

An anti–HIV-1 drug is found to destroy leukemia cells in adults.
Keywords: Adult T-cell leukemia, Abacavir, nucleoside-analog reverse-transcriptase inhibitor, tyrosyl-DNA-phosphodiesterase 1, DNA repair
Abstract
Adult T cell leukemia (ATL) is an aggressive T cell malignancy caused by human T cell leukemia virus type 1 (HTLV-1) and has a poor prognosis. We analyzed the cytotoxic effects of various nucleoside analog reverse transcriptase inhibitors (NRTIs) for HIV-1 on ATL cells and found that abacavir potently and selectively kills ATL cells. Although NRTIs have minimal genotoxicities on host cells, the therapeutic concentration of abacavir induced numerous DNA double-strand breaks (DSBs) in the chromosomal DNA of ATL cells. DSBs persisted over time in ATL cells but not in other cell lines, suggesting impaired DNA repair. We found that the reduced expression of tyrosyl-DNA phosphodiesterase 1 (TDP1), a repair enzyme, is attributable to the cytotoxic effect of abacavir on ATL cells. We also showed that TDP1 removes abacavir from DNA ends in vitro. These results suggest a model in which ATL cells with reduced TDP1 expression are unable to excise abacavir incorporated into genomic DNA, leading to irreparable DSBs. On the basis of the above mechanism, we propose abacavir as a promising chemotherapeutic agent for ATL.
INTRODUCTION
Adult T cell leukemia (ATL) is caused by human T cell leukemia virus type 1 (HTLV-1) (1–3). Because of resistance to most cytotoxic drugs, patients suffering from ATL have an extremely poor prognosis with conventional chemotherapy [3-year overall survival rate of 24% (4, 5)]. Even allogeneic hematopoietic stem cell transplantation only increases the 3-year overall survival rate to a modest 33% (6). The combination of azidothymidine (AZT, also called zidovudine) and interferon-α (IFN-α), first proposed by Gill et al. (7) and Hermine et al. (8), is currently considered the most effective therapy, with an overall 5-year survival rate that reaches 46% (9). Some speculate that this combination therapy represses ATL cells by activating the immune response against HTLV-1–infected ATL cells as well as by interfering with the productive replication of HTLV-1 (10). However, the molecular mechanism underlying the cytotoxic effect of AZT against ATL remains elusive.
AZT is a first-generation nucleoside analog reverse transcriptase inhibitor (NRTI) (11) and has been widely used for HIV-1 patients. NRTI is efficiently incorporated into newly synthesizing viral DNA by HIV-1 reverse transcriptase, but not by replicative DNA polymerases in host cells. Efficient incorporation by reverse transcriptase is attributable to a lack of proofreading activity as well as to less stringent discrimination (12). Incorporated NRTIs prevent further extension of DNA synthesis, thereby inhibiting the productive replication of HIV-1. NRTIs approved for the treatment of HIV-1 are believed to have a minimal genotoxic effect on host cells.
Tyrosyl-DNA phosphodiesterase 1 (TDP1) was initially identified as a key enzyme for eliminating covalently bound topoisomerase I (TopI) from cleaved DNA ends (13). A mutation in the TDP1 gene causes spinocerebellar ataxia with axonal neuropathy (SCAN1). A defect in TDP1 causes hypersensitivity to camptothecin (CPT11), a chemotherapeutic TopI poison, which stabilizes the covalent binding of TopI to cleaved DNA ends and kills cancer cells by inducing double-strand breaks (DSBs) during DNA replication, as shown in both SCAN1 patient–derived cell lines and knockout mouse models (14, 15).
Here, we found that a therapeutic concentration of abacavir (ABC), an NRTI, induces chromosomal DSBs and thereby kills ATL cells but not non–HTLV-1–infected cells. ABC is widely used as a key drug for the treatment of HIV-1 infection because it is well tolerated by >90% of patients with few adverse effects and has a desirable bioavailability (16). Once ABC is incorporated into the cells, it is phosphorylated in a unique stepwise anabolism to be converted to the triphosphated guanine analog carbovir (CBV) (17). The therapeutic concentration (16, 18, 19) of ABC causes a marked increase in the number of DSBs. We here demonstrate that this pronounced accumulation of DSBs caused by ABC results primarily from the down-regulation of TDP1 in ATL cells. We thus propose ABC as a novel and potent therapeutic agent for ATL.
RESULTS
ABC selectively kills HTLV-1–infected cells, including ATL cells
We first examined the cytotoxic effects of six NRTIs (ddI, d4T, 3TC, AZT, TDF, and ABC), all of which are current therapeutic agents for HIV-1, on ATL cell lines [ED-40515(−), ED-40515(+), SYK-11L(+), and ATL-43T] and on HTLV-1–infected cell lines (MT-2 and Hut-102). AZT was toxic to the ED-40515(−), SYK-11L(+), ATL-43T, and MT-2 cell lines but not to the others, whereas ddI, d4T, 3TC, and TDF showed poor cytotoxicity (Fig. 1A). Strikingly, the clinically relevant concentration of ABC was highly toxic to all ATL- and HTLV-1–infected cell lines but not to non–HTLV-1–infected cell lines (Jurkat, H9, and SU-DHL-6) (Fig. 1, A and B, fig. S1, and table S1). We confirmed the cytotoxicity of ABC to MT-2 cells by cell counting assays as well as MTS assay (fig. S2) because the mitochondrial toxicity of the NRTIs (20) could have affected the MTS assay. Because IFN-α is combined with AZT to treat ATL patients (7–9), we also tested the synergistic effect of IFN-α with AZT and ABC. IFN-α did not enhance the lethality of either AZT or ABC in either MT-2 or ED-40515(−) cells (Fig. 1C), suggesting that the cytotoxic effect of IFN-α in vivo may not be by a direct action (10). We therefore conclude that ABC potently and selectively kills HTLV-1–infected cells, including ATL cells, in vitro.
Fig. 1. ABC selectively kills HTLV-1–infected and ATL cell lines.
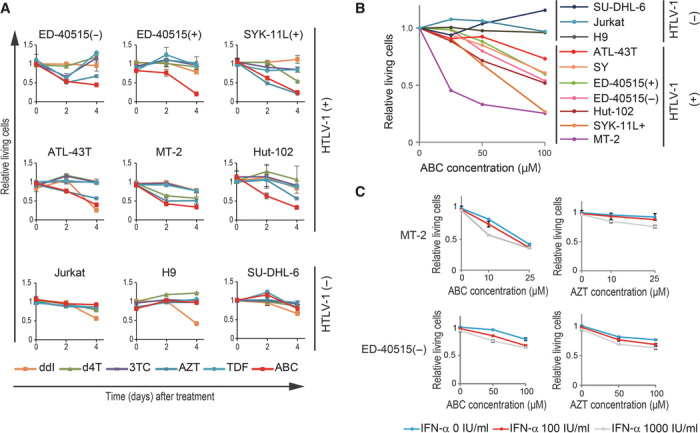
(A) Viability of the indicated cells after treatment (2 and 4 days) with 100 μM of the indicated NRTI. MTS values of treated cells relative to untreated cells are shown. Results are expressed as means ± SD of three independent experiments. (B) Viability of the indicated cells after treatment (2 days) with the indicated ABC dose. MTS values of treated cells relative to untreated cells are shown. (C) MT-2 and ED-40515(−) cells were treated with ABC or AZT together with IFN-α (0, 100, or 1000 IU/ml) for 2 days and analyzed with an MTS assay. MTS values of treated cells relative to untreated cells are shown.
We next examined the effect of ABC on cell cycle and apoptosis in ATL cells. ABC induced S/G2-phase arrest and apoptosis in ED-40515(−) cells, but not in Jurkat cells (Fig. 2, A to C, and fig. S3). This finding suggests that ABC may cause DNA damage by prematurely terminating the replication of host chromosomal DNA, thereby activating the DNA damage checkpoint to induce transient S/G2-phase arrest and apoptosis.
Fig. 2. ABC induced S/G2-phase arrest and apoptosis.
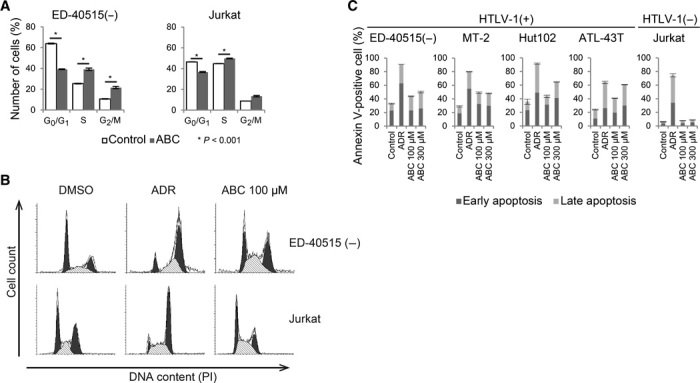
(A) Flow cytometry was used to analyze cell cycle status after propidium iodide (PI) staining. ED-40515(−) and Jurkat cells were treated with and without 100 μM ABC for 2 days and analyzed using flow cytometry. Results are expressed as means ± SD of three independent experiments. (B) Representation of flow cytometric analysis of cell cycle after PI staining. (C) Flow cytometric analysis of apoptosis after annexin V and PI double staining. ED-40515(−), MT-2, Hut-102, ATL-43T, and Jurkat cells were treated with the indicated dose of ABC or Adriamycin (ADR; 1 μg/ml) for 48 hours. Dark and light bars represent early and late apoptosis, respectively. Results are expressed as means ± SD of three independent experiments.
