

Notch inhibitors cause cancer growth in tumors expressing PIGFβ protein.
Keywords: Angiogenesis, Notch signaling, tumor growth, PlGF, vascular, remodeling
Abstract
Inhibition of Dll4 (delta-like ligand 4)–Notch signaling–mediated tumor angiogenesis is an attractive approach in cancer therapy. However, inhibition of Dll4-Notch signaling has produced different effects in various tumors, and no biomarkers are available for predicting the anti–Dll4-Notch–associated antitumor activity. We show that human and mouse tumor cell–derived placental growth factor (PlGF) is a key determinant of the Dll4-Notch–induced vascular remodeling and tumor growth. In natural PlGF-expressing human tumors, inhibition of Dll4-Notch signaling markedly accelerated tumor growth by increasing blood perfusion in nonleaking tumor vasculatures. Conversely, in PlGF-negative tumors, Dll4 inhibition suppressed tumor growth by the formation of nonproductive and leaky vessels. Surprisingly, genetic inactivation of vascular endothelial growth factor receptor 1 (VEGFR1) completely abrogated the PlGF-modulated vascular remodeling and tumor growth, indicating a crucial role for VEGFR1-mediated signals in modulating Dll4-Notch functions. These findings provide mechanistic insights on PlGF-VEGFR1 signaling in the modulation of the Dll4-Notch pathway in angiogenesis and tumor growth, and have therapeutic implications of PlGF as a biomarker for predicting the antitumor benefits of Dll4 and Notch inhibitors.
INTRODUCTION
Tumor vasculatures exhibit distinctive structural and functional features (1, 2) that include (i) high leakiness, (ii) poor coverage of perivascular cells, (iii) irregular vascular networks, (iv) excessive vascular sprouting and fusion, (v) sluggish blood flow, (vi) mosaic cell types in the vessel wall, and (vii) incomplete basement membrane around the endothelium. These irregular vascular networks constantly experience growth, remodeling, and regression depending on the presence of various angiogenic factors and cytokines whose expression levels are coordinately regulated by tumor growth, necrosis, and regression. In the tumor microenvironment, vascular density, structure, architecture, and function are the key determinants of tumor growth, invasion, metastasis, and antiangiogenic drug responses (3–5).
Vascular endothelial growth factor (VEGF) is highly expressed in almost all tumors as compared with their healthy counterpart tissues (6). In addition to genetic mutations of oncogenes and tumor suppressor genes, tissue hypoxia, stromal fibroblasts, and infiltration of inflammatory cells are crucial regulatory elements for high VEGF expression in tumors. It is generally believed that VEGF binds to VEGF receptor 2 (VEGFR2) to trigger angiogenic and vascular permeability responses (7, 8). Despite copious knowledge about the VEGF-VEGFR2 signaling pathway in tumor angiogenesis, little is known about the functions of the two other members of the VEGF family, that is, placental growth factor (PlGF) and VEGF-B, in the regulation of tumor angiogenesis. PlGF and VEGF-B are VEGFR1 binding ligands, and they lack the ability to interact with VEGFR2 (9, 10). Both positive and negative functions of PlGF and VEGF-B in the regulation of tumor angiogenesis have been described. On the basis of numerous experimental data, it is generally concluded that complex mechanisms are involved in the functional activity of VEGFR1-exclusive binding ligands in different model systems (9, 10). The spatiotemporal relation between VEGFR1-exclusive binding ligands and other angiogenic factors is probably the key determinant of their modes of actions. For example, positive and negative modulations of VEGF functions by PlGF have been ascribed in various tumor models (11–17).
VEGF stimulates angiogenic sprouts in close collaboration with the Notch-signaling pathways (18–20). Whereas VEGF stimulates vessel growth, delta-like ligand 4 (Dll4)–Notch signaling prevents excessive sprouting by switching endothelial cells to a quiescent phenotype (18–20). Inhibition of Dll4-Notch signaling increases the density and tortuosity of tumor microvessels that are poorly perfused (20). Despite increases in the number of nonproductive vessels, Dll4-Notch inhibitors suppress tumor growth. Thus, Notch inhibition provides an attractive approach in cancer therapy (19–22). Paradoxically, activation of Dll4-Notch signaling has also been demonstrated to inhibit angiogenesis and tumor growth (23, 24). Although these controversial claims even in the same study need to be resolved, the relation between Dll4-Notch signaling and other factors has not been carefully studied. Here, we report our unexpected findings that PlGF serves as a biomarker that predicts therapeutic outcome with Dll4-Notch inhibitors. In PlGF-expressing human and mouse tumors, inhibition of Dll4 and Notch signaling accelerates tumor growth by increasing blood perfusion in relatively normalized angiogenic vessels. Inversely, inhibition of Dll4-Notch signaling suppresses tumor growth in PlGF-negative tumors. Using genetic mouse model approaches, we further demonstrate that activation of VEGFR1 is critically required for the PlGF-modulated opposing effects of Notch inhibition on vascular functions and tumor growth.
