

Two cell entry mechanisms cooperate in peptide-mediated intracellular delivery.
Keywords: cell-penetrating peptide, CendR pathway, neuropilin-1, intracellular delivery, vascular permeability, synchronized ultrastructural imaging
Abstract
Cell-penetrating peptides (CPPs) have been widely used to deliver nanomaterials and other types of macromolecules into mammalian cells for therapeutic and diagnostic use. Cationic CPPs that bind to heparan sulfate (HS) proteoglycans on the cell surface induce potent endocytosis; however, the role of other surface receptors in this process is unclear. We describe the convergence of an HS-dependent pathway with the C-end rule (CendR) mechanism that enables peptide ligation with neuropilin-1 (NRP1), a cell surface receptor known to be involved in angiogenesis and vascular permeability. NRP1 binds peptides carrying a positive residue at the carboxyl terminus, a feature that is compatible with cationic CPPs, either intact or after proteolytic processing. We used CPP and CendR peptides, as well as HS- and NRP1-binding motifs from semaphorins, to explore the commonalities and differences of the HS and NRP1 pathways. We show that the CendR-NRP1 interaction determines the ability of CPPs to induce vascular permeability. We also show at the ultrastructural level, using a novel cell entry synchronization method, that both the HS and NRP1 pathways can initiate a macropinocytosis-like process and visualize these CPP-cargo complexes going through various endosomal compartments. Our results provide new insights into how CPPs exploit multiple surface receptor pathways for intracellular delivery.
INTRODUCTION
Cellular membranes pose a formidable barrier to the delivery of hydrophilic drugs, proteins, and nanoscopic carriers. Cationic cell-penetrating peptides (CPPs) have been widely exploited to overcome this barrier and deliver various types of cargo into mammalian cells (1). A large body of research has elucidated certain aspects of how cationic CPPs deliver nanosized cargo (20 nm to more than 150 nm) into cells; receptor-mediated endocytosis is thought to be the prevailing mechanism (2, 3). A notable example is the transactivator of transcription (TAT) peptide isolated from human immunodeficiency virus. It is well appreciated that TAT uses heparan sulfate (HS) proteoglycans as cell surface receptors, and that TAT binding to them initiates macropinocytosis, a type of endocytotic process (4–7). On the other hand, neuropilin-1 (NRP1) was recently suggested to play a role in the cell entry of TAT-bound cargo (8).
NRP1 is a transmembrane protein and a co-receptor of various ligands, such as semaphorins and vascular endothelial growth factors (VEGFs), and thus is involved in a variety of biological functions including cell migration, angiogenesis, and vascular permeability. NRP1 has a short cytoplasmic domain containing a PSD-95/DLG/ZO-1 (PDZ) binding motif (SEA-COOH), which interacts with the cytoplasmic PDZ domain–containing protein GIPC1/synectin (9). NRP1 regulates internalization and trafficking through endosomal transport pathways of a number of cell surface receptors (for example, plexins, VEGF receptor 2, and integrins) and their receptor-ligand complexes (10). Because it is common for growth factors to have HS-binding domains that increase affinity to extracellular matrix and cell surface receptors (11, 12), a conundrum arises in distinguishing the roles of NRP1 and HS in the internalization of these ligands. Distinguishing between the NRP1 and HS roles is further complicated by the fact that cell surface receptor proteins, including NRP1, frequently carry HS chains (13).
We recently discovered a class of peptides that can bind to NRP1 through a motif of arginine (R) or lysine (K) at their C terminus (C-end rule, or CendR), contained preferably within an R/KXXR/K motif (14). The strict requirement for CendR-NRP1 interaction is that the C terminus of the peptide, besides being an R or K, ends with the carboxyl group (-COOH) (14). This motif binds to a shallow pocket in NRP1 b1b2 domain (15). The CendR motif has been observed in NRP1-bound natural ligands (semaphorins and VEGF) as well as viruses (14, 16). Conversely, short CendR peptides acquire some properties of these ligands, such as the induction of vascular permeability (14). Although the precise mechanism is not completely clear, NRP1 engagement by CendR ligands induces their rapid endocytosis, and this process resembles macropinocytosis at the ultrastructural level (17). The most common CPP used in intracellular delivery applications, TAT, often terminates with an R (18, 19). Yet, the involvement of the CendR-NRP1 interaction remains undefined. In previous studies, a synthetic TAT was often chosen arbitrarily to be synthetically blocked by amidation or be followed by another motif or cargo at the C terminus. Such peptides would only meet the CendR requirement if deamidated or proteolytically cleaved after one of the arginine residues. Tumor-penetrating CendR peptides contain a cryptic CendR motif, which is activated by proteolysis after the peptide has homed to a primary receptor in a tumor (20, 21).
Here, we examine the relative roles of the CendR and HS pathways in TAT cell entry and vascular permeability. We also developed a method to synchronize the cell entry of CPP-nanoparticles and examined the transport route of CPP-nanoparticles from the cell surface to intracellular organelles at the ultrastructural level. These studies reveal multiple mechanisms through which cationic CPPs transport cargo into cells.
RESULTS
We first investigated whether cationic CPPs use their CendR motif for cell entry. The minimal sequence of TAT, YGRKKRRQRRR-COOH (18), was used as the prototype of cationic CPPs. A prototypic CendR peptide, RPARPAR (14), was used to exclusively trace the CendR-NRP1 interaction. We coated these peptides onto silver-based nanoparticles (AgNPs) as a model nanomaterial. AgNPs greatly amplify the intensity of fluorescence dyes attached to them (22). Furthermore, we used a cell-impermeable etching solution to dissolve nanoparticles that remain outside of cells, unprotected by a cellular membrane (22). The etching was carried out on live cells to avoid the fixation-induced artifacts reported to be a potential problem in TAT cell entry studies (23). Variants of TAT were synthesized to render the CendR motif cryptic while only minimally changing the structure of the peptide. Table 1 lists the peptides used in this study.
