signaling by IGF-1 is blocked by expression of mutant forms of FoxG1 (FoxG1172–481-Flag, FoxG137–481-Flag, FoxG1N219A-Flag, and FoxG1T271A-Flag), but not by expressing wild-type FoxG1 or GFP in LK media supplemented with IGF-1. In support of the conclusion that FoxG1 is a downstream mediator of IGF-1-mediated neuronal survival is the finding that treatment of CGNs with IGF-1 inhibits the downregulation of FoxG1 expression that occurs during LK-induced apoptosis (Fig. 7B).
Figure 7.

FoxG1 is a downstream effector of IGF-1 signaling. A, CGNs were transfected with plasmids as indicated in the graph and then switched to LK media supplemented with 25 ng/ml IGF-1. Viability of transfected neurons was quantified after immunocytochemistry using GFP/Flag antibody and normalized to the viability of GFP-transfected neurons in LK with IGF-1. IGF-1 was able to rescue GFP-transfected neurons in LK. IGF-1 was not able to rescue CGNs transfected with FoxG1 172–481 Flag, FoxG137–481-Flag, Flag-FoxG1N219A, and Flag-FoxG1T271A. ***p < 0.001 as compared to FoxG1-transfected CGNs in LK media supplemented with IGF1. Results were obtained from four independent experiments performed in duplicate. B, Six-day-old CGN cultures were treated with HK, LK, LK with IGF-1 (25 ng/ml), and LK with IGF-1 plus PI-3 kinase/Akt pathway inhibitors (wortmannin, 200 nm and Akt inhibitor X, 5 μm). RNA or protein lysates were prepared 6 h after treatment and analyzed for FoxG1 expression. Upper panel shows results of RT-PCR analysis of FoxG1 and actin mRNA. Lower panel shows results of Western blot analysis performed using FoxG1 and tubulin antibodies. IGF-1 inhibits the LK-induced reduction of FoxG1 expression. However, inhibitors of PI-3 kinase–Akt signaling inhibit the ability of IGF1 to maintain elevated FoxG1 expression.
Given that the activation of Akt occurs soon after exposure to IGF-1 (within 10 min in cultured CGNs) (D'Mello et al., 1997; Dudek et al., 1997), it was likely that the stimulatory effect of IGF-1 on FoxG1 expression is subsequent to Akt activation. To examine this issue, we treated CGNs with IGF-1 in the presence of wortmannin or Akt-X. As shown in Figure 7B, these inhibitors of PI-3 kinase and Akt inhibited the ability of IGF-1 to maintain elevated FoxG1 expression in LK-treated neurons. Together with the finding that mutation of the Akt phosphorylation site in FoxG1 is essential for its survival-promoting effect, these results indicate that Akt lies upstream of FoxG1 in the signaling pathway activated by IGF-1.
FoxG1 is directly phosphorylated by Akt
A previous study has suggested that FoxG1 is phosphorylated by Akt at Thr271 (Regad et al., 2007). To confirm this finding, we immunoprecipitated FoxG1-Flag and Flag-FoxG1T271A from HEK293T cells that were treated with IGF-1 and performed Western blot analysis of the pulled-down protein using a phospho-Akt substrate antibody. As shown in Figure 8A, the wild-type FoxG1 is phosphorylated by Akt even in the absence of IGF-1, perhaps by growth factors in serum, including IGF-1. The level of phosphorylation is enhanced in cultures treated with IGF-1. In contrast to wild-type FoxG1, phosphorylation of FoxG1T271A-Flag is barely detectable either basally or after addition of IGF-1 (Fig. 8A), confirming that phosphorylation of FoxG1 by Akt occurs mainly at T271. To confirm that Akt phosphorylates FoxG1, we performed an in vitro kinase assay in which FoxG1 immunoprecipitated from HEK293T cells was incubated with active Akt protein. Again, a basal level of FoxG1 phosphorylation is observed even in the absence of exogenous Akt, but addition of Akt enhances the level of FoxG1 phosphorylation (Fig. 8B). Pharmacological inhibition of Akt abolishes FoxG1 phosphorylation to a level even lower than basal levels. The lowering of phosphorylation to a level that was lower than that observed in the absence of exogenously added Akt raised the possibility that Akt was being pulled down along with FoxG1 and was contributing to phosphorylation in the in vitro kinase assay. To examine whether FoxG1 interacts with Akt, it was overexpressed in HEK293T cells and then immunoprecipitated. Western blot analysis of the immunoprecipitate with an Akt antibody does reveal interaction between the two proteins (Fig. 8C). In contrast to wild-type FoxG1, interaction between Akt and FoxG1T271A was almost undetectable (Fig. 8C).