ABC induces DSBs due to impaired DNA repair in ATL cells
To characterize DNA damage, we measured chromosome breaks in mitotic ATL cells after exposure to ABC. The exposure significantly increased the number of chromosome breaks in ED-40515(−) cells (Fig. 3A, arrow) but not in Jurkat cells (Fig. 3B). The number of chromosome breaks induced by AZT in ED-40515(−) cells was several times smaller than that induced by ABC (Fig. 3B, left panel), which corresponds with the data in Fig. 1A. We therefore conclude that the therapeutic concentration of ABC as well as that of AZT efficiently induces DSBs in the chromosomal DNA of ATL cells, as does ionizing radiation.
Fig. 3. ABC-induced DSB accumulation in ATL cells as a result of the impairment of DNA repair.
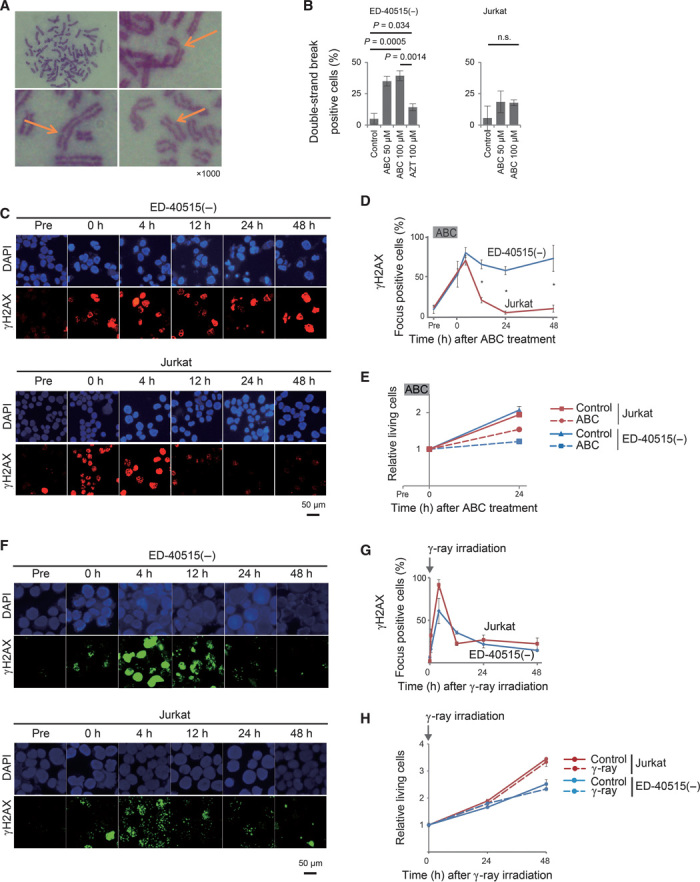
(A) Chromosome breaks induced by ABC. ED-40515(−) cells treated with 100 μM ABC for 2 days were evaluated for chromosome breaks (yellow arrow). (B) ED-40515(−) and Jurkat cells were treated with the indicated doses of ABC and AZT for 2 days. Percentages of chromosome break–positive cells are shown. Results are expressed as means ± SD of three independent experiments. n.s., not significant. (C) Expression kinetics of γH2AX foci after acute exposure to ABC. ED-40515(−) and Jurkat cells were treated with 500 μM ABC for 12 hours. After treatment, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) and anti-γH2AX at the indicated times. (D) Percentages of γH2AX focus–positive cells at the indicated times. Focus-positive cells are those containing more than five γH2AX foci. (E) Cell proliferation of ED-40515(−) and Jurkat cells based on an MTS assay. MTS values relative to time zero are shown. (F) Expression kinetics of γH2AX foci at the indicated times after 2-Gy γ-irradiation. Irradiated ED-40515(−) and Jurkat cells were stained with DAPI and γH2AX. (G) Percentages of γH2AX focus–positive cells at the indicated times. (H) Cell proliferation of ED-40515(−) and Jurkat cells evaluated by an MTS assay. Data shown are as in (E).
To monitor the kinetics of DSB repair, we transiently exposed cells to a high dose of ABC for 12 hours and then monitored the number of unrepaired DSBs over time by counting the number of subnuclear foci containing the phosphorylated H2A histone variant (γH2AX). ED-40515(−) and Jurkat cells displayed comparable numbers of γH2AX foci after 4 hours, whereas resolution of γH2AX focus formation was indeed delayed in ED-40515(−) cells, but not in Jurkat cells, even after 48 hours (Fig. 3, C and D). Cellular proliferation was also reduced in ED-40515(−) cells (Fig. 3E). Thus, ATL cells are severely deficient in repairing ABC-induced DSBs. We next assessed the performance of the two major DSB repair pathways, homologous recombination (HR) and nonhomologous end-joining, in ATL cells. To this end, we monitored the kinetics of DSB repair after ionizing radiation. ED-40515(−) and Jurkat cells displayed very similar kinetics for DSB repair, indicating the competence of the canonical DSB repair pathways in these cells (Fig. 3, F to H). Because HR is responsible for repairing DSBs generated during DNA replication (21), we examined ongoing HR by measuring the polymerization of RAD51 recombinase, the essential HR factor, at DSB sites in ABC-treated cells. Treatment with ABC induced RAD51 focus formation colocalizing with γH2AX foci in ED-40515(−) cells (Fig. 4, A and B). This finding suggests that frequent premature termination of DNA replication resulted in both the formation of DSBs and the activation of HR-dependent DSB repair to a significantly higher extent in ATL cells than in non-ATL cells.
Fig. 4. ABC-induced prolonged activation of homologous recombination–dependent repair in ATL cells.
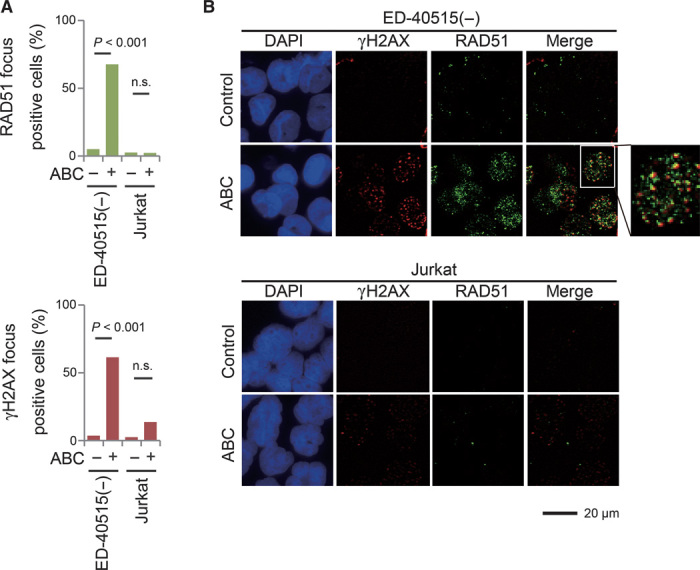
(A) Immunocytochemical staining with DAPI, anti-γH2AX, and anti-RAD51 recombinase. ED-40515(−) and Jurkat cells were treated with or without 100 μM ABC for 2 days. The percentages of γH2AX and RAD51 focus–positive cells, defined as cells containing more than five foci per cell, are shown. (B) Immunocytochemical staining with DAPI, anti-γH2AX, and anti-RAD51. ED-40515(−) and Jurkat cells were treated with or without 100 μM ABC for 2 days. The merged images display frequent colocalization of γH2AX and RAD51.
A defect in TDP1 is responsible for the cytotoxic effect of ABC on ATL cells
To identify the factors required for efficient repair of ABC-induced DNA DSBs, we screened a panel of 48 isogenic mutant chicken DT40 cell lines deficient in DNA repair factors (22) for sensitivity to ABC (table S2). The result (fig. S4) was a telling sensitivity profile; the sensitivity of the five mutant cell lines (Tdp1, Poll, Usp1, Gen1, and Rad18) to ABC was more than twofold greater than that of the wild-type DT40 cell line (Fig. 5, A and B, and fig. S5A). Furthermore, the relative sensitivity of Tdp1-deficient DT40 cells to the six individual NRTIs correlated with their cytotoxic effect on ATL cells (Figs. 1A and 5B). These results suggest that a defect in TDP1 might account for the cytotoxic effect of ABC on ATL cells.
Fig. 5. Impaired TDP1 responsible for ATL cell sensitivity to ABC.
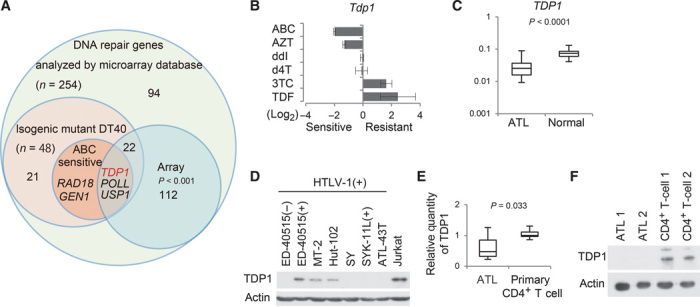
(A) Venn diagram of DNA repair genes defective in mutant chicken DT40 cell lines sensitive to ABC and down-regulated DNA repair genes in ATL cells. (B) Relative sensitivities of Tdp1-deficient DT40 cells treated with six NRTIs. Results are expressed as means ± SD of three independent experiments. (C) Analysis of mRNA expression levels based on microarray data from the GEO database (available at www.ncbi.nlm.nih.gov/geo/; accession number GSE33615). The relative expression levels of TDP1 in peripheral blood cells obtained from ATL patients and normal healthy donor controls are shown. The box plot uses the median, the approximate quartiles, and the lowest and highest data points to convey the level, spread, and symmetry of the distribution of data values. Statistical analysis was performed using Student’s t test to compare the mRNA expression levels between the ATL cases (n = 52) and the normal controls (n = 21). (D) Western blot analysis of TDP1 expression in HTLV-1–infected cell lines and in Jurkat cells. (E) Quantitative reverse transcription–polymerase chain reaction (RT-PCR) for TDP1 in primary ATL cells from 10 patients and in primary CD4+ T cells from 5 healthy donors. Data shown as in (C). (F) Western blot analysis of TDP1 in primary ATL cells from two patients and primary CD4+ T cells from two healthy donors.