Table 1. Abbreviations and sequences (or chemical composition) of the peptides used in this study.
| Peptide | Sequence |
| TAT-OH | YGRKKRRQRRR-COOH |
| TAT-NH2 | YGRKKRRQRRR-CONH2 |
| d-TAT | YGRKKRRQRRR-CONH2 (d–amino acid) |
| RPARPAR | RPARPAR-COOH |
| Sema-OH | GNKKGRNRR-COOH |
| Sema-NH2 | GNKKGRNRR-CONH2 |
| AA-Sema-OH | GNAAGRNRR-COOH |
Cells expressing both NRP1 and HS proteoglycans robustly internalized AgNP-TAT-OH, and neither blocking NRP1 binding with an antibody nor blocking HS binding with soluble HS caused a significant reduction in the internalization of AgNPs (Fig. 1A). However, combining NRP1 blockade and HS competition completely abolished the internalization (Fig. 1A). To investigate the role of the CendR-NRP1 interaction in this process, we disabled the CendR motif of TAT-OH by synthetically placing an amide group at the C terminus (TAT-NH2) or by synthesizing the peptide using d–amino acids (d-TAT). The internalization of AgNPs coated with either TAT-NH2 or d-TAT was only sensitive to soluble HS competition, not to NRP1 blockade (Fig. 1A and fig. S1A). TAT-OH was resistant to HS competition at concentrations at least 100-fold higher than those needed for TAT-NH2 inhibition (Fig. 1B). This result suggests that the active CendR motif greatly reduces the HS dependence of TAT-OH delivery of nanomaterials into cells. As expected, the internalization of AgNP-RPARPAR was insensitive to HS competition at all concentrations tested but was completely inhibited by NRP1 blockade (Fig. 1, A and B). At higher concentrations of NRP1 blocking antibody, AgNP-TAT-NH2 and AgNP-TAT-OH were more resistant to NRP1 blockade than was AgNP-RPARPAR (Fig. 1C). Additionally, we knocked down NRP1 expression with small interfering RNA (siRNA) (fig. S1B), which had little effect on cellular morphology (fig. S1C). In addition to abolishing the cellular uptake of AgNP-RPARPAR, knocking down NRP1 caused a much smaller reduction of AgNP-TAT-OH and even slightly increased AgNP-TAT-NH2 uptake (Fig. 1D). Binding assays showed that AgNP-TAT-OH interacts with both purified NRP1 protein and HS, whereas RPARPAR only binds to NRP1, and AgNP-TAT-NH2 and AgNP-d-TAT only bind to HS (Fig. 1, E and F).
Fig. 1. Utilization of NRP1- and HS-dependent pathways by TAT variants.
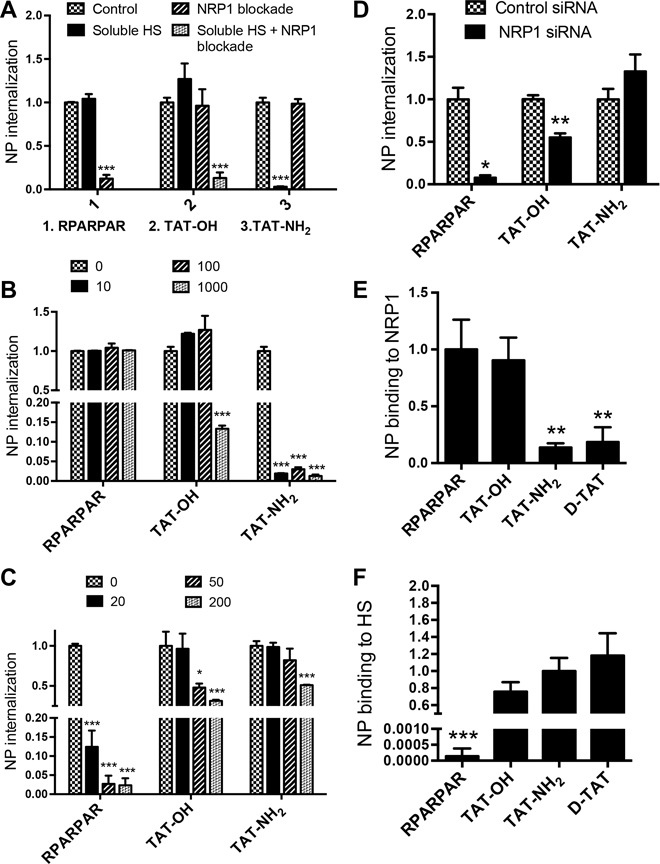
(A) PPC1 cells were incubated with NRP1 blocking antibody or soluble HS (100 μg/ml) before the addition of AgNPs coated with the indicated peptides to measure NP internalization. The quantity of internalized AgNPs as fluorescence intensity per cell was normalized to that of control (no NRP1 blockade or soluble HS) and plotted on the y axis. (B and C) PPC1 cells were incubated at different concentrations of soluble HS (0, 10, 100, and 1000 μg/ml; B) or NRP1 blocking antibody (0, 20, 50, and 200 μg/ml; C) before the addition of AgNPs coated with the indicated peptides. The quantity of internalized AgNPs as fluorescence intensity per cell was normalized to that of control [no soluble HS (B) or NRP1 blocking antibody (C)] and plotted on the y axis. (D) PPC1 cells were treated with control or NRP1 siRNA before the addition of AgNPs coated with the indicated peptides for internalization. The quantity of internalized AgNPs as fluorescence intensity per cell was normalized to that of control (control siRNA) and plotted on the y axis. (E and F) The binding of AgNPs coated with the indicated peptides to immobilized NRP1 (E) or HS (F) was quantified as described in Materials and Methods. The fluorescence intensity of bound AgNPs was normalized to the average value of the control peptide [AgNP-RPARPAR (E) or AgNP-TAT-NH2 (F)] and plotted on the y axis. All experiments were independently carried out at least three times. The error bars indicate the SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t test (compared to the control group of the indicated peptide).