Figure 8.
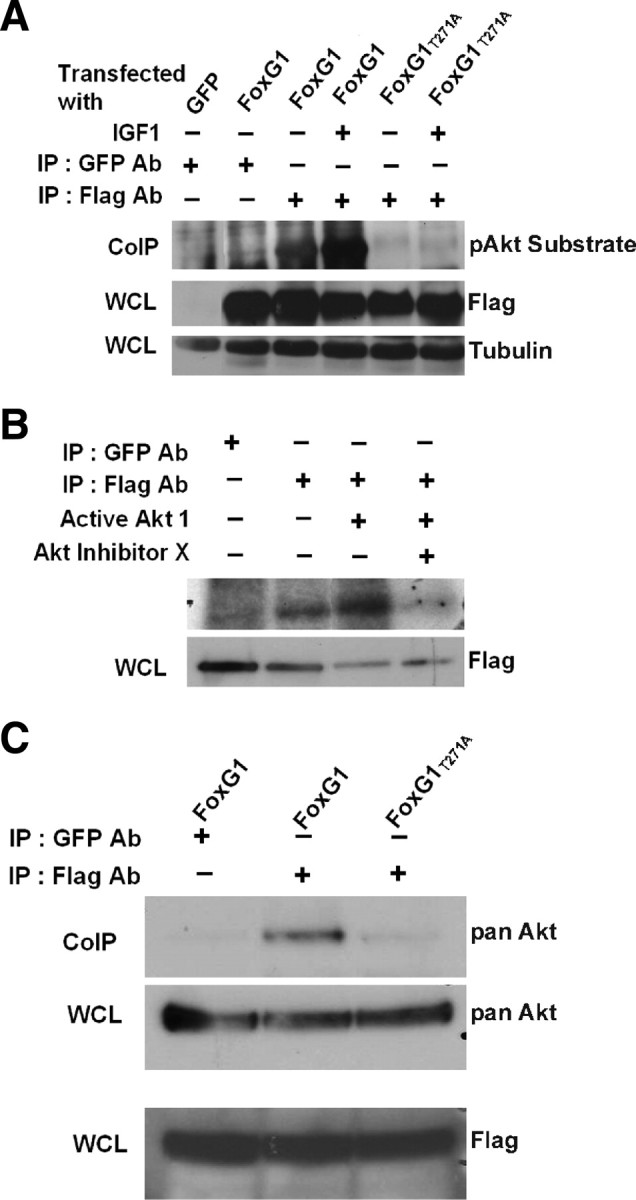
FoxG1 is a substrate of Akt. A, HEK293T cells were transfected with Flag-FoxG1 or Flag-FoxG1T271A and then treated for 15 min with IGF-1. Then FoxG1 was immunoprecipitated from the cultures using a Flag antibody, and the immunoprecipitate was subjected to Western blotting using a phospho-Akt substrate antibody. Lysates from GFP and Flag-FoxG1-transfected cells immunoprecipitated with a GFP antibody served as negative controls. Preimmunoprecipitation aliquots of the whole-cell lysates (WCLs) were subjected to Western blotting using Flag and Tubulin antibody to verify that similar amounts of lysates were used in each lane. Wild-type FoxG1 is phosphorylated in serum-containing medium not supplemented with IGF-1. The extent of phosphorylation is higher after exposure to IGF-1. FoxG1T271A is not phosphorylated even in the presence of IGF-1. B, Lysates from HEK293T cells transfected with Flag-FoxG1 were immunoprecipitated with antibodies to Flag or GFP (negative control) as indicated in the figure. The immunoprecipitate was used in an in vitro kinase assay performed in the presence or absence of active Akt enzyme. In one sample, a chemical inhibitor of Akt, Akt-X, was added to the kinase reaction mixture 15 min before addition of active Akt. Preimmunoprecipitation WCL was analyzed by Western blotting using a Flag antibody to verify that similar levels of FoxG1 were produced by transfection. C, HEK293T cells were transfected with Flag-FoxG1 or Flag-FoxG1T271A and pulled down with Flag or GFP antibody as indicated in the figure. Western blot analysis was performed with an Akt antibody. Flag-FoxG1 interacts with endogenous Akt, whereas Flag-FoxG1T271A fails to do so. Preimmunoprecipitation WCLs were analyzed by Western blotting using a Flag antibody to verify that similar levels of wild-type and mutant FoxG1 were produced by transfection.