RESULTS
PlGF expression determines opposing effects of Dll4-Notch inhibition on human tumor growth and vascular remodeling
PlGF and Dll4 have been known to remodel disorganized tumor vasculatures toward a normal phenotype (17, 25–27). To study the functional interactions between PlGF and the Dll4-Notch signaling pathway, we investigated tumor growth and vascular structures in PlGF-positive and PlGF-negative human tumors. Natural JE-3 and BeWo choriocarcinomas expressed high levels of PlGF protein, whereas the human MDA-MB-231 breast carcinoma lacked a detectable level of PlGF expression (fig. S1A). Similar to human MDA-MB-231 breast carcinomas, human Hep3B hepatocellular carcinomas (HCCs) also lacked a detectable level of PlGF expression (fig. S1A). Surprisingly, treatment of JE-3 and BeWo tumor–bearing mice with a γ-secretase inhibitor (DAPT) or an anti-human Dll4 neutralizing antibody (Dll4 blockade) resulted in accelerated rather than delayed tumor growth (Fig. 1, A and B). In contrast, treated MDA-MB-231 and Hep3B tumor–bearing mice showed an inhibition of tumor growth (Fig. 1C and fig. S2). These findings show that PlGF expression might facilitate the growth of Dll4 and Notch inhibitor–treated tumors. Thus, PlGF in tumors not only confer drug resistance to Dll4-Notch inhibitors but also accelerates tumor growth.
Fig. 1. Dll4-Notch inhibition–altered vascular changes and tumor growth in high PlGF– and low PlGF–expressing human cancers.
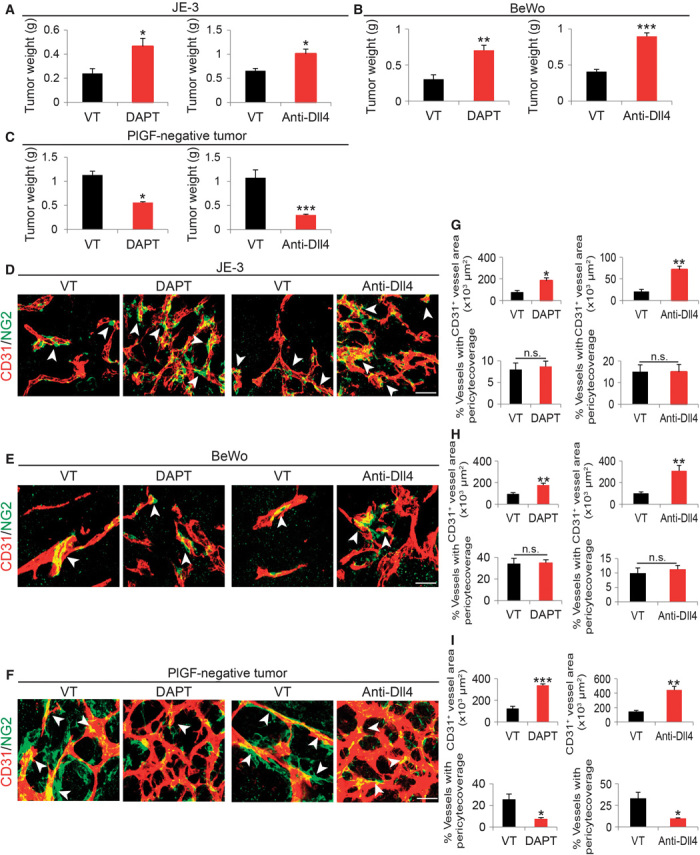
(A to C) Tumor weight of vehicle (VT)–, DAPT-, and Dll4 blockade–treated JE-3, BeWo, and MDA-MB-231 tumors (n = 4 to 6 mice per group). (D to F) Representative images of CD31+ tumor vessels in vehicle-, DAPT-, and anti-Dll4 antibody–treated JE-3, BeWo, and MDA-MB-231 tumors. Red, CD31-positive signals; green, NG2-positive signals; yellow, overlapping signals. Arrowheads point to microvessel-associated pericytes. Scale bar, 50 μm. (G to I) Quantification of microvessel density and pericyte coverage in vehicle-, DAPT-, and anti-Dll4 antibody–treated JE-3, BeWo, and MDA-MB-231 tumors (5 to 10 random fields per group); data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
We next analyzed the vasculatures of Dll4 and Notch inhibitor–treated PlGF-positive and PlGF-negative tumors. Treatments with DAPT and Dll4 blockade resulted in marked increases in microvessel density in both PlGF-positive and PlGF-negative human tumors (Fig. 1, D to I). Quantitative analyses showed that Dll4 blockade produced slightly more potent proangiogenic effects than did DAPT in these human tumor models (Fig. 1, G to I). Perhaps, the longer half-life of the antibody-based drugs was responsible for this slight difference as compared with DAPT. Notably, DAPT and Dll4 blockade did not affect NG2+ pericyte coverages in PlGF-positive JE-3 and BeWo tumors (Fig. 1, G and H). However, these Notch and Dll4 inhibitors significantly ablated pericyte coverage of tumor microvessels in PlGF-negative human MDA-MB-231 and Hep3B tumors (Fig. 1, F and I, and fig. S2, B to D). These data indicate that PlGF opposes the antitumor activities of Dll4 and Notch inhibitors by remodeling tumor vasculatures.