To examine the possible effect of HS on the CendR peptide–NRP1 interaction, we used a mutant CHO (Chinese hamster ovary) cell line, CHO pgs745. These cells express no glycosaminoglycans, including HS (5), and they also lack NRP1 (fig. S2A). None of our test peptides promoted any entry of AgNPs into these cells, reinforcing the notion that TAT-OH requires either NRP1 or HS chains to deliver nanomaterials into cells (fig. S2, A and B). Forced expression of NRP1 rendered the pgs745 cells capable of taking up AgNPs coated with TAT-OH and RPARPAR (Fig. 2, A and B). In contrast, AgNPs coated with the non-CendR TAT variants (TAT-NH2 and d-TAT) failed to be internalized by these cells despite the NRP1 expression (Fig. 2, A and B). The internalization of AgNP-TAT-OH and AgNP-RPARPAR into NRP1-expressing pgs745 cells was abolished by NRP1 blockade (fig. S2, C and D). Together, our results suggest that (i) TAT-OH, which is equipped with an active CendR motif, uses both NRP1 and HS interactions to deliver NPs to cells; (ii) CendR/NRP1- and HS-driven pathways function independently because the cellular uptake through one pathway remains unchanged when the other is silenced; (iii) we can trace these two pathways separately using peptides designed to engage one pathway or the other.
Fig. 2. NRP1 dependence of NP internalization into HS-deficient cells.
(A) CHO pgs745 cells transfected with NRP1 expression vector were incubated with AgNPs coated with the indicated peptides (red) in regular culture medium. Extracellular NPs were dissolved by etching, and live cells were stained for surface NRP1 (green) and nuclei (blue). Representative immunofluorescence images are shown. Scale bars, 10 μm. (B) The Ag intensity per cell in NRP1-positive cells was quantified using ImageJ for samples from (A). The values were normalized to that of Ag-RPARPAR (y axis). Error bars, SEM. **P < 0.01 and ***P < 0.001, Student’s t test. At least three independent experiments were carried out for each condition.
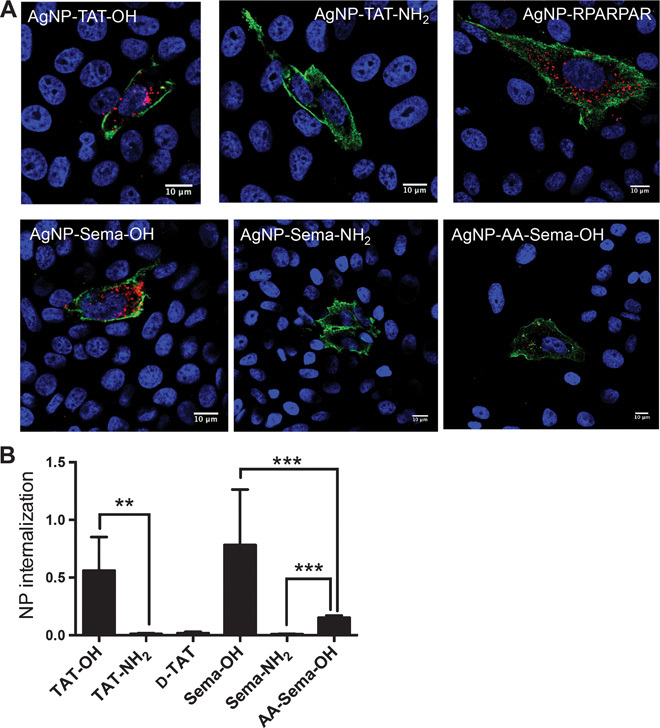
To explore whether this dual-entry mode may be exploited by natural ligands, we synthesized a peptide from the C terminus of furin-processed semaphorin-3A (Sema) with an exposed CendR motif (Table 1) (24, 25). Similar to AgNP-TAT-OH, AgNP-Sema-OH bound to both purified NRP1 and HS (Fig. 3, A and B), and its cell entry was resistant to either NRP1 blockade or HS competition alone (Fig. 3C). NPs coated with a CendR-inactive variant, Sema-NH2, only bound to HS and were sensitive to HS competition similarly to TAT-NH2 (Fig. 3, A to C). Noting that the KKGR internal stretch resembles a known HS-binding motif, BBXB, where B is a basic residue, we tested a mutant Sema peptide, in which we substituted the two lysines with alanines (AA-Sema-OH) (Table 1). This change abolished HS binding but preserved the NRP1 interaction (Fig. 3, A to C). AgNP-AA-Sema-OH displayed a much reduced cell entry activity, and the addition of free HS to the culture media greatly increased its internalization (Fig. 3D). Finally, Sema variants failed to be internalized by CHO pgs745 cells (fig. S2A). AgNP-Sema-OH and, to a lesser extent, AgNP-AA-Sema-OH entered the NRP1-transfected CHO pgs745 cells, whereas AgNP-Sema-NH2 failed to do so (Fig. 2, A and B).
Fig. 3. Utilization of NRP1 and HS pathways by Sema C-terminal peptide variants.