We next investigated TDP1 expression in ATL cells [Gene Expression Omnibus (GEO) database] according to the Joint Study on Prognostic Factors of ATL Development (23), and found that the level of TDP1 expression is indeed down-regulated in ATL cells relative to primary CD4+ T cells (Fig. 5, A and C, and fig. S5B). Likewise, the TDP1 protein and mRNA levels in ATL cell lines (Fig. 5D) and in primary ATL cells (Fig. 5, E and F, and table S3) were significantly lower than those in the noninfected T cells and primary CD4+ T cells, respectively. These observations suggest that a defect in TDP1 causes the sensitivity to ABC in ATL cells.
TDP1 removes ABC from DNA ends in vitro
To confirm that TDP1 plays a crucial role in ABC-induced DNA damage repair, we first examined whether human TDP1 (hTDP1) removed CBV that was covalently associated with the 3′ end of a DNA oligonucleotide (Fig. 6A). We incubated total cell lysates from DT40 Tdp1−/− cells or Tdp1−/− cells complemented with hTDP1, with two substrate oligonucleotides containing either 3′-CBV or 3′-Tyr. The latter cellular lysate, but not the former one, efficiently generated the product that migrated one nucleotide shorter than the substrate (Fig. 6B). The processing efficacy of TDP1 for 3′-CBV increased in a dose-dependent manner (Fig. 6C). These biochemical results demonstrate that TDP1 removes 3′-blocking CBV efficiently in vitro.
Fig. 6. TDP1 removes ABC from DNA ends in vitro.
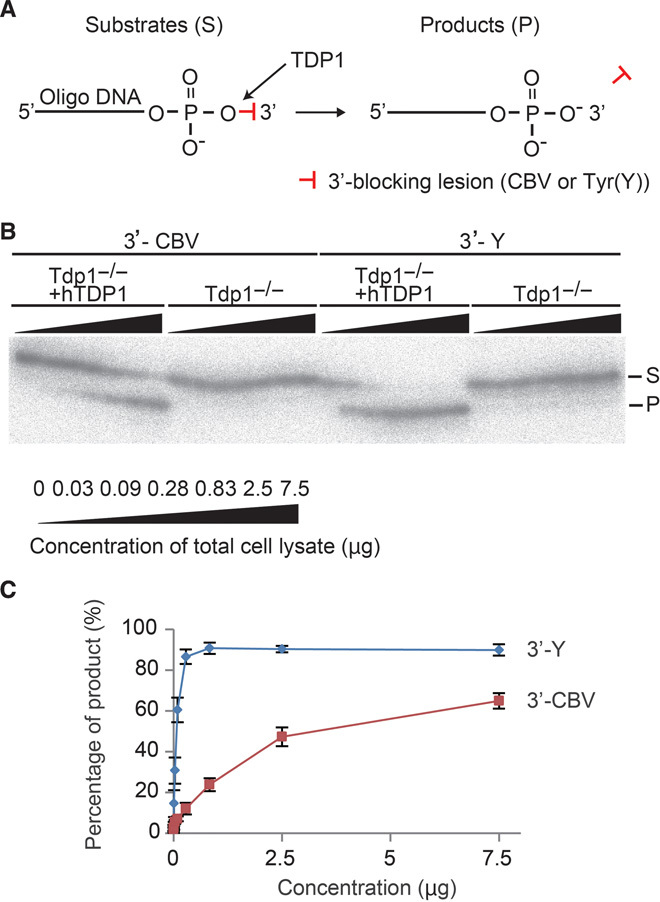
(A) Schematic diagram of in vitro biochemical assays for TDP1 activity. Both substrates contained the same sequence and conjugated CBV or tyrosine as a 3′-blocking lesion via a phosphodiester linkage. The substrates (S) were radiolabeled at the 5′ end with 32P. The products that are removed from the 3′-blocking lesion from the substrates by TDP1 are labeled “P.” Y: Tyr. (B) A representative gel demonstrating the processing of the indicated substrates by increasing amount of total cell lysates from Tdp1−/− DT40 cells or Tdp1−/− DT40 cells stably transfected with hTDP1 transgene. The substrates were incubated with serially diluted total cell lysates ranging from 0.03 to 7.5 μg. Reaction proceeded for 15 min at 25°C before being quenched and analyzed on 16% denaturing gels. (C) The percentage of product yield is plotted against increasing lysate concentration. Results are expressed as means ± SD of three independent experiments.
TDP1 catalytic activity is required for cellular tolerance to ABC
To verify the importance of TDP1 in cellular tolerance to ABC, we depleted TDP1 and reconstituted TDP1-deficient cells with a TDP1 transgene. TDP1 small interfering RNA (siRNA)–treated Jurkat cells were more sensitive to ABC than the control siRNA–treated Jurkat cells (Fig. 7A, lanes 4 and 2, respectively). Conversely, reconstitution of MT-2 cells with human wild-type TDP1 markedly increased cellular tolerance to ABC (Fig. 7B and fig. S6, A to C). These data indicate that TDP1 plays an important role in cellular tolerance to ABC. The ectopic expression of TDP1WT did not reverse the sensitivity of MT-2 cells to AZT (fig. S7), suggesting that hTDP1 eliminates AZT less efficiently than ABC. We conclude that the attenuated functionality of TDP1 is responsible for a high sensitivity to ABC in ATL cells.
Fig. 7. TDP1 catalytic activity requirement for cellular tolerance to ABC.
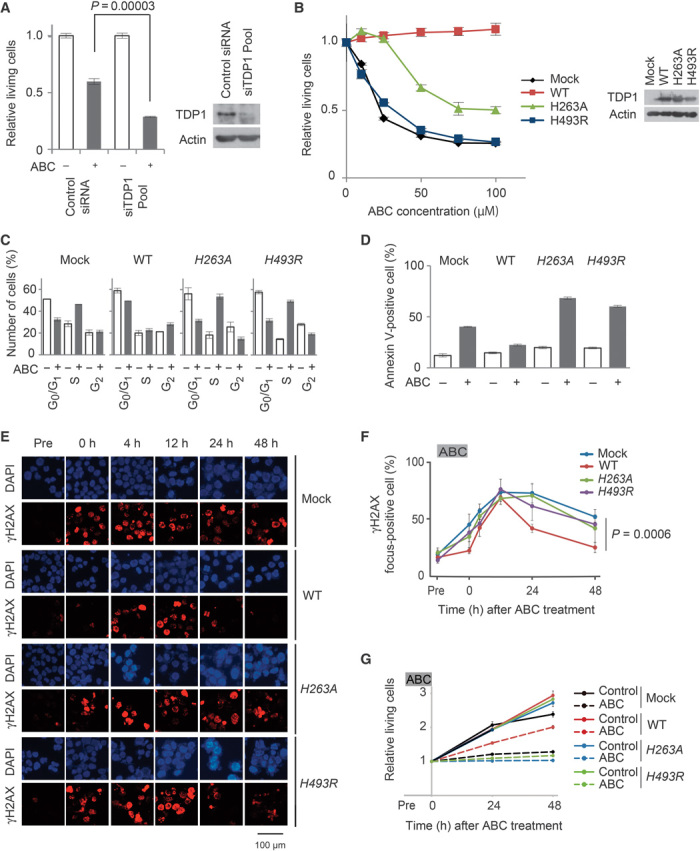
(A) Control siRNA or siTDP1 mixture was transfected into Jurkat cells. Depletion of TDP1 expression was confirmed by Western blot analysis 48 hours after transfection (right panel). Jurkat cells transfected with control siRNA or siTDP1 were treated with or without 300 μM ABC for 48 hours. MTS values of treated relative to untreated cells are shown (left panel). Results are expressed as means ± SD of three independent experiments. (B) MT-2 cells stably transfected with either wild-type (WT) TDP1, H263A TDP1, or H493R TDP1 transgenes. Stably transfected clones and control cells (mock) were treated with the indicated dose of ABC for 48 hours. Western blot analysis was conducted for expression of transgenes (right panel). Data shown as in (A, left panel). (C) Cell cycle analysis is shown as in Fig. 2A. MT-2/TDP1WT, MT-2/TDP1H263A, and MT-2/TDP1H493R cells were treated with or without 100 μM ABC for 24 hours and subjected to flow cytometry. (D) The extent of apoptosis is shown as the percentage of annexin V–positive cells (y axis). Cells were treated as in (C). (E) The indicated cells were treated as in Fig. 3C. Data shown as in Fig. 3C. (F) Statistical analysis of data in (E). MT-2/TDP1WT cells, but not MT-2/TDP1H263A or MT-2/TDP1H493R cells, are resistant to ABC. (G) Proliferation of the indicated cells is shown as in Fig. 3E.
To confirm that TDP1 catalytic activity is required for cellular tolerance to ABC, we prepared two mutant TDP1 transgenes [H263A and H493R (13, 24)] and a wild-type TDP1 transgene (25) and ectopically expressed them in MT-2 cells to generate MT-2/TDP1H263A, MT-2/TDP1H493R, and MT-2/TDP1WT cells, respectively (fig. S6, A to C). Ectopic expression of TDP1WT greatly increased MT-2 cell tolerance to ABC, but not that of the two mutant transgenes (Fig. 7B). Likewise, the phenotypes of the MT-2/TDP1H263A and MT-2/TDP1H493R cells were very similar to that of the parent MT-2 cells in terms of cellular response to ABC, including cell cycle arrest at the S phase (Fig. 7C), apoptosis (Fig. 7D), prolonged γH2AX focus accumulation (Fig. 7, E and F), and inhibition of cellular proliferation (Fig. 7G). We therefore conclude that TDP1 catalytic activity is required for cellular tolerance to ABC. This conclusion is supported by our data shown above and the previous reports showing that TDP1 can remove NRTIs localized at the 3′ ends of single-stranded oligonucleotides in vitro, because TDP1 eliminates trapped TopI from the 3′ ends of DNA strands in vivo (13, 26, 27).