Discussion
FoxG1 is best known for its essential role in the development of the embryonic telencephalon. It is also involved in the generation of neurons in the hippocampal formation and in other neurodevelopmental processes such as forebrain patterning and the development of the retina and the inner ear. Although FoxG1 is expressed in the postnatal and adult brain, its role in postmitotic neurons is not known. We show that FoxG1 is involved in maintaining the survival of mature neurons. Indeed, FoxG1 expression is reduced in neurons induced to undergo apoptosis. Reestablishment of elevated FoxG1 protects neurons from death, whereas shRNA-mediated suppression of FoxG1 expression promotes apoptosis in otherwise healthy neuronal cultures. Although evidence for a survival-promoting action of FoxG1 in mature neurons in vivo is lacking, analysis of FoxG1+/− mice has suggested such a role in proliferating neuroepithelial cells and developing neuronal populations (Ahlgren et al., 2003; Shen et al., 2006). Indeed, we have found that forced expression of FoxG1 protects HT22 neuroblastoma cells from HCA-induced death (data not shown), confirming that the survival-promoting action of FoxG1 extends beyond mature neurons. In humans, mutations in the FoxG1 locus have been found to be associated with Rett syndrome as well as other neurodevelopmental disorders such as microcephaly and epilepsy. It is tempting to speculate that reduced expression of FoxG1 or mutations affecting its survival-promoting function could contribute to the pathogenesis of neurodegenerative diseases also.
Using deletion mutants of FoxG1, we find that a construct containing only the first 275 residues of the 481 aa protein is as capable of maintaining survival as the full-length form of FoxG1. Within this 275-residue region, the N-terminal 36 aa CS domain plays an essential role. Interestingly, deletion of this CS domain not only eliminates survival-promoting activity, but also has a dominant-negative effect on endogenous FoxG1 as evidenced by its ability to induce death in otherwise healthy cultures. It is likely that this domain associates with other proteins that participate in promoting neuronal survival. Given the high degree of conservation of the CS region through vertebrate evolution (Obendorf et al., 2007), it is likely that such associations as well as the neuroprotective function of FoxG1 are evolutionarily conserved. Several FoxG1-interacting proteins have been identified, including the Groucho-related proteins TLE1 and Grg6, members of the Smad family (Smad1–4), and the H3K4 demethylase/transcriptional repressor, PLU-1 (Dou et al., 2000; Rodriguez et al., 2001; Yao et al., 2001; Tan et al., 2003; Marçal et al., 2005). However, all these proteins bind to regions within the C-terminal half of FoxG1. In the case of TLE1, a second interaction site within FoxG1 has been proposed to lie in the N-terminus region, although its precise location has not been mapped (Yao et al., 2001; Buscarlet et al., 2008). FoxG1 also interacts with the androgen receptor at a site(s) between residues 175–481 (Obendorf et al., 2007). Thus, if the CS region acts via protein-protein interactions, the FoxG1-interacting protein necessary for the survival-promoting action of FoxG1 remains to be identified.