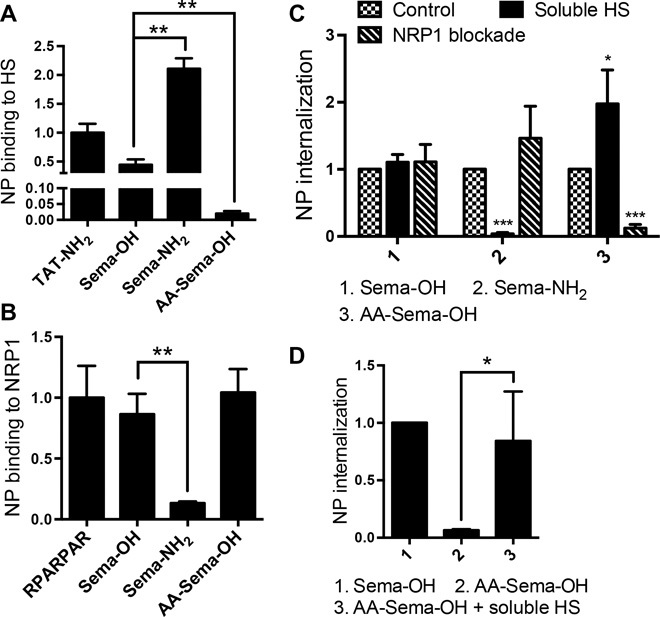
(A and B) The binding of AgNPs coated with the indicated peptides to immobilized NRP1 (A) or HS (B) was quantified as described in Materials and Methods. The fluorescence intensity of bound AgNPs was normalized to the average value of AgNP-RPARPAR (A) or AgNP-TAT-NH2 (B) and plotted on the y axis. (C) Internalization of AgNPs coated with the indicated peptides into PPC1 cells preincubated with NRP1 blocking antibody or soluble HS (100 μg/ml). After etching to dissolve extracellular NPs, the fluorescence intensity of the internalized NPs per cell was normalized to that of control (no NRP1 blockade, no soluble HS) and plotted on the y axis. (D) PPC1 cells were incubated with AgNPs coated with the indicated peptides in the absence or presence of soluble HS (100 μg/ml). After etching, AgNP internalization per cell was quantified, normalized to that of AgNP-Sema-OH, and plotted on the y axis. At least three independent experiments were carried out for each condition. Error bars, SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t test [compared to the control group of the indicated peptide in (C)].
VEGF and semaphorins, as well as CendR peptides, are known to induce vascular permeability through the CendR motif (14, 26). This property is particularly useful in macromolecule drugs and nanoparticles because it overcomes the transport barrier presented by blood vessel walls (1, 21, 27, 28). TAT protein and TAT peptides have been reported to induce vascular permeability (29), but the contribution of CendR and HS-binding motifs to this activity remains to be addressed. We found that TAT-OH, but not TAT-NH2, induces vascular permeability in a mouse skin model (Fig. 4A). This result confirms the dependence of the permeability-inducing activity on the CendR motif (14). As was the case with TAT, Sema-OH, but not Sema-NH2, induced vascular permeability (Fig. 4B). Paralleling the weaker cell entry activity, the ability of AA-Sema-OH to induce vascular permeability was also significantly lower than that of Sema-OH (Fig. 4B).
Fig. 4. Vascular permeability induced by peptide-neutravidin complexes.
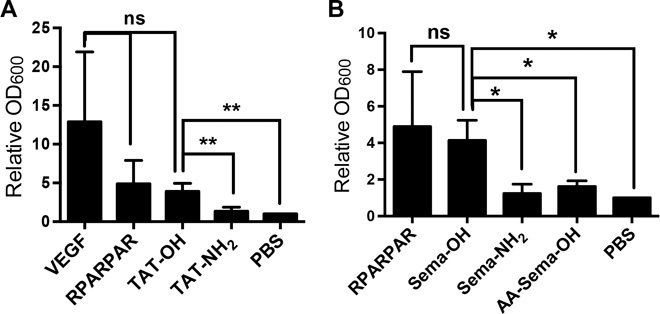
(A and B) Skin vascular permeability assay was carried out with VEGF or neutravidin complexed with the indicated peptides as described in Materials and Methods. The 600-nm absorbance was normalized to that of PBS control and plotted on the y axis. At least three independent experiments were carried out for each condition. Error bars, SEM. ns, not significant; *P < 0.05 and **P < 0.01, Student’s t test.
To perform ultrastructural studies on TAT entry into cells, we coated gold (Au) NPs with RPARPAR or d-TAT to trace the NRP1- and HS-mediated cell entry pathways, respectively. Transmission electron microscopy (TEM) can provide direct evidence of cell entry and give insight to the transport route inside the cells (30). To examine the cellular compartments carrying TAT cargo as a function of time, we synchronized the cell entry of the nanoparticles. Low-temperature incubation has been the most commonly used method to synchronize the cell entry process. However, we found the method unsatisfactory and chose to adapt a hypo-K+ buffer system for this purpose (31). Incubation of cells with the hypo-K+ buffer had little effect on the cell surface binding of the NPs but immediately blocked their internalization (Fig. 5A). Because hypo-K+ treatment occurs at 37°C, it allowed much more efficient binding to cell surface receptors than that obtained at low temperatures (fig. S3A). Replacing the hypo-K+ buffer with regular tissue culture medium reactivated the cellular internalization of the NPs (fig. S3B). This method enabled us to exclusively investigate the affinity of peptides to their receptors in the cellular context, without interference from internalization events. In wild-type CHO cells expressing glycosaminoglycans, AgNPs bearing all TAT variants, but not RPARPAR, bound to the cell surface (fig. S4A). In contrast, only CendR-active peptides bound to the surface of NRP1-transfected pgs745 cells (fig. S4B). These results agree with the plate binding tests (Fig. 1, E and F). Similar results were obtained using PPC1 cells (fig. S4C).
Fig. 5. Synchronization of cell entry and stepwise transport of peptide-coated AuNPs.