CPT11 and veliparib enhance the lethality of ABC on ATL cells
CPT11 sensitivity is the classical hallmark of TDP1 loss. We verified a strong cytotoxic effect of CPT11 on ED-40515(−) cells (fig. S8A). We then tested whether a minimal dose of CPT11 synergistically increased the lethality of ABC on ATL cells. CPT11 significantly enhanced the lethality of ABC against ATL cells, with a low combination index (CI < 0.7; fig. S8B).
Furthermore, TDP1 removes trapped TopI from DNA by collaborating with poly(adenosine diphosphate–ribose) polymerase (PARP) (28). We thus theorized that combining ABC with PARP inhibitors might have a synergic cytotoxic effect on ATL cells by completely wiping out the residual activity of TDP1 in ATL cells. We then tested minimum doses of veliparib, which inhibits the catalytic activity of PARP without trapping PARP at DNA damage sites (29). Although veliparib alone had no obvious toxic effect on ATL cells, it significantly enhanced the lethality of ABC on ATL cells, with a low CI (<0.7; fig. S8C). These results suggest that a combination of ABC with CPT11 and/or PARP inhibitors could be a potent new therapeutic strategy against ATL.
ABC inhibits the growth of ATL cell xenografts in NOD/SCID mice
Finally, we tested the in vivo effect of ABC on ATL cell growth using an ATL xenograft model. Nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice were inoculated with ED-40515(−) cells and treated daily with ABC via oral gavage at a dose of 75 mg/kg per day, which is comparable to 410 mg once daily of orally administered ABC in humans. Orally administered ABC significantly inhibited the growth of ATL tumor grafts (Fig. 8A) and improved survival (Fig. 8B).
Fig. 8. ABC inhibits the growth of ATL cell xenografts in NOD/SCID mice.
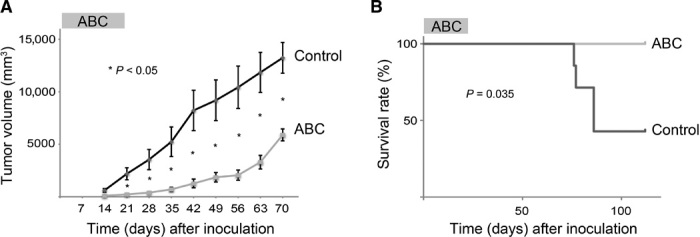
(A) Tumor sizes of xenografted NOD/SCID mice treated with or without ABC. The mean sizes of individual tumors were plotted (control group, n = 7; ABC group, n = 6). P values were determined using Student’s t test (*P < 0.05). Error bars represent the SEM. (B) Kaplan-Meier survival curves for the ABC-treated and control mice. P values were determined using the Wilcoxon test.
DISCUSSION
In this report, we demonstrate that ABC specifically and potently kills ATL cells. ABC triggers apoptosis by inducing DSBs in host chromosomal DNA. The most likely mechanism for DSB induction is that ABC is converted to the triphosphated CBV in cells and then incorporated into host chromosomal DNA by replicative DNA polymerases, leading to premature termination of DNA replication, collapse of the replication fork, and DSB formation (Fig. 9). Thus, ABC is an effective chain terminator not only in the reverse transcription of retroviral DNA but also in the replication of host chromosomal DNA in ATL cells. This is totally unexpected because long-term treatment with ABC is well tolerated, causing only few side effects in HIV-1 patients. ABC has also been shown to be virtually nontoxic to bone marrow progenitor cells and leukemic cell lines (17). Our study identifies efficient DSB formation as the unique mechanism underlying the specific killing of ATL cells by the antiretroviral agent ABC.
Fig. 9. Specific lethality of ABC on ATL cells due to a defect in TDP1.
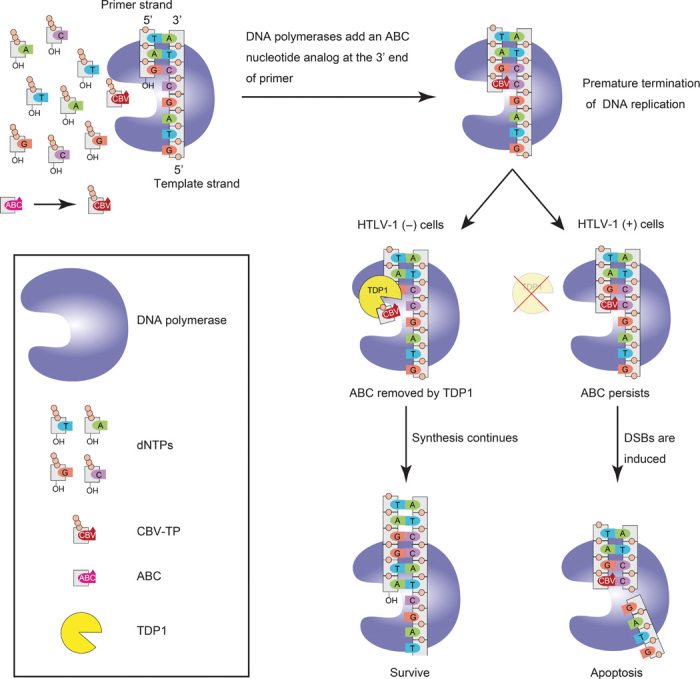
ABC is phosphorylated in a unique stepwise anabolism and is converted to the triphosphate of CBV. During DNA synthesis, triphosphorylated ABC was incorporated into host chromosomal DNA by replicative DNA polymerases, leading to premature termination of DNA replication. In normal cells, TDP1 removes ABC quickly and DNA synthesis continues. However, in HTLV-1(+) cells, the collapse of the replication fork is induced because of the deficiency of TDP1, leading to DSB formation and apoptosis.
Three mechanisms can inhibit NRTI genotoxicity. First, NRTIs are not potent inhibitors of replicative DNA polymerases (30, 31). Second, the catalytic pocket of replicative DNA polymerases is able to stringently discriminate between triphosphated NRTIs and normal nucleotides, with proofreading activities considered also as a contributing factor. Third, host DNA repair factors may be able to remove NRTIs when they are misincorporated into the 3′ end of primers by replicative DNA polymerases. The third mechanism accounts for the high toxicity of ABC to ATL cells, which, unlike non–HTLV-1–infected cells, are almost entirely unable to repair ABC-induced DSBs (Fig. 3, C and D). Moreover, our study demonstrates that a TDP1 defect is primarily responsible for the potent and selective cytotoxic effect of ABC on ATL cells. TDP1 can eliminate CBV misincorporated by replicative DNA polymerases into the 3′ ends (Fig. 6) because TDP1 eliminates TopI trapped at the 3′ ends of DNA cleavage (13) and oxidative 3′-blocking lesions (32). It is surprising that TDP1 could remove the 3′-blocking nucleotide analog that was not eliminated by the proofreading nuclease of replicative DNA polymerases. TDP1 seems to remove AZT from 3′ ends with considerably less efficiency than ABC because ectopic expression of a TDP1 transgene in MT-2 cells greatly increased cellular tolerance to ABC but not to AZT (Fig. 7B and fig. S7). We thus propose that ABC is a more potent chemotherapeutic agent against ATL than AZT. The current study also suggests that a therapy that combines ABC and CPT11 or a PARP inhibitor such as veliparib would provide a novel therapeutic strategy against ATL by enhancing the cytotoxic effect of ABC with minimum side effects on normal cells (fig. S8).
The molecular mechanism underlying reduced TDP1 gene expression is unclear. However, the current study reveals that the transcriptional status of the TDP1 gene may be a reliable biomarker for predicting the efficacy of ABC in anti-malignancy therapy. In addition, a recent report showing the defect of TDP1 in several lung cancer cell lines suggests the much broader indication of ABC to other cancers (33).
Finally, we demonstrate that ABC efficiently inhibits the growth of ATL cell xenografts in NOD/SCID mice. We administered ABC at 75 mg/kg per day orally. The plasma concentration of ABC in the mice was comparable to that in HIV-1 patients taking orally 410 mg of ABC once daily (34). Thus, the daily ABC dose given to HIV-1 patients might be sufficient for treating ATL, and our findings establish a rationale to expand its use for ATL patients. Moreover, ABC might prove useful for treating HTLV-1 asymptomatic carriers. A high HTLV-1 proviral load in the asymptomatic carrier promotes the progression to ATL and other HTLV-1–associated diseases (35). We here show that ABC is useful for suppressing HTLV-1–infected cells other than ATL cells (Fig. 1). In addition, ABC has very few side effects, even during long-term treatment. Thus, ABC would be an effective prophylactic agent in both HTLV-1 carriers with a high proviral load and patients with HTLV-1–associated myelopathy/tropical spastic paraparesis (HAM/TSP), preventing the progression of these diseases.