In addition to the CS domain, mutation of either Asp219 or Thr271 destroys the survival-promoting activity and has a dominant-negative effect. In the developing telencephalon, FoxG1 inhibits neuronal differentiation through a DNA binding-dependent mechanism, whereas its regulation of cell proliferation is independent of DNA binding (Hanashima et al., 2002). Asp219 is required for DNA binding of FoxG1, suggesting that the promotion of neuronal survival is mediated by binding to and repressing the activity of specific promoters. Thr271 is phosphorylated by Akt. It was previously reported that in cultured cortical progenitors FoxG1 resides in the nucleus, but translocates to the cytoplasm when these cells are induced to differentiate by treatment with FGF2. Translocation is blocked by pharmacological inhibition of Akt (Obendorf et al., 2007; Regad et al., 2007). A similar Akt-mediated nucleus to cytoplasm translocation of ectopically expressed FoxG1 occurs in HeLa cells (Regad et al., 2007). Further analysis by Regad et al. (2007) revealed that the translocation out of the nucleus was dependent on the phosphorylation of Thr271 by Akt. Mutation of this residue to alanine blocks translocation out of the nucleus in HeLa cells (Regad et al., 2007). We find that in postmitotic cortical neurons and CGNs, as well as the HT22 cells and HEK293T cell lines, FoxG1 is exclusively in the nucleus. At least in neurons, the nuclear localization of FoxG1 is maintained even when constitutively active Akt is overexpressed (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Inhibition of Akt with pharmacological inhibitors or through expression of a dominant-negative form of the enzyme also does not alter the nuclear localization pattern of FoxG1. Based on these observations, we conclude that Akt is not generally involved in regulating the subcellular localization of FoxG1, and that FoxG1 is predominantly a nuclear protein. Akt does, however, play a critical role in regulating the expression and survival-promoting activity of FoxG1. Stimulation of endogenous Akt activity by IGF-1 prevents the apoptosis-associated downregulation of IGF-1 expression, whereas the inhibition of Akt by pharmacological agents or by expression of a dominant-negative construct blocks FoxG1-mediated survival of neurons. Although it is possible that other residues within FoxG1 are phosphorylated by Akt, our results suggest that phosphorylation at Thr271 plays an essential role in the promotion of survival. Moreover, while our results suggest that FoxG1 is a direct target of Akt, it is conceivable that IGF-1 and the PI-3 kinase–Akt pathway act indirectly by modulating the activity of other molecules that are required for FoxG1-mediated promotion of neuronal survival. The finding that inhibition of the PI-3 kinase–Akt pathway blocks the ability of FoxG1 to promote neuronal survival argues against the possibility that FoxG1 and IGF-1/Akt act through independent mechanisms. Further studies are necessary to determine how Akt phosphorylation regulates the neuroprotective activity of FoxG1. One possibility is that phosphorylation of FoxG1 might stimulate its transcriptional activity through modulation of its interaction with other proteins and/or its DNA-binding activity.
The C-terminal half of the FoxG1 protein is not required for it survival-promoting activity in neurons. This is surprising given that this part of the protein is highly conserved at the amino acid level between vertebrates ranging from frogs and chicken to humans (>90% sequence identity). It is likely that the C-terminal half is involved in other important functions distinct from the regulation of neuronal survival. For example, the well established role of FoxG1 in the promotion of neuronal progenitor proliferation or the inhibition of their differentiation into neurons could require this region. FoxG1 has also been shown to repress TGFβ signaling by interacting with Smad proteins. This activity is independent of DNA binding but requires the C-terminus region (Dou et al., 2000; Rodriguez et al., 2001).
A significant finding in this report is that FoxG1 is required for the survival-promoting effect of IGF-1. In addition to CGNs, IGF-1 maintains the survival of a variety of other neuronal types in vitro and in vivo, and delivery of this neurotrophic factor has been considered as a therapeutic approach for several neurodegenerative pathologies (Torres-Aleman, 2000; Trejo et al., 2004; Cohen and Dillin, 2008). The survival-promoting effect of IGF-1 in CGNs is mediated through IRS-1 and the activation of the PI-3 kinase–Akt pathway (D'Mello et al., 1997; Brunet et al., 1999; Dudek et al., 1997). Once activated, Akt phosphorylates and inactivates specific apoptosis-promoting molecules, one of which is FoxO1. FoxO1 phosphorylation by Akt leads to its sequestration in the cytoplasm by 14-3-3 (Brunet et al., 1999). In neurons deprived of trophic factor support, FoxO is not phosphorylated, permitting its translocation to the nucleus, an event that is critical for the induction of neuronal death (Brunet et al., 1999). We present evidence that FoxG1 is another member of the Forkhead family regulated by IGF-1. In contrast to the death-promoting activity of FoxO1, however, FoxG1 plays a survival-promoting role. Besides FoxO1, the phosphorylation of several other proteins (including CREB, Bad, IKK, and GSK3β) by Akt contributes to the potent survival-promoting effect of this kinase in neurons and non-neuronal cells (D'Mello and Chin, 2005). Our studies identify FoxG1 as a new target of Akt-mediated promotion of neuronal survival.