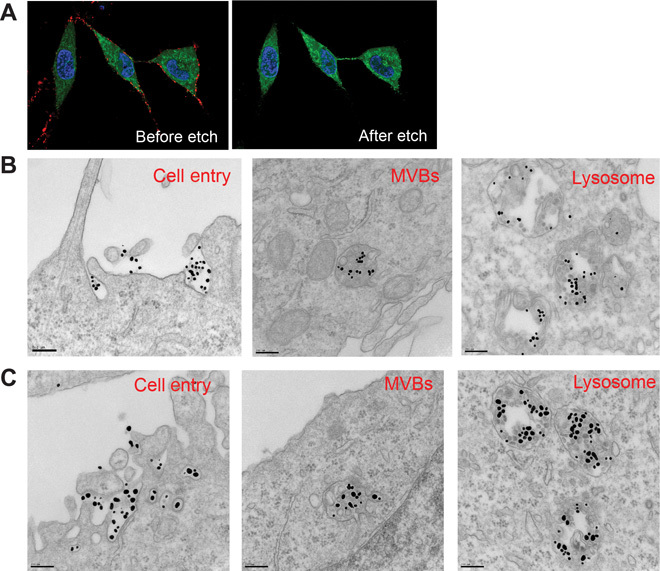
(A) PPC1–green fluorescent protein (GFP) cells (green) were incubated with AgNP-RPARPAR (red) in hypo-K+ buffer for 30 min at 37°C. The same cells were imaged for surface-bound Ag (before etch) and their internalization (after etch). Representative images are shown. (B) PPC1 cells were incubated with AuNP-d-TAT (~17 nm in diameter) in hypo-K+ buffer for 30 min at 37°C. After washing to remove unbound particles, the cells were incubated in regular culture medium for 30, 60, and 240 min before subcellular transport was stopped by fixation. Representative TEM images at each of these time points (30 min, left; 60 min, middle; 240 min, right) are shown to indicate the major intracellular compartments containing NPs. MVBs, multivesicular bodies. Scale bars, 200 nm. (C) PPC1 cells were incubated with AuNP-d-TAT (~17 nm in diameter) and AuNP-RPARPAR (~50 nm in diameter) and processed as in (B). Scale bars, 200 nm.
TEM of the synchronized cells revealed that both Au-RPARPAR and Au-d-TAT were engulfed in structures of macropinosome size (>150 nm) (Fig. 5B) (17). Subsequently, 30, 60, and 240 min after initiating the cell entry, we observed these particles sequentially in early endosomes, late endosomes/multivesicular bodies, and eventually lysosomes (Fig. 5B). No sign of endosomal escape into cytosol was seen even up to 24 hours of cellular incubation (fig. S5), and Au-RPARPAR and Au-d-TAT were colocalized in some, but not all, vesicles (Fig. 5C).
DISCUSSION
The positively charged amino acids hold the key to the cell-penetrating activity of cationic CPPs and various bioactive proteins. Strong electrostatic interactions between the positive residues and the anionic HS chains of proteoglycans have been commonly thought as the primary mechanism in initiating the endocytosis of CPPs and their macromolecular cargo. Here, we describe an HS-independent pathway for cellular uptake of the TAT peptide that is triggered by the binding of TAT to NRP1.
NRP1 binding requires a peptide to have a C-terminal arginine (CendR motif) and the natural l conformation. Moreover, the α carboxyl group of the arginine has to be free of substituents (14). In agreement with these results, we found that AgNPs coated with l–amino acid TAT peptide containing a free C-terminal arginine (TAT-OH) bound to purified NRP1 protein and, when incubated with cells, entered the NRP1 pathway. Evidence for the NRP1-dependent uptake of AgNP-TAT-OH included the use of cells that are devoid of NRP1 and HS; forced expression of NRP1 rendered these cells capable of taking up AgNP-TAT-OH. AgNPs coated with TAT variants that do not conform to the CendR requirements did not bind to NRP1 or enter the NRP1-positive, HS-negative cells. Thus, TAT-OH cargo can use the NRP1-triggered CendR pathway to enter cells.
In addition to using the CendR pathway, AgNP-TAT-OH also enters cells in an HS-dependent manner because blocking NRP1 revealed robust cellular uptake that was inhibited by soluble HS. Inhibiting NRP1 alone or HS alone caused no significant inhibition of AgNP-TAT-OH uptake, but using both inhibitors simultaneously resulted in complete inhibition. A likely explanation for these results is that neither pathway was saturated at the NP dose we used, and that if prevented from using one pathway, the NPs entered through the other. Inhibition experiments with anti-NRP1 showed that TAT peptides with a blocked C terminus or composed of d–amino acids do not enter the CendR pathway to a significant extent; the uptake of AgNPs coated with these TAT variants was completely HS-dependent. However, a C-terminal block by an amide group or by additional amino acids could be reversed through deamidation or a protease cleavage, particularly in vivo, which would activate the possibility of entry into the CendR pathway. Although NRP1 is often associated with other cell surface receptors such as integrins, peptides with an active CendR motif displayed no affinity for integrins (20), and we previously found no requirement for integrins in the CendR endocytotic process (17). Therefore, we focused this study on NRP1.
The receptor binding and cell entry properties of Sema C-terminal peptides largely reproduced the findings with the TAT peptide, but with some differences. Sema-OH, while gaining affinity to NRP1, bound to HS at a lower efficiency than Sema-NH2. The difference in HS binding between TAT-OH and TAT-NH2 was much smaller, likely because a greater number of positive residues in TAT make it better able to tolerate a small change in the charge of the peptide. The efficiency of receptor binding largely correlated with the cell entry activity. The ability to bind to NRP1 determined whether a peptide could use the CendR pathway. However, we found that although AA-Sema-OH has a similar affinity to purified NRP1 as Sema-OH, the loss of the HS-binding motif greatly decreased the cell entry efficiency of the peptide. The addition of free HS restored most of the lost cell entry activity. These results suggest that HS binding has a synergistic effect on the CendR motif–NRP1 interaction at the cell membrane, as has been shown for HS-binding growth factors (11, 12). Accordingly, AA-Sema-OH also exhibited significantly lower activity in inducing vascular permeability, which largely depends on the ligation with NRP1 on the cellular context.