MATERIALS AND METHODS
Cell culture
ED-40515(+) is an interleukin-2 (IL-2)–dependent T cell line of leukemic cell origin established from an ATL patient (36). ED-40515(−) is an IL-2–independent subclone of ED-40515(+) (37). SY is an IL-2–dependent HTLV-1–infected T cell line derived from a nonleukemic cell clone of ED-40515(+) from an ATL patient. ATL-43T is an IL-2–dependent T cell line established from double-negative (CD4−, CD8−) leukemic cells from a patient with ATL (38). SYK-11L(+) is a leukemic cell line from ATL tumor cells in an in vivo cell proliferation model using SCID mice (39). Hut-102 is an IL-2–independent T cell line from a patient with mycosis fungoides (40). MT-2 is a stably transformed HTLV-1–infected cell line, as previously reported (36). A summary of HTLV-1–infected and ATL cell lines is presented in table S4. Jurkat and H9 are IL-2–independent non–HTLV-1–infected human T cell lines. SU-DHL-6 is a non–HTLV-1–infected human B cell line. These cells were maintained in RPMI 1640 medium (Nacalai Tesque) containing 10% fetal bovine serum (FBS) and penicillin (100 U/ml), streptomycin (100 μg/ml), and l-glutamine (0.292 mg/ml) (PSG) (Invitrogen). The IL-2–dependent cell lines were maintained in the same medium with 0.5 nM recombinant human IL-2 (a gift from Shionogi Pharma). Human embryonic kidney (HEK) 293T cells were maintained in Dulbecco’s modified Eagle’s medium (Nacalai Tesque) containing 10% FBS and 1% PSG. The chicken DT40 cell lines used in this study were obtained from the Laboratory of Radiation Genetics, Graduate School of Medicine, Kyoto University (Kyoto, Japan). All mutant cell lines were previously authenticated (references provided in table S2). DT40 Tdp1−/− and Tdp1−/− hTDP1 cells (32) were gifts from Y. Pommier [Laboratory of Molecular Pharmacology, Center for Cancer Research, National Institutes of Health (NIH), Bethesda, MD]. The DT40 cells were cultured at 39.5°C with 5% CO2 in RPMI 1640 medium supplemented with 1% chicken serum (Gibco BRL), 10% FBS (Sigma-Aldrich), 10−5 M β-mercaptoethanol (Nacalai Tesque), penicillin (100 U/ml), streptomycin (100 μg/ml; Nacalai Tesque), and l-glutamine (0.292 mg/ml; Nacalai Tesque).
Reagents and antibodies
ABC, AZT, ddI, d4T, 3TC, and TDF were donated by the NIH through the AIDS reagent program. ABC was purchased from Sigma-Aldrich (SML0089) and Carbosynth (NA10019). Camptothecin (TG4110) and veliparib (ABT-888) were purchased from TopoGEN and Selleck Chemicals, respectively. These chemicals were dissolved in 100% dimethyl sulfoxide (DMSO, Nacalai Tesque) to a stock concentration of 10 to 500 mM and were stored at −20°C. Natural-type human IFN-α (Sumiferon) was purchased from Dainippon Sumitomo Pharma. Mouse anti-γH2AX (JBW301) was purchased from Merck Millipore, rabbit anti-RAD51 (H-92) was purchased from Santa Cruz Biotechnology, and rabbit anti-RAD51 (70–001) and mouse anti-RAD51 (B01P) were purchased from BioAcademia and Abnoba, respectively. Phycoerythrin–annexin V was purchased from BD Biosciences, and fluorescein isothiocyanate (FITC)–annexin V from BioLegend. PI (Nacalai Tesque) was dissolved in phosphate-buffered saline (PBS) to a stock concentration of 2 mg/ml. Rabbit anti-TDP1 (ab4166) was purchased from Abcam. Anti–β-actin (AC-15, A5441) was purchased from Sigma-Aldrich.
Human cell killing assay
To assess the sensitivity of human cell lines to the NRTIs with or without IFN-α or veliparib, 1 × 104 to 2 × 104 cells were seeded in 96-well flat-bottomed culture plates (final volume of 100 μl per well) and cultured for 0 to 96 hours at 37°C. Twenty microliters of CellTiter 96 AQueous One solution from a cell proliferation assay kit (Promega) was added to each well, after which cell survival was determined by measuring the absorbance intensity at 490 nm using an ALVO X-3 (PerkinElmer). The absorbance values were normalized to those of controls.
Cell cycle analysis
The pretreated cells were fixed with chilled 70% ethanol for 30 min at 4°C and then stained with PI (5 μg/ml; Nacalai Tesque) in the presence of 1% bovine serum albumin (BSA; Nacalai Tesque) and ribonuclease (100 μg/ml; Sigma-Aldrich) for 30 min at room temperature. Immediately after staining, evaluation was performed using a FACSCalibur analyzer, and the data were analyzed using ModFit software. DMSO treatment was used as a negative control, and Adriamycin (1 μg/ml) treatment was used as a positive control.
Apoptosis assay
The pretreated cells were stained with FITC–annexin V (BioLegend) and PI for 10 min at 4°C. After staining, analysis was performed immediately using a FACSCalibur analyzer. DMSO treatment was used as a negative control, and Adriamycin (1 μg/ml) treatment was used as a positive control.
Chromosomal analysis
The cells were cultured with colcemid (50 ng/ml) for 2 hours at 37°C, fixed with Carnoy’s solution, and placed on a glass slide. The slide was stained with 3.5% Giemsa (Nacalai Tesque) for 10 min, and the cells were examined by microscopy. Cells containing one chromosome break were defined as chromosome break–positive.
Cell irradiation
Cells were irradiated with 2 Gy of 135Cs γ-radiation using a Gammacell 40 Exactor (Best Theratronics).
Immunofluorescence staining
Prepared cells were placed on a glass slide via Cytospin. The cells were fixed with 4% formaldehyde (Nacalai Tesque) and washed with PBS. Next, the fixed cells were permeabilized using 0.1% NP-40/PBS for 10 min. After blocking with 3% BSA/PBS–Tween 20 for 15 min, the cells were incubated with the primary antibody at 37°C. Cells were then stained with Alexa Fluor 594 goat anti-mouse immunoglobulin G (IgG) (H+L) antibody, Alexa Fluor 488 goat anti-rabbit IgG (H+L) antibody, or Alexa Fluor 488 goat anti-mouse IgG (H+L) antibody (Molecular Probes) and DAPI (H-1200, Vector Laboratories) and observed using a fluorescence microscope (BZ-8100, Keyence).
Evaluation of relative cellular sensitivity to NRTIs
To assess the sensitivity of DT40 cells to various NRTIs, 1 × 103 DT40 cells were seeded in 96-well black dishes (final volume of 100 μl per well) and continuously exposed to 25 μM of the indicated drug for 48 hours. CellTiter-Glo Luminescent Cell Viability Assay solution (100 μl; Promega) was directly added to each well, and cell survival was determined by measuring the luminescence intensity using ALVO X-3 (PerkinElmer). Wild-type cells were examined in each assay. To evaluate the cellular sensitivity of each mutant relative to wild-type cells, the sensitivity of each mutant was normalized to that of the untreated cells. The values of the mutant and wild-type cell lines were converted to a logarithmic scale (base 2). Each value was plotted on a bar graph.
Real-time PCR
Total RNA was isolated from human samples using a High Pure RNA Isolation Kit (Roche). First-strand complementary DNA (cDNA) was generated using the PrimeScript II 1st strand cDNA Synthesis Kit (Takara). cDNA was amplified in a Thermal Cycler Dice Real Time System II (Takara) using SYBR Premix Ex Taq II (Takara). The data were normalized to the corresponding values for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The expression levels of the cells relative to normal cells (=1) are shown in Fig. 5 and fig. S5. Error bars represent SDs. The primers used were TDP1 forward, 5′-AGGCAGCCTTGGACAGATT-3′; TDP1 reverse, 5′-GGTCAGCTGAGACTTCTGGC-3′; GAPDH forward, 5′-GAAGGTGAAGGTCGGAGTC-3′; and GAPDH reverse, 5′-GAAGATGGTGATGGGATTTC-3′.
Western blot
A total of 5 × 106 cells were lysed using ice-cold lysis buffer (MPER, Thermo Scientific) containing 1 mM phenylmethylsulfonyl fluoride, phosphatase inhibitor cocktail (Roche), and protease inhibitor cocktail (Nacalai Tesque), followed by centrifugation at 13,000 rpm for 15 min at 4°C. The cell lysates were mixed with an equal volume of twofold concentrated sample buffer (Bio-Rad Laboratories) containing β-mercaptoethanol (Nacalai Tesque) and were treated for 5 min at 100°C. Western blot was performed as described previously (41).
Isolation of primary CD4 T cell and ATL cell samples
This project was approved by the Institutional Review Board of Kyoto University Hospital, and all participants provided written informed consent in accordance with the Declaration of Helsinki. Peripheral blood mononuclear cells were isolated from healthy donors and 10 ATL patients through Ficoll gradient centrifugation. Peripheral blood mononuclear cells isolated from two acute-type ATL patients through Ficoll gradient centrifugation were used for Western blot (Fig. 5F). These two patients’ characteristics are provided in table S3. The CD4+ T cell population was isolated using an EasySep Human CD4+ T Cell Enrichment Kit (STEMCELL Technologies).
In vitro biochemical assays for TDP1 activity
All single-standard DNA substrates share the commons sequence of 5′-TCCGTTGAAGCCTGCTTT-3′. Oligonucleotides were synthesized in the reversed direction, as described previously (42). CBV was prepared according to the literature (43). Total cell lysates from DT40 Tdp1−/− and Tdp1−/− hTDP1 cells were prepared in the same manner as previously described (44). The protein concentration of lysates was determined by Bradford protein assay (Quick Start protein assay, Bio-Rad Laboratories). To prepare DNA substrate, oligonucleotides with 3-phosphotyrosine and CBV linkages synthesized were incubated with [γ-32P]adenosine triphosphate (NEG502A, PerkinElmer Life Sciences) and T4 polynucleotide kinase (Takara) for 5′-end labeling. After being purified using a nucleotide removal kit (Qiagen), 1 nM labeled oligonucleotide was incubated with cell lysates at 25°C for 15 min. Samples were separated by 16% denaturing polyacrylamide gel electrophoresis (7 M, urea). Dried gel was exposed on the imaging plate (Fujifilm) and then scanned in Fuji Bas 2500 system. Quantification was performed by Image Gauge software (Fujifilm).