In conclusion, we have identified a new and important function for FoxG1 in mature neurons—the promotion of survival. The deficiency of this function could contribute to some of the neurodevelopmental abnormalities observed in FoxG1 knock-out and haplodeficient mice. Indeed, it has been suggested that FoxG1+/− mice have an increased level of cell death in developing neuronal populations of the CNS (Shen et al., 2006; Obendorf et al., 2007). We report that survival-promoting action of FoxG1 is critically dependent on Akt activity, and that that FoxG1 is a downstream effector of IGF-1 signaling. IGF-1 maintains an elevated level of FoxG1 expression. It is possible that a reduction of FoxG1 also contributes to neuronal death in neurodegenerative disorders. We show that ectopic expression of FoxG1 can prevent neuronal death induced by oxidative stress. Increased oxidative stress is believed to contribute to cell death in a variety of neurodegenerative disorders. Increasing the expression or activity of FoxG1 could represent a novel therapeutic approach to reduce neuronal loss in these pathologies.
Footnotes
This work was supported by grants from the National Institute of Neurological Diseases and Stroke (NS40408 and NS058462) to S.R.D. We thank Nazanin Majdzadeh and Matthew Jamison for their preliminary observations on the role of FoxG1 in the promotion of neuronal survival. We thank Dr. Stefano Stifani (Center for Neuronal Survival, Montreal Neurological Institute, McGill University, Montreal, Quebec, Canada) for the Flag-FoxG1 construct.
References
- Ahlgren S, Vogt P, Bronner-Fraser M. Excess FoxG1 causes overgrowth of the neural tube. J Neurobiol. 2003;57:337–349. doi: 10.1002/neu.10287. [DOI] [PubMed] [Google Scholar]
- Bahi-Buisson N, Nectoux J, Girard B, Van Esch H, De Ravel T, Boddaert N, Plouin P, Rio M, Fichou Y, Chelly J, Bienvenu T. Revisiting the phenotype associated with FOXG1 mutations: two novel cases of congenital Rett variant. Neurogenetics. 2010;11:241–249. doi: 10.1007/s10048-009-0220-2. [DOI] [PubMed] [Google Scholar]
- Bartlett WP, Li XS, Williams M, Benkovic S. Localization of insulin-like growth factor-1 mRNA in murine central nervous system during postnatal development. Dev Biol. 1991;147:239–250. doi: 10.1016/s0012-1606(05)80021-1. [DOI] [PubMed] [Google Scholar]
- Bondy CA. Transient IGF-I gene expression during the maturation of functionally related central projection neurons. J Neurosci. 1991;11:3442–3455. doi: 10.1523/JNEUROSCI.11-11-03442.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy CA, Lee WH. Patterns of insulin-like growth factor and IGF receptor gene expression in the brain. functional implications. Ann N Y Acad Sci. 1993;692:33–43. doi: 10.1111/j.1749-6632.1993.tb26203.x. [DOI] [PubMed] [Google Scholar]
- Borodezt K, D'Mello SR. Decreased expression of the metabotropic glutamate receptor-4 gene is associated with neuronal apoptosis. J Neurosci Res. 1998;53:531–541. doi: 10.1002/(SICI)1097-4547(19980901)53:5<531::AID-JNR3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Bourguignon C, Li J, Papalopulu N. XBF-1, a winged helix transcription factor with dual activity, has a role in positioning neurogenesis in Xenopus competent ectoderm. Development. 1998;125:4889–4900. doi: 10.1242/dev.125.24.4889. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Kienlen-Campard P, Loeffler JP. Depolarization regulates cyclin D1 degradation and neuronal apoptosis: a hypothesis about the role of the ubiquitin/proteasome signalling pathway. Eur J Neurosci. 1999;11:441–448. doi: 10.1046/j.1460-9568.1999.00451.x. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Trinh E, Loeffler JP. Constitutive repression of E2F1 transcriptional activity through HDAC proteins is essential for neuronal survival. Ann N Y Acad Sci. 2002;973:438–442. doi: 10.1111/j.1749-6632.2002.tb04679.x. [DOI] [PubMed] [Google Scholar]