There is a consensus that TAT-conjugated macromolecules and nanoparticles enter HS proteoglycan–expressing cells through endocytosis, primarily by macropinocytosis (7). Although extensive evidence supports this model, to our knowledge, no direct visualization of this process by ultrastructural imaging has been reported. Here, we provide the first electron microscopy (EM) images of the endocytotic process induced by TAT through HS binding and compare the process with the CendR pathway. Because EM only captures static images of cellular transport, we developed a methodology that synchronizes the cell entry process more accurately than the commonly used low-temperature treatment. The method enables us to visualize the engulfment of nanoparticle cargo and the subsequent formation of the intracellular compartments containing the cargo step by step. Allowing cells to simultaneously take up the CendR probe Au-RPARPAR and the HS-dependent Au-d-TAT showed that these probes are taken up together and that the resulting endocytotic structures resemble macropinosomes, as has previously been shown for Au-RPARPAR (16). It is important to note that we focused this study on the uptake of peptide-nanoparticle complexes into cells. Thus, our results are likely to pertain only to nanoparticle cargo, and perhaps macromolecular cargo, such as proteins, which has also been shown to be an endocytotic process (5). CPPs as free peptides and as conjugates with small molecules may use nonendocytotic pathways such as direct membrane translocation (7, 19). Further study is needed for a complete understanding of the roles played by different entry pathways in CPP activities.
The set of peptide tools we have established here should aid further studies of the endocytotic machineries involved in the uptake of extracellular cargo including drugs and other therapeutics. The specificity of these peptides for a given receptor makes it possible to trace the subsequent endocytotic process exclusively. In contrast, natural ligands often have multiple functional units and interact with more than one cell surface receptor, making it difficult to clearly interpret experimental results. In addition, peptides can be more easily combined with delivery vehicles such as nanoparticles, imaging probes, etc., than natural proteins.
Finally, TAT and perhaps other CPPs that bind to both NRP1 and HS may offer some advantages as drug delivery tools over peptides that only rely on the HS pathway. The CendR pathway serves as an alternative route of cell entry and thus makes TAT less dependent on HS. More importantly, the NRP1 interaction renders the peptide capable of inducing extravasation and tissue penetration through the CendR pathway, which is particularly useful in the delivery of macromolecules and nanomaterials in vivo. Thus, a CPP that has dual binding activity can be more potent and versatile than a peptide that only interacts with HS.
MATERIALS AND METHODS
Peptides and reagents
All peptides used in this study were purchased from LifeTein and contained a biotin tag at the N terminus, and 6-aminohexanoic acid and GGS (glycine-glycine-serine) as a spacer. Peptide-coated AgNPs and AuNPs and neutravidin complexes were assembled through biotin-neutravidin interaction as previously described by Braun et al. (22). AgNPs also carried CF555 fluorescent dye (Biotium) for quantification by flow cytometry and for imaging (22). PPC1, CHO wild-type, and pgs745 cell lines were purchased from the American Type Culture Collection. PPC1 cells labeled with GFP have been previously described (17). PPC1 and PPC1-GFP cells were cultured in Dulbecco’s modified Eagle’s medium (Thermo Scientific) plus 10% fetal bovine serum (FBS) with penicillin/streptomycin. CHO wild-type and pgs745 cells were cultured in F-12K medium plus FBS with penicillin/streptomycin.
The chemicals and antibodies used in this study were heparin [Sigma-Aldrich, dissolved in phosphate-buffered saline (PBS) at the indicated concentrations]; rabbit anti-human NRP1 blocking antibody, which was previously generated (17) and used at a final concentration of 20 μg/ml unless otherwise indicated; and siRNA for NRP1 knockdown, which was purchased from Ambion (cat. no. AM51331). As a negative control, we used a mix of nonspecific siRNAs (32) provided by the Sanford Burnham Prebys Functional Genomics Core. Mouse anti-human NRP1 antibody (Miltenyi Biotec, cat. no. 130-090-693) was used in immunofluorescence studies.
NP internalization into cells
Cell uptake assay was performed as previously described by Pang et al. (17). Briefly, cells were grown in 48-well plates containing 500 μl of culture medium per well for 24 hours. At about 80% confluency, 5 μl of Ag [optical density (OD), 300] coated with the indicated peptides was added. After a 1-hour incubation at 37°C, etching buffer [at a final concentration of 10 mM Na2S2O3 and 10 mM K3Fe(III)CN63− in PBS] was added into the solution, and incubation was continued for 5 s to eliminate extracellular and surface-bound Ag. Live cells were then stained with Hoechst 33342 (1.25 μg/ml; Life Technologies) and quantified for internalized AgNP fluorescence intensity per cell using either flow cytometry or confocal imaging. To express NRP1 in CHO pgs745 cells, the cells were transfected with a plasmid NRP1 expression vector (a gift from M. Klagsbrun), using FuGENE 6 (Promega) according to the manufacturer’s instructions.
To inhibit cell surface HS binding or block NRP1, soluble heparin or anti-NRP1 blocking antibody was added into the cell culture medium at the indicated concentrations, followed 5 min later by the addition of peptide-coated AgNPs. To knock down NRP1 expression with siRNA, cells were seeded 1 day earlier and transfected with 10 nM siRNA for 72 hours, using RNAiMAX (Life Technologies) according to the manufacturer’s instruction. mRNA extraction and quantitative polymerase chain reaction were performed as previously described by Pang et al. (17).