RNA interference
To evaluate the role of TDP1 in the cell sensitivity to ABC, we knocked down TDP1 in Jurkat cells using specific siRNA. Transient TDP1 knockdown was achieved using the siGENOME SMARTpool (Thermo Scientific). A total of 5 × 106 Jurkat cells were electroporated with either nontargeted siRNA (Stealth RNAi NEGATIVE CONTROL, Invitrogen) or hTDP1-specific siRNA (20 pmol/μl) using the Amaxa Cell Nucleofector System (Lonza). Knockdown efficiency was determined by quantitative PCR 24 hours after transfection and Western blot 48 hours after transfection.
Plasmid constructs
Plasmids encoding FLAG-TDP1 wild type, FLAG-TDP1 H263A, and FLAG-TDP1 H493R were a gift from Y. Pommier (NIH). H263A and H493R are inactive TDP1 mutants; the latter causes spinocerebellar ataxia with axonal neuropathy (SCAN1). Wild-type, H263A, and H493R FLAG-TDP1 were cloned into the lentiviral vector CSII-CMV-MCS-IRES-hrGFP.
Generation of MT-2 cells reconstituted with hTDP1
Wild-type, H263A, and H493R FLAG-TDP1–expressing lentiviral vectors were prepared as described previously (41). MT-2 cells were infected with each viral vector. Confirmation of infectivity was based on human recombinant green fluorescent protein (hrGFP) expression, and the expression level of TDP1 in each group of infected MT-2 cells was verified by Western blot with rabbit anti-TDP1.
Preparation of ATL model mice
The rapid tumor formation by the ATL cell line in NOD/SCID mice has been previously established (38, 45). Four-week-old NOD/SCID mice were purchased from CLEA Japan. Fourteen mice were anesthetized with ether, and 4 × 107 ED-40515(−) cells were subcutaneously inoculated into the posterior cervical lesion. Beginning 2 weeks after inoculation, the long and short axes were measured weekly. The tumor volume was approximated as (long axis) × (short axis)2. All experiments were performed under the approved protocols of the Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University.
Administration of ABC
ABC dissolved in water was administered orally to mice at a dose of 75 mg/kg per day on weekdays from day 0 to day 20. The control mice received an equivalent amount of water.
Analysis of combined treatment effects
The synergistic effects of the combined treatments were analyzed using the Chou-Talalay method (46). The CI of each combined treatment was calculated using CompuSyn software, and CIs of 0.3 to 0.7, 0.1 to 0.3, and <0.1 were defined as synergism, strong synergism, and very strong synergism, respectively (47).
Statistical analysis
The statistical significance of the differences between the drug-treated and control cultures was determined using Student’s t test or the χ2 test. The statistical significance of the survival rates was determined using the Wilcoxon test.
Supplementary Material
Acknowledgments
We thank M. Maeda for providing the ED-40515(−), ED-40515(+), and ATL-43T cells, and M. Takada (Kyoto University) for providing the Fancc−/−, Fancg−/−, Snm1a1b−/−, and Artemis−/− DT40 cells. We also thank K. Imada for his suggestion regarding the use of the mouse xenograft model. We are grateful to H. Mitsuya, T. Watanabe, and J. Murai for helpful discussions. We also thank J. Hejna for English language editing. Funding: This work was partially supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI program; grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; and Health and Labour Sciences research grants from the Ministry of Health, Labour, and Welfare of Japan. Author contributions: K.T. and M.K. designed and performed most of the experiments and wrote the manuscript. Y.T., F.I., T.S., K.N., M.S., K.H., J.Y., S.I., and K.I. performed some of the experiments, and K.S., M.H., K.S., and N.K. analyzed the data and interpreted the results. H.S. and S.T. participated in the experimental design and provided materials. A.T.-K. participated in the experimental design, supervised and funded the project, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/3/e1400203/DC1
Fig. S1. ABC selectively kills HTLV-1–infected and ATL cell lines.
Fig. S2. Cell counting assays showing ABC cytotoxicity to MT-2 cells.
Fig. S3. ABC-induced apoptosis.
Fig. S4. Relative sensitivities to six NRTIs of DT40 cells deficient in the indicated gene.
Fig. S5. The relative sensitivity of candidate gene-deficient DT40 cells to the six individual NRTIs and the level of the mRNA expression in ATL cells.
Fig. S6. Ectopic expression of TDP1WT, TDP1H263A, and TDP1H493R in MT-2 cells.
Fig. S7. ABC and AZT cytotoxicity in MT-2/TDP1WT cells.
Fig. S8. CPT11 and veliparib enhance the lethality of ABC on ATL cells.
Table S1. Inhibitory concentration (IC50) values of ABC for each cell line.
Table S2. Isogenic mutant chicken DT40 cell lines used in this study.
Table S3. Characteristics of two ATL patients.
Table S4. List of HTLV-1–infected and ATL cell lines.
REFERENCES AND NOTES
- 1.Uchiyama T., Yodoi J., Sagawa K., Takatsuki K., Uchino H., Adult T-cell leukemia: Clinical and hematologic features of 16 cases. Blood 50, 481–492 (1977). [PubMed] [Google Scholar]
- 2.Tsukasaki K., Hermine O., Bazarbachi A., Ratner L., Ramos J. C., Harrington W. Jr, O’Mahony D., Janik J. E., Bittencourt A. L., Taylor G. P., Yamaguchi K., Utsunomiya A., Tobinai K., Watanabe T., Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: A proposal from an international consensus meeting. J. Clin. Oncol. 27, 453–459 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsuoka M., Jeang K. T., Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 7, 270–280 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Yamada Y., Tomonaga M., Fukuda H., Hanada S., Utsunomiya A., Tara M., Sano M., Ikeda S., Takatsuki K., Kozuru M., Araki K., Kawano F., Niimi M., Tobinai K., Hotta T., Shimoyama M. The Lymphoma Study Group of The Japan Clinical Onocology Group (1994–96) , A new G-CSF-supported combination chemotherapy, LSG15, for adult T-cell leukaemia-lymphoma: Japan Clinical Oncology Group Study 9303. Br. J. Haematol. 113, 375–382 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Tsukasaki K., Utsunomiya A., Fukuda H., Shibata T., Fukushima T., Takatsuka Y., Ikeda S., Masuda M., Nagoshi H., Ueda R., Tamura K., Sano M., Momita S., Yamaguchi K., Kawano F., Hanada S., Tobinai K., Shimoyama M., Hotta T., Tomonaga M. Japan Clinical Oncology Group Study JCOG9801 , VCAP-AMP-VECP compared with biweekly CHOP for adult T-cell leukemia-lymphoma: Japan Clinical Oncology Group Study JCOG9801. J. Clin. Oncol. 25, 5458–5464 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Hishizawa M., Kanda J., Utsunomiya A., Taniguchi S., Eto T., Moriuchi Y., Tanosaki R., Kawano F., Miyazaki Y., Masuda M., Nagafuji K., Hara M., Takanashi M., Kai S., Atsuta Y., Suzuki R., Kawase T., Matsuo K., Nagamura-Inoue T., Kato S., Sakamaki H., Morishima Y., Okamura J., Ichinohe T., Uchiyama T., Transplantation of allogeneic hematopoietic stem cells for adult T-cell leukemia: A nationwide retrospective study. Blood 116, 1369–1376 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Gill P. S., Harrington W. Jr, Kaplan M. H., Ribeiro R. C., Bennett J. M., Liebman H. A., Bernstein-Singer M., Espina B. M., Cabral L., Allen S., Kornblau S., Pike M. C., Levine A. M., Treatment of adult T-cell leukemia–lymphoma with a combination of interferon alfa and zidovudine. N. Engl. J. Med. 332, 1744–1748 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Hermine O., Bouscary D., Gessain A., Turlure P., Leblond V., Franck N., Buzyn-Veil A., Rio B., Macintyre E., Dreyfus F., Bazarbachi A., Brief report: Treatment of adult T-cell leukemia–lymphoma with zidovudine and interferon alfa. N. Engl. J. Med. 332, 1749–1751 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Bazarbachi A., Plumelle Y., Carlos Ramos J., Tortevoye P., Otrock Z., Taylor G., Gessain A., Harrington W., Panelatti G., Hermine O., Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J. Clin. Oncol. 28, 4177–4183 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Bazarbachi A., Nasr R., El-Sabban M. E., Mahe A., Mahieux R., Gessain A., Darwiche N., Dbaibo G., Kersual J., Zermati Y., Dianoux L., Chelbi-Alix M. K., de The H., Hermine O., Evidence against a direct cytotoxic effect of α interferon and zidovudine in HTLV-I associated adult T cell leukemia/lymphoma. Leukemia 14, 716–721 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Mitsuya H., Weinhold K. J., Furman P. A., Clair M. H. St, Lehrman S. N., Gallo R. C., Bolognesi D., Barry D. W., Broder S., 3′-Azido-3′-deoxythymidine (BW A509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. U.S.A. 82, 7096–7100 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts J. D., Bebenek K., Kunkel T. A., The accuracy of reverse transcriptase from HIV-1. Science 242, 1171–1173 (1988). [DOI] [PubMed] [Google Scholar]