- Bredenkamp N, Seoighe C, Illing N. Comparative evolutionary analysis of the FoxG1 transcription factor from diverse vertebrates identifies conserved recognition sites for microRNA regulation. Dev Genes Evol. 2007;217:227–233. doi: 10.1007/s00427-006-0128-x. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Buscarlet M, Perin A, Laing A, Brickman JM, Stifani S. Inhibition of cortical neuron differentiation by Groucho/TLE1 requires interaction with WRPW, but not Eh1, repressor peptides. J Biol Chem. 2008;283:24881–24888. doi: 10.1074/jbc.M800722200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HM, Wang L, D'Mello SR. A chemical compound commonly used to inhibit PKR, {8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one}, protects neurons by inhibiting cyclin-dependent kinase. Eur J Neurosci. 2008;28:2003–2016. doi: 10.1111/j.1460-9568.2008.06491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin PC, Liu L, Morrison BE, Siddiq A, Ratan RR, Bottiglieri T, D'Mello SR. The c-raf inhibitor GW5074 provides neuroprotection in vitro and in an animal model of neurodegeneration through a MEK-ERK and akt-independent mechanism. J Neurochem. 2004;90:595–608. doi: 10.1111/j.1471-4159.2004.02530.x. [DOI] [PubMed] [Google Scholar]
- Clark KL, Halay ED, Lai E, Burley SK. Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature. 1993;364:412–420. doi: 10.1038/364412a0. [DOI] [PubMed] [Google Scholar]
- Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008;9:759–767. doi: 10.1038/nrn2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danesin C, Peres JN, Johansson M, Snowden V, Cording A, Papalopulu N, Houart C. Integration of telencephalic wnt and hedgehog signaling center activities by Foxg1. Dev Cell. 2009;16:576–587. doi: 10.1016/j.devcel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- D'Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–255. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Chin PC. Treating neurodegenerative conditions through the understanding of neuronal apoptosis. Curr Drug Targets CNS Neurol Disord. 2005;4:3–23. doi: 10.2174/1568007053005118. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou C, Lee J, Liu B, Liu F, Massague J, Xuan S, Lai E. BF-1 interferes with transforming growth factor beta signaling by associating with smad partners. Mol Cell Biol. 2000;20:6201–6211. doi: 10.1128/mcb.20.17.6201-6211.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Duggan CD, DeMaria S, Baudhuin A, Stafford D, Ngai J. Foxg1 is required for development of the vertebrate olfactory system. J Neurosci. 2008;28:5229–5239. doi: 10.1523/JNEUROSCI.1134-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham J, Eilers A, Whitfield J, Neame SJ, Shah B. c-jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol. 2000;60:1015–1021. doi: 10.1016/s0006-2952(00)00372-5. [DOI] [PubMed] [Google Scholar]
- Hanashima C, Shen L, Li SC, Lai E. Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J Neurosci. 2002;22:6526–6536. doi: 10.1523/JNEUROSCI.22-15-06526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanashima C, Li SC, Shen L, Lai E, Fishell G. Foxg1 suppresses early cortical cell fate. Science. 2004;303:56–59. doi: 10.1126/science.1090674. [DOI] [PubMed] [Google Scholar]
- Hetman M, Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur J Biochem. 2004;271:2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x. [DOI] [PubMed] [Google Scholar]
- Hwang CH, Simeone A, Lai E, Wu DK. Foxg1 is required for proper separation and formation of sensory cristae during inner ear development. Dev Dyn. 2009;238:2725–2734. doi: 10.1002/dvdy.22111. [DOI] [PubMed] [Google Scholar]