To synchronize cell entry, the culture medium was aspirated and the cells were washed twice with PBS. The cells were then incubated with the hypo-K+ buffer (71 mM KCl, 1.8 mM CaCI2, 10 mM Hepes, 0.17 mM K2HPO4, 0.175 mM KH2PO4, and 0.405 mM MgSO4) together with the indicated AgNPs or AuNPs for 30 min at 37°C. After washing, prewarmed regular culture medium was added to the cells to initiate endocytosis. After the indicated length of incubation, endocytosis and subcellular transport were stopped by fixing the cells with 4% formaldehyde (Sigma-Aldrich).
Receptor binding
One hundred microliters of PBS containing NRP1 b1b2 protein (5 μg/ml) was added into each well of high-binding plates (Corning, Costar, cat. no. 3590) for immobilization through overnight incubation at 4°C. HS plates were purchased from bioWORLD (cat. no. 20140005-3). For NRP1 binding, the wells were blocked with 1% bovine serum in PBS (PBS-B) for 1 hour at room temperature. Two microliters of AgNPs coated with the indicated peptides (OD, 300) was then added to 200 μl of PBS-B per well for 1 hour at room temperature. Similarly, the HS plate was first washed three times with 100 μl of PBS with 0.05% Tween 20 (PBS-T), and then 2 μl of AgNPs coated with the indicated peptides was added to 200 μl of PBS-T per well for 1 hour at room temperature. After washing, AgNP binding was measured with a fluorescent plate reader. To perform the cell surface binding assay, the indicated cells were washed with PBS twice before the addition of the hypo-K+ buffer containing peptide-AgNPs. After a 30-min incubation at 37°C, the cells were washed three times with PBS to remove unbound particles and then subjected to confocal imaging.
Transmission electron microscopy
After synchronized peptide-AuNP internalization, cells were processed by the Electron Microscopy Facility of the University of California, San Diego. Images were taken with a JEOL 1200 EX II transmission electron microscope (JEOL) according to the manufacturer’s instruction.
Statistics
We calculated all P values using Student’s t test by comparing the indicated sample and their corresponding control group. A value of P < 0.05 was considered significant, and different significance levels are indicated with asterisks. We give the lower P values when applicable to indicate the degree of statistical confidence in the results.
Vascular permeability assay
A modified Miles assay to measure skin vascular permeability was carried out as previously described by Teesalu et al. (14). Briefly, athymic nu/nu female mice were intravenously injected with 100 μl of PBS containing 1% Evans blue dye. After 10 min, the mice were injected intradermally on the ventral skin with 40 μl of PBS containing VEGF (50 ng) or tetrameric neutravidin-peptide complexes (final peptide concentration, 8 μM). The mice were perfused through the heart with PBS-B to eliminate the dye and peptide-neutravidin remaining in the blood. The skin in the injection area was cut out, and 4-mm-diameter samples containing the site of injection were punched out. The samples were photographed and then incubated in N,N-dimethylformamide for 48 hours at 37°C to extract the dye. The amount of dye per unit weight of skin was quantified by measuring the absorbance at 600 nm in a spectrophotometer. Animal experimentation was performed according to procedures approved by the Institutional Animal Care and Use Committee at the Sanford Burnham Prebys Medical Discovery Institute.
Acknowledgments
We thank T. Teesalu and K. N. Sugahara for contributions to the early stages of this work and for discussions. Funding: This work was supported by Cancer Center Support Grant CA CA30199 and National Cancer Institute grant CA152327. G.B.B. was supported by NIH T32 fellowship (CA121949). Author contributions: H.-B.P., G.B.B., and E.R. designed the experiments; H.-B.P. and G.B.B. performed all experiments and analyzed the results; H.-B.P. and E.R. wrote the manuscript. Competing interests: NIH author disclaimer: The views and opinions of the authors expressed on OER Web sites do not necessarily state or reflect those of the U.S. Government, and they may not be used for advertising or product endorsement purposes. E.R. has ownership interest (including patents) in CendR Therapeutics Inc. E.R. is the founder, chairman of the board, consultant/advisory board member, and major shareholder of CendR Therapeutics Inc. and has ownership interest (including patents) in the same. E.R. is also a consultant/advisory board member and major shareholder of EnduRx Pharmaceuticals Inc. and has ownership interest (including patents) in the same. No potential conflicts of interest were disclosed by the other authors. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/10/e1500821/DC1
Fig. S1. Use of NRP1- and HS-dependent pathways by TAT variants.
Fig. S2. AgNP internalization into HS-deficient cells.
Fig. S3. Synchronization of NP cell entry using the hypo-K+ buffer.
Fig. S4. Peptide-AgNP binding to cell surface receptors.
Fig. S5. Subcellular localization of peptide-Au complexes.