- 13.Caldecott K. W., Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Miao Z. H., Agama K., Sordet O., Povirk L., Kohn K. W., Pommier Y., Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes. DNA Repair 5, 1489–1494 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Katyal S., el-Khamisy S. F., Russell H. R., Li Y., Ju L., Caldecott K. W., McKinnon P. J., TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 26, 4720–4731 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P. N., Sweet D. E., McDowell J. A., Symonds W., Lou Y., Hetherington S., LaFon S., Safety and pharmacokinetics of abacavir (1592U89) following oral administration of escalating single doses in human immunodeficiency virus type 1-infected adults. Antimicrob. Agents Chemother. 43, 603–608 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faletto M. B., Miller W. H., Garvey E. P., Clair M. H. St, Daluge S. M., Good S. S., Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob. Agents Chemother. 41, 1099–1107 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDowell J. A., Chittick G. E., Ravitch J. R., Polk R. E., Kerkering T. M., Stein D. S., Pharmacokinetics of [14C]abacavir, a human immunodeficiency virus type 1 (HIV-1) reverse transcriptase inhibitor, administered in a single oral dose to HIV-1-infected adults: A mass balance study. Antimicrob. Agents Chemother. 43, 2855–2861 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDowell J. A., Lou Y., Symonds W. S., Stein D. S., Multiple-dose pharmacokinetics and pharmacodynamics of abacavir alone and in combination with zidovudine in human immunodeficiency virus-infected adults. Antimicrob. Agents Chemother. 44, 2061–2067 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis W., Kohler J. J., Hosseini S. H., Haase C. P., Copeland W. C., Bienstock R. J., Ludaway T., McNaught J., Russ R., Stuart T., Santoianni R., Antiretroviral nucleosides, deoxynucleotide carrier and mitochondrial DNA: Evidence supporting the DNA pol γ hypothesis. AIDS 20, 675–684 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodarzi A. A., Jeggo P. A., The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 82, 1–45 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Maede Y., Shimizu H., Fukushima T., Kogame T., Nakamura T., Miki T., Takeda S., Pommier Y., Murai J., Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 13, 214–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamagishi M., Nakano K., Miyake A., Yamochi T., Kagami Y., Tsutsumi A., Matsuda Y., Sato-Otsubo A., Muto S., Utsunomiya A., Yamaguchi K., Uchimaru K., Ogawa S., Watanabe T., Polycomb-mediated loss of miR-31 activates NIK-dependent NF-κB pathway in adult T cell leukemia and other cancers. Cancer Cell 21, 121–135 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Comeaux E. Q., Cuya S. M., Kojima K., Jafari N., Wanzeck K. C., Mobley J. A., Bjornsti M. A., van Waardenburg R. C., Tyrosyl-DNA phosphodiesterase I catalytic mutants reveal an alternative nucleophile that can catalyze substrate cleavage. J. Biol. Chem. 290, 6203–6214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lebedeva N. A., Rechkunova N. I., El-Khamisy S. F., Lavrik O. I., Tyrosyl-DNA phosphodiesterase 1 initiates repair of apurinic/apyrimidinic sites. Biochimie 94, 1749–1753 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pommier Y., Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 8, 82–95 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang S. Y., Murai J., Dalla Rosa I., Dexheimer T. S., Naumova A., Gmeiner W. H., Pommier Y., TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 41, 7793–7803 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das B. B., Huang S. Y., Murai J., Rehman I., Ame J. C., Sengupta S., Das S. K., Majumdar P., Zhang H., Biard D., Majumder H. K., Schreiber V., Pommier Y., PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res., 42, 4435–4449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murai J., Zhang Y., Morris J., Ji J., Takeda S., Doroshow J. H., Pommier Y., Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 349, 408–416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parker W. B., White E. L., Shaddix S. C., Ross L. J., Buckheit R. W. Jr, Germany J. M., Secrist J. A. III, Vince R., Shannon W. M., Mechanism of inhibition of human immunodeficiency virus type 1 reverse transcriptase and human DNA polymerases α, β, and γ by the 5′-triphosphates of carbovir, 3′-azido-3′-deoxythymidine, 2′,3′-dideoxyguanosine and 3′-deoxythymidine. A novel RNA template for the evaluation of antiretroviral drugs. J. Biol. Chem. 266, 1754–1762 (1991). [PubMed] [Google Scholar]
- 31.Nickel W., Austermann S., Bialek G., Grosse F., Interactions of azidothymidine triphosphate with the cellular DNA polymerases α, δ, and ϵ and with DNA primase. J. Biol. Chem. 267, 848–854 (1992). [PubMed] [Google Scholar]
- 32.Murai J., Huang S. Y., Das B. B., Dexheimer T. S., Takeda S., Pommier Y., Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem. 287, 12848–12857 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao R., Das B. B., Chatterjee R., Abaan O. D., Agama K., Matuo R., Vinson C., Meltzer P. S., Pommier Y., Epigenetic and genetic inactivation of tyrosyl-DNA-phosphodiesterase 1 (TDP1) in human lung cancer cells from the NCI-60 panel. DNA Repair 13, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar P. N., Sweet D. E., McDowell J. A., Symonds W., Lou Y., Hetherington S., LaFon S., Safety and pharmacokinetics of abacavir (1592U89) following oral administration of escalating single doses in human immunodeficiency virus type 1-infected adults. Antimicrob. Agents Chemother. 43, 603–608 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwanaga M., Watanabe T., Utsunomiya A., Okayama A., Uchimaru K., Koh K. R., Ogata M., Kikuchi H., Sagara Y., Uozumi K., Mochizuki M., Tsukasaki K., Saburi Y., Yamamura M., Tanaka J., Moriuchi Y., Hino S., Kamihira S., Yamaguchi K., Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: A nationwide prospective study in Japan. Blood 116, 1211–1219 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Maeda M., Shimizu A., Ikuta K., Okamoto H., Kashihara M., Uchiyama T., Honjo T., Yodoi J., Origin of human T-lymphotrophic virus I-positive T cell lines in adult T cell leukemia. Analysis of T cell receptor gene rearrangement. J. Exp. Med. 162, 2169–2174 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arima N., Kamio M., Imada K., Hori T., Hattori T., Tsudo M., Okuma M., Uchiyama T., Pseudo-high affinity interleukin 2 (IL-2) receptor lacks the third component that is essential for functional IL-2 binding and signaling. J. Exp. Med. 176, 1265–1272 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imada K., Takaori-Kondo A., Akagi T., Shimotohno K., Sugamura K., Hattori T., Yamabe H., Okuma M., Uchiyama T., Tumorigenicity of human T-cell leukemia virus type I-infected cell lines in severe combined immunodeficient mice and characterization of the cells proliferating in vivo. Blood 86, 2350–2357 (1995). [PubMed] [Google Scholar]
- 39.Koga H., Imada K., Ueda M., Hishizawa M., Uchiyama T., Identification of differentially expressed molecules in adult T-cell leukemia cells proliferating in vivo. Cancer Sci. 95, 411–417 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poiesz B. J., Ruscetti F. W., Gazdar A. F., Bunn P. A., Minna J. D., Gallo R. C., Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 77, 7415–7419 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakamoto T., Kobayashi M., Tada K., Shinohara M., Io K., Nagata K., Iwai F., Takiuchi Y., Arai Y., Yamashita K., Shindo K., Kadowaki N., Koyanagi Y., Takaori-Kondo A., CKIP-1 is an intrinsic negative regulator of T-cell activation through an interaction with CARMA1. PLOS One 9, e85762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Claeboe C. D., Gao R., Hecht S. M., 3′-Modified oligonucleotides by reverse DNA synthesis. Nucleic Acids Res. 31, 5685–5691 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vince R., Hua M., Synthesis of carbovir and abacavir from a carbocyclic precursor. Curr. Protoc. Nucleic Acid Chem. Chapter 14, Unit 14.4 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Marchand C., Huang S. Y., Dexheimer T. S., Lea W. A., Mott B. T., Chergui A., Naumova A., Stephen A. G., Rosenthal A. S., Rai G., Murai J., Gao R., Maloney D. J., Jadhav A., Jorgensen W. L., Simeonov A., Pommier Y., Biochemical assays for the discovery of TDP1 inhibitors. Mol. Cancer Ther. 13, 2116–2126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kondo A., Imada K., Hattori T., Yamabe H., Tanaka T., Miyasaka M., Okuma M., Uchiyama T., A model of in vivo cell proliferation of adult T-cell leukemia. Blood 82, 2501–2509 (1993). [PubMed] [Google Scholar]
- 46.Chou T. C., Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Chou T. C., Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58, 621–681 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Takata M., Sasaki M. S., Sonoda E., Morrison C., Hashimoto M., Utsumi H., Yamaguchi-Iwai Y., Shinohara A., Takeda S., Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 17, 5497–5508 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adachi N., Ishino T., Ishii Y., Takeda S., Koyama H., DNA ligase IV-deficient cells are more resistant to ionizing radiation in the absence of Ku70: Implications for DNA double-strand break repair. Proc. Natl. Acad. Sci. U.S.A. 98, 12109–12113 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukushima T., Takata M., Morrison C., Araki R., Fujimori A., Abe M., Tatsumi K., Jasin M., Dhar P. K., Sonoda E., Chiba T., Takeda S., Genetic analysis of the DNA-dependent protein kinase reveals an inhibitory role of Ku in late S–G2 phase DNA double-strand break repair. J. Biol. Chem. 276, 44413–44418 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Andoh T., Ishida R., Catalytic inhibitors of DNA topoisomerase II. Biochim. Biophys. Acta 1400, 155–171 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Nakamura K., Sakai W., Kawamoto T., Bree R. T., Lowndes N. F., Takeda S., Taniguchi Y., Genetic dissection of vertebrate 53BP1: A major role in non-homologous end joining of DNA double strand breaks. DNA Repair 5, 741–749 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Takao N., Kato H., Mori R., Morrison C., Sonada E., Sun X., Shimizu H., Yoshioka K., Takeda S., Yamamoto K., Disruption of ATM in p53-null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene 18, 7002–7009 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Saberi A., Nakahara M., Sale J. E., Kikuchi K., Arakawa H., Buerstedde J. M., Yamamoto K., Takeda S., Sonoda E., The 9-1-1 DNA clamp is required for immunoglobulin gene conversion. Mol. Cell. Biol. 28, 6113–6122 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sonoda E., Zhao G. Y., Kohzaki M., Dhar P. K., Kikuchi K., Redon C., Pilch D. R., Bonner W. M., Nakano A., Watanabe M., Nakayama T., Takeda S., Takami Y., Collaborative roles of γH2AX and the Rad51 paralog Xrcc3 in homologous recombinational repair. DNA Repair 6, 280–292 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Yamaguchi-Iwai Y., Sonoda E., Buerstedde J. M., Bezzubova O., Morrison C., Takata M., Shinohara A., Takeda S., Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 18, 6430–6435 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bezzubova O., Silbergleit A., Yamaguchi-Iwai Y., Takeda S., Buerstedde J. M., Reduced x-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell 89, 185–193 (1997). [DOI] [PubMed] [Google Scholar]