- Jacob FD, Ramaswamy V, Andersen J, Bolduc FV. Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet. 2009;17:1577–1581. doi: 10.1038/ejhg.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Santos R, Kim J, Hollenbeck PL, Murray RC, Calof AL. The role of foxg1 in the development of neural stem cells of the olfactory epithelium. Ann N Y Acad Sci. 2009a;1170:21–27. doi: 10.1111/j.1749-6632.2009.04372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi S, Kim J, Santos R, Wu HH, Lander AD, Calof AL. Foxg1 promotes olfactory neurogenesis by antagonizing Gdf11. Development. 2009b;136:1453–1464. doi: 10.1242/dev.034967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulich E, Nguyen T, Johnson K, Giardina C, D'mello S. NF-kappaB is involved in the survival of cerebellar granule neurons: association of IkappaBbeta [correction of ikappabeta] phosphorylation with cell survival. J Neurochem. 2001;76:1188–1198. doi: 10.1046/j.1471-4159.2001.00134.x. [DOI] [PubMed] [Google Scholar]
- Li J, Chang HW, Lai E, Parker EJ, Vogt PK. The oncogene qin codes for a transcriptional repressor. Cancer Res. 1995;55:5540–5544. [PubMed] [Google Scholar]
- Linseman DA, Bartley CM, Le SS, Laessig TA, Bouchard RJ, Meintzer MK, Li M, Heidenreich KA. Inactivation of the myocyte enhancer factor-2 repressor histone deacetylase-5 by endogenous ca(2+)//calmodulin-dependent kinase II promotes depolarization-mediated cerebellar granule neuron survival. J Biol Chem. 2003;278:41472–41481. doi: 10.1074/jbc.M307245200. [DOI] [PubMed] [Google Scholar]
- Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D'Mello SR. HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev Neurobiol. 2008;68:1076–1092. doi: 10.1002/dneu.20637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçal N, Patel H, Dong Z, Belanger-Jasmin S, Hoffman B, Helgason CD, Dang J, Stifani S. Antagonistic effects of Grg6 and Groucho/TLE on the transcription repression activity of brain factor 1/FoxG1 and cortical neuron differentiation. Mol Cell Biol. 2005;25:10916–10929. doi: 10.1128/MCB.25.24.10916-10929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B, Morrison H, Price DJ, Mason JO. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev Biol. 2005;283:113–127. doi: 10.1016/j.ydbio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Mencarelli MA, Kleefstra T, Katzaki E, Papa FT, Cohen M, Pfundt R, Ariani F, Meloni I, Mari F, Renieri A. 14q12 microdeletion syndrome and congenital variant of Rett syndrome. Eur J Med Genet. 2009;52:148–152. doi: 10.1016/j.ejmg.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Mencarelli MA, Spanhol-Rosseto A, Artuso R, Rondinella D, De Filippis R, Bahi-Buisson N, Nectoux J, Rubinsztajn R, Bienvenu T, Moncla A, Chabrol B, Villard L, Krumina Z, Armstrong J, Roche A, Pineda M, Gak E, Mari F, Ariani F, Renieri A. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet. 2010;47:49–53. doi: 10.1136/jmg.2009.067884. [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D'Mello SR. Neuroprotection by histone deacetylase-related protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DB, Wiese S, Burfeind P, Schmundt D, Mattei MG, Schulz-Schaeffer W, Thies U. Human brain factor 1, a new member of the fork head gene family. Genomics. 1994;21:551–557. doi: 10.1006/geno.1994.1313. [DOI] [PubMed] [Google Scholar]
- Nardi N, Avidan G, Daily D, Zilkha-Falb R, Barzilai A. Biochemical and temporal analysis of events associated with apoptosis induced by lowering the extracellular potassium concentration in mouse cerebellar granule neurons. J Neurochem. 1997;68:750–759. doi: 10.1046/j.1471-4159.1997.68020750.x. [DOI] [PubMed] [Google Scholar]
- Obendorf M, Meyer R, Henning K, Mitev YA, Schröder J, Patchev VK, Wolf SS. FoxG1, a member of the forkhead family, is a corepressor of the androgen receptor. J Steroid Biochem Mol Biol. 2007;104:195–207. doi: 10.1016/j.jsbmb.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Pauley S, Lai E, Fritzsch B. Foxg1 is required for morphogenesis and histogenesis of the mammalian inner ear. Dev Dyn. 2006;235:2470–2482. doi: 10.1002/dvdy.20839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe C, Amsallem D, Francannet C, Lambert L, Saunier A, Verneau F, Jonveaux P. Phenotypic variability in Rett syndrome associated with FOXG1 mutations in females. J Med Genet. 2010;47:59–65. doi: 10.1136/jmg.2009.067355. [DOI] [PubMed] [Google Scholar]