REFERENCES AND NOTES
- 1.Bertrand N., Wu J., Xu X., Kamaly N., Farokhzad O. C., Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 66, 2–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruan G., Agrawal A., Marcus A. I., Nie S., Imaging and tracking of tat peptide-conjugated quantum dots in living cells: New insights into nanoparticle uptake, intracellular transport, and vesicle shedding. J. Am. Chem. Soc. 129, 14759–14766 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Madani F., Lindberg S., Langel Ü., Futaki S., Gräslund A., Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 414729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan I. M., Wadia J. S., Dowdy S. F., Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 102, 247–253 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Tyagi M., Rusnati M., Presta M., Giacca M., Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 276, 3254–3261 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Console S., Marty C., García-Echeverría C., Schwendener R., Ballmer-Hofer K., Antennapedia and HIV transactivator of transcription (TAT) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J. Biol. Chem. 278, 35109–35114 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Gump J. M., Dowdy S. F., TAT transduction: The molecular mechanism and therapeutic prospects. Trends Mol. Med. 13, 443–448 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Kadonosono T., Yamano A., Goto T., Tsubaki T., Niibori M., Kuchimaru T., Kizaka-Kondoh S., Cell penetrating peptides improve tumor delivery of cargos through neuropilin-1-dependent extravasation. J. Control. Release 201, 14–21 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Ellis L. M., The role of neuropilins in cancer. Mol. Cancer Ther. 5, 1099–1107 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Salikhova A., Wang L., Lanahan A. A., Liu M., Simons M., Leenders W. P. J., Mukhopadhyay D., Horowitz A., Vascular endothelial growth factor and semaphorin induce neuropilin-1 endocytosis via separate pathways. Circ. Res. 103, e71–e79 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vander Kooi C. W., Jusino M. A., Perman B., Neau D. B., Bellamy H. D., Leahy D. J., Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. U.S.A. 104, 6152–6157 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teran M., Nugent M. A., Synergistic binding of vascular endothelial growth factor-A and its receptors to heparin selectively modulates complex affinity. J. Biol. Chem. 290, 16451–16462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shintani Y., Takashima S., Asano Y., Kato H., Liao Y., Yamazaki S., Tsukamoto O., Seguchi O., Yamamoto H., Fukushima T., Sugahara K., Kitakaze M., Hori M., Glycosaminoglycan modification of neuropilin-1 modulates VEGFR2 signaling. EMBO J. 25, 3045–3055 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teesalu T., Sugahara K. N., Kotamraju V. R., Ruoslahti E., C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. U.S.A. 106, 16157–16162 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zanuy D., Kotla R., Nussinov R., Teesalu T., Sugahara K. N., Alemán C., Haspel N., Sequence dependence of C-end rule peptides in binding and activation of neuropilin-1 receptor. J. Struct. Biol. 182, 78–86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugahara K. N., Braun G. B., de Mendoza T. H., Kotamraju V. R., French R. P., Lowy A. M., Teesalu T., Ruoslahti E., Tumor-penetrating iRGD peptide inhibits metastasis. Mol. Cancer Ther. 14, 120–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pang H.-B., Braun G. B., Friman T., Aza-Blanc P., Ruidiaz M. E., Sugahara K. N., Teesalu T., Ruoslahti E., An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat. Commun. 5, 4904 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vivès E., Brodin P., Lebleu B., A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 272, 16010–16017 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Heitz F., Morris M. C., Divita G., Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 157, 195–206 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugahara K. N., Teesalu T., Prakash Karmali P., Ramana Kotamraju V., Agemy L., Girard O. M., Hanahan D., Mattrey R. F., Ruoslahti E., Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 16, 510–520 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teesalu T., Sugahara K. N., Ruoslahti E., Tumor-penetrating peptides. Front. Oncol. 3, 216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun G. B., Friman T., Pang H.-B., Pallaoro A., de Mendoza T. H., Willmore A.-M. A., Ramana Kotamraju V., Mann A. P., She Z.-G., Sugahara K. N., Reich N. O., Teesalu T., Ruoslahti E., Etchable plasmonic nanoparticle probes to image and quantify cellular internalization. Nat. Mater. 13, 904–911 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lundberg M., Wikström S., Johansson M., Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther. 8, 143–150 (2003). [DOI] [PubMed] [Google Scholar]
- 24.He Z., Tessier-Lavigne M., Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 90, 739–751 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Parker M. W., Linkugel A. D., Vander Kooi C. W., Effect of C-terminal sequence on competitive semaphorin binding to neuropilin-1. J. Mol. Biol. 425, 4405–4414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Acevedo L. M., Barillas S., Weis S. M., Göthert J. R., Cheresh D. A., Semaphorin 3A suppresses VEGF-mediated angiogenesis yet acts as a vascular permeability factor. Blood 111, 2674–2680 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugahara K. N., Teesalu T., Karmali P. P., Kotamraju V. R., Agemy L., Greenwald D. R., Ruoslahti E., Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 328, 1031–1035 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chauhan V. P., Stylianopoulos T., Boucher Y., Jain R. K., Delivery of molecular and nanoscale medicine to tumors: Transport barriers and strategies. Annu. Rev. Chem. Biomol. Eng. 2, 281–298 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Arese M., Ferrandi C., Primo L., Camussi G., Bussolino F., HIV-1 Tat protein stimulates in vivo vascular permeability and lymphomononuclear cell recruitment. J. Immunol. 166, 1380–1388 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Hirose H., Takeuchi T., Osakada H., Pujals S., Katayama S., Nakase I., Kobayashi S., Haraguchi T., Futaki S., Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol. Ther. 20, 984–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward D. M., Perou C. M., Lloyd M., Kaplan J., “Synchronized” endocytosis and intracellular sorting in alveolar macrophages: The early sorting endosome is a transient organelle. J. Cell Biol. 129, 1229–1240 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J., Lee J. E., Heynen-Genel S., Suyama E., Ono K., Lee K., Ideker T., Aza-Blanc P., Gleeson J. G., Functional genomic screen for modulators of ciliogenesis and cilium length. Nature 464, 1048–1051 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/10/e1500821/DC1
Fig. S1. Use of NRP1- and HS-dependent pathways by TAT variants.
Fig. S2. AgNP internalization into HS-deficient cells.
Fig. S3. Synchronization of NP cell entry using the hypo-K+ buffer.
Fig. S4. Peptide-AgNP binding to cell surface receptors.
Fig. S5. Subcellular localization of peptide-Au complexes.