- 58.Nakahara M., Sonoda E., Nojima K., Sale J. E., Takenaka K., Kikuchi K., Taniguchi Y., Nakamura K., Sumitomo Y., Bree R. T., Lowndes N. F., Takeda S., Genetic evidence for single-strand lesions initiating Nbs1-dependent homologous recombination in diversification of Ig V in chicken B lymphocytes. PLOS Genet. 5, e1000356 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takata M., Sasaki M. S., Tachiiri S., Fukushima T., Sonoda E., Schild D., Thompson L. H., Takeda S., Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol. 21, 2858–2866 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamaguchi-Iwai Y., Sonoda E., Sasaki M. S., Morrison C., Haraguchi T., Hiraoka Y., Yamashita Y. M., Yagi T., Takata M., Price C., Kakazu N., Takeda S., Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 18, 6619–6629 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin R. W., Orelli B. J., Yamazoe M., Minn A. J., Takeda S., Bishop D. K., RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 67, 9658–9665 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Hatanaka A., Yamazoe M., Sale J. E., Takata M., Yamamoto K., Kitao H., Sonoda E., Kikuchi K., Yonetani Y., Takeda S., Similar effects of Brca2 truncation and Rad51 paralog deficiency on immunoglobulin V gene diversification in DT40 cells support an early role for Rad51 paralogs in homologous recombination. Mol. Cell. Biol. 25, 1124–1134 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ross W., Rowe T., Glisson B., Yalowich J., Liu L., Role of topoisomerase II in mediating epipodophyllotoxin-induced DNA cleavage. Cancer Res. 44, 5857–5860 (1984). [PubMed] [Google Scholar]
- 64.Yin Y., Seifert A., Chua J. S., Maure J. F., Golebiowski F., Hay R. T., SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 26, 1196–1208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oestergaard V. H., Pentzold C., Pedersen R. T., Iosif S., Alpi A., Bekker-Jensen S., Mailand N., Lisby M., RNF8 and RNF168 but not HERC2 are required for DNA damage-induced ubiquitylation in chicken DT40 cells. DNA Repair 11, 892–905 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Yamashita Y. M., Okada T., Matsusaka T., Sonoda E., Zhao G. Y., Araki K., Tateishi S., Yamaizumi M., Takeda S., RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. EMBO J. 21, 5558–5566 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fortune J. M., Osheroff N., Topoisomerase II as a target for anticancer drugs: When enzymes stop being nice. Prog. Nucleic Acid Res. Mol. Biol. 64, 221–253 (2000). [DOI] [PubMed] [Google Scholar]
- 68.Zeng Z., Cortes-Ledesma F., El Khamisy S. F., Caldecott K. W., TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 286, 403–409 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nakamura K., Kogame T., Oshiumi H., Shinohara A., Sumitomo Y., Agama K., Pommier Y., Tsutsui K. M., Tsutsui K., Hartsuiker E., Ogi T., Takeda S., Taniguchi Y., Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLOS Genet. 6, e1000828 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hirano S., Yamamoto K., Ishiai M., Yamazoe M., Seki M., Matsushita N., Ohzeki M., Yamashita Y. M., Arakawa H., Buerstedde J. M., Enomoto T., Takeda S., Thompson L. H., Takata M., Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. EMBO J. 24, 418–427 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshikiyo K., Kratz K., Hirota K., Nishihara K., Takata M., Kurumizaka H., Horimoto S., Takeda S., Jiricny J., KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc. Natl. Acad. Sci. U.S.A. 107, 21553–21557 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamamoto K., Ishiai M., Matsushita N., Arakawa H., Lamerdin J. E., Buerstedde J. M., Tanimoto M., Harada M., Thompson L. H., Takata M., Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol. Cell. Biol. 23, 5421–5430 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Murai J., Yang K., Dejsuphong D., Hirota K., Takeda S., D’Andrea A. D., The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Mol. Cell. Biol. 31, 2462–2469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishiai M., Kimura M., Namikoshi K., Yamazoe M., Yamamoto K., Arakawa H., Agematsu K., Matsushita N., Takeda S., Buerstedde J. M., Takata M., DNA cross-link repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Mol. Cell. Biol. 24, 10733–10741 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tano K., Nakamura J., Asagoshi K., Arakawa H., Sonoda E., Braithwaite E. K., Prasad R., Buerstedde J. M., Takeda S., Watanabe M., Wilson S. H., Interplay between DNA polymerases β and lambda in repair of oxidation DNA damage in chicken DT40 cells. DNA Repair 6, 869–875 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoshimura M., Kohzaki M., Nakamura J., Asagoshi K., Sonoda E., Hou E., Prasad R., Wilson S. H., Tano K., Yasui A., Lan L., Seki M., Wood R. D., Arakawa H., Buerstedde J. M., Hochegger H., Okada T., Hiraoka M., Takeda S., Vertebrate POLQ and POLβ cooperate in base excision repair of oxidative DNA damage. Mol. Cell 24, 115–125 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kawamoto T., Araki K., Sonoda E., Yamashita Y. M., Harada K., Kikuchi K., Masutani C., Hanaoka F., Nozaki K., Hashimoto N., Takeda S., Dual roles for DNA polymerase η in homologous DNA recombination and translesion DNA synthesis. Mol. Cell 20, 793–799 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Sonoda E., Okada T., Zhao G. Y., Tateishi S., Araki K., Yamaizumi M., Yagi T., Verkaik N. S., van Gent D. C., Takata M., Takeda S., Multiple roles of Rev3, the catalytic subunit of polζ in maintaining genome stability in vertebrates. EMBO J. 22, 3188–3197 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matsuzaki Y., Adachi N., Koyama H., Vertebrate cells lacking FEN-1 endonuclease are viable but hypersensitive to methylating agents and H2O2. Nucleic Acids Res. 30, 3273–3277 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Okada T., Sonoda E., Yamashita Y. M., Koyoshi S., Tateishi S., Yamaizumi M., Takata M., Ogawa O., Takeda S., Involvement of vertebrate Polκ in Rad18-independent postreplication repair of UV damage. J. Biol. Chem. 277, 48690–48695 (2002). [DOI] [PubMed] [Google Scholar]
- 81.Kikuchi K., Taniguchi Y., Hatanaka A., Sonoda E., Hochegger H., Adachi N., Matsuzaki Y., Koyama H., van Gent D. C., Jasin M., Takeda S., Fen-1 facilitates homologous recombination by removing divergent sequences at DNA break ends. Mol. Cell. Biol. 25, 6948–6955 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kohzaki M., Hatanaka A., Sonoda E., Yamazoe M., Kikuchi K., Trung N. Vu, Szuts D., Sale J. E., Shinagawa H., Watanabe M., Takeda S., Cooperative roles of vertebrate Fbh1 and Blm DNA helicases in avoidance of crossovers during recombination initiated by replication fork collapse. Mol. Cell. Biol. 27, 2812–2820 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Imamura O., Fujita K., Shimamoto A., Tanabe H., Takeda S., Furuichi Y., Matsumoto T., Bloom helicase is involved in DNA surveillance in early S phase in vertebrate cells. Oncogene 20, 1143–1151 (2001). [DOI] [PubMed] [Google Scholar]
- 84.Imamura O., Fujita K., Itoh C., Takeda S., Furuichi Y., Matsumoto T., Werner and Bloom helicases are involved in DNA repair in a complementary fashion. Oncogene 21, 954–963 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/3/e1400203/DC1
Fig. S1. ABC selectively kills HTLV-1–infected and ATL cell lines.
Fig. S2. Cell counting assays showing ABC cytotoxicity to MT-2 cells.
Fig. S3. ABC-induced apoptosis.
Fig. S4. Relative sensitivities to six NRTIs of DT40 cells deficient in the indicated gene.
Fig. S5. The relative sensitivity of candidate gene-deficient DT40 cells to the six individual NRTIs and the level of the mRNA expression in ATL cells.
Fig. S6. Ectopic expression of TDP1WT, TDP1H263A, and TDP1H493R in MT-2 cells.
Fig. S7. ABC and AZT cytotoxicity in MT-2/TDP1WT cells.
Fig. S8. CPT11 and veliparib enhance the lethality of ABC on ATL cells.
Table S1. Inhibitory concentration (IC50) values of ABC for each cell line.
Table S2. Isogenic mutant chicken DT40 cell lines used in this study.
Table S3. Characteristics of two ATL patients.
Table S4. List of HTLV-1–infected and ATL cell lines.