- Picker A, Cavodeassi F, Machate A, Bernauer S, Hans S, Abe G, Kawakami K, Wilson SW, Brand M. Dynamic coupling of pattern formation and morphogenesis in the developing vertebrate retina. PLoS Biol. 2009;7:e1000214. doi: 10.1371/journal.pbio.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regad T, Roth M, Bredenkamp N, Illing N, Papalopulu N. The neural progenitor-specifying activity of FoxG1 is antagonistically regulated by CKI and FGF. Nat Cell Biol. 2007;9:531–540. doi: 10.1038/ncb1573. [DOI] [PubMed] [Google Scholar]
- Rodriguez C, Huang LJ, Son JK, McKee A, Xiao Z, Lodish HF. Functional cloning of the proto-oncogene brain factor-1 (BF-1) as a smad-binding antagonist of transforming growth factor-beta signaling. J Biol Chem. 2001;276:30224–30230. doi: 10.1074/jbc.M102759200. [DOI] [PubMed] [Google Scholar]
- Salminen A, Tapiola T, Korhonen P, Suuronen T. Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res Mol Brain Res. 1998;61:203–206. doi: 10.1016/s0169-328x(98)00210-1. [DOI] [PubMed] [Google Scholar]
- Selimi F, Doughty M, Delhaye-Bouchaud N, Mariani J. Target-related and intrinsic neuronal death in lurcher mutant mice are both mediated by caspase-3 activation. J Neurosci. 2000;20:992–1000. doi: 10.1523/JNEUROSCI.20-03-00992.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Nam HS, Song P, Moore H, Anderson SA. FoxG1 haploinsufficiency results in impaired neurogenesis in the postnatal hippocampus and contextual memory deficits. Hippocampus. 2006;16:875–890. doi: 10.1002/hipo.20218. [DOI] [PubMed] [Google Scholar]
- Tan K, Shaw AL, Madsen B, Jensen K, Taylor-Papadimitriou J, Freemont PS. Human PLU-1 has transcriptional repression properties and interacts with the developmental transcription factors BF-1 and PAX9. J Biol Chem. 2003;278:20507–20513. doi: 10.1074/jbc.M301994200. [DOI] [PubMed] [Google Scholar]
- Tao W, Lai E. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron. 1992;8:957–966. doi: 10.1016/0896-6273(92)90210-5. [DOI] [PubMed] [Google Scholar]
- Thimmaiah KN, Easton JB, Germain GS, Morton CL, Kamath S, Buolamwini JK, Houghton PJ. Identification of N10-substituted phenoxazines as potent and specific inhibitors of akt signaling. J Biol Chem. 2005;280:31924–31935. doi: 10.1074/jbc.M507057200. [DOI] [PubMed] [Google Scholar]
- Torres-Aleman I. Serum growth factors and neuroprotective surveillance: focus on IGF-1. Mol Neurobiol. 2000;21:153–160. doi: 10.1385/mn:21:3:153. [DOI] [PubMed] [Google Scholar]
- Trejo JL, Carro E, Garcia-Galloway E, Torres-Aleman I. Role of insulin-like growth factor I signaling in neurodegenerative diseases. J Mol Med. 2004;82:156–162. doi: 10.1007/s00109-003-0499-7. [DOI] [PubMed] [Google Scholar]
- Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–1152. doi: 10.1016/0896-6273(95)90262-7. [DOI] [PubMed] [Google Scholar]
- Yalcin A, Koulich E, Mohamed S, Liu L, D'Mello SR. Apoptosis in cerebellar granule neurons is associated with reduced interaction between CREB-binding protein and NF-kappaB. J Neurochem. 2003;84:397–408. doi: 10.1046/j.1471-4159.2003.01540.x. [DOI] [PubMed] [Google Scholar]
- Yao J, Lai E, Stifani S. The winged-helix protein brain factor 1 interacts with groucho and hes proteins to repress transcription. Mol Cell Biol. 2001;21:1962–1972. doi: 10.1128/MCB.21.6.1962-1972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ghetti B, Lee WH. Decreased IGF-I gene expression during the apoptosis of Purkinje cells in pcd mice. Brain Res Dev Brain Res. 1997;98:164–176. doi: 10.1016/s0165-3806(96)00168-x. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Hu MC, Miller SA, Yu Z, Xia W, Lin SY, Hung MC. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J Biol Chem. 2000;275:8027–8031. doi: 10.1074/jbc.275.11.8027. [DOI] [PubMed] [Google Scholar]