PlGF prevents Dll4-Notch inhibitor–induced vascular disorganization and increases blood perfusion in tumors
In DAPT- and Dll4-treated JE-3 and BeWo tumors, we noticed that the tumor vasculature maintained a relatively normal architecture (Fig. 1, D and E). In sharp contrast, treatment with these inhibitors of PlGF-negative MDA-MB-231 and Hep3B tumors further increased disorganization and tortuosity of tumor microvessels (Fig. 1F and fig. S2B). These findings suggested that PlGF might modulate vascular functions in the tumor microenvironment. To study vascular functions, we injected fluorescently labeled and fixable lysinated dextrans into tumor-bearing mice. A rhodamine-labeled 70-kD dextran was used for measurement of vascular leakage, and the 2000-kD dextran molecules were used for measurement of blood perfusion. Both PlGF-expressing JE-3 and BeWo tumors contained leaky tumor vasculatures, and a substantial amount of 70-kD dextran was extravasated in nontreated tumor tissues (Fig. 2, A and B). Surprisingly, treatments of JE-3 and BeWo tumors with DAPT and Dll4 blockade markedly prevented extravasation of 70-kD dextran in these PlGF-expressing human tumors (Fig. 2, A and B). These findings are consistent with the DAPT- and Dll4-induced vascular normalization in PlGF-expressing tumors. In contrast to JE-3 and BeWo tumors, DAPT and Dll4 blockade drastically induced vascular tortuosity and disorganization in PlGF-negative MDA-MB-231 and Hep3B tumors, leading to significant increases in vascular leakage (Fig. 2C and fig. S2, B and E).
Fig. 2. Blood perfusion and leakiness of Dll4-Notch inhibition–treated human PlGF+ and PlGF– tumors.

(A to C) Left: Representative images of leakage of 70-kD LRD (lysinated rhodamine-labeled dextran) in vehicle (VT)–, DAPT-, and anti-Dll4 antibody–treated human JE-3, BeWo, and MDA-MB-231 tumors. Red, CD31-positive signals; green, extravasated 70-kD LRD; yellow, intravascular 70-kD LRD. Arrowheads indicate extravasated 70-kD LRD. Scale bars, 50 μm. Right: Quantification of extravasated 70-kD LRD (n = 4 to 6 random fields per group); data are presented as means ± SEM. *P < 0.05; **P < 0.01. (D to F) Left: Representative images of perfusion of 2000-kD LRD in vehicle-, DAPT-, and anti-Dll4 antibody–treated human JE-3, BeWo, and MDA-MB-231 tumors. Red, CD31-positive signals; yellow, perfused 2000-kD LRD. Arrowheads indicate positive signals of 2000-kD LRD. Scale bars, 50 μm. Right: Quantification of perfused 2000-kD LRD (n = 4 to 6 random fields per group); data are presented as means ± SEM.
Notably, DAPT and Dll4 blockade treatments significantly increased blood perfusion in PlGF-expressing JE-3 and BeWo tumors, whereas these same drugs induced a nonperfused vasculature in PlGF-negative MDA-MB-231 and Hep3B tumors (Fig. 2, D to F, and fig. S2, B and F). The induction of nonproductive tumor angiogenesis by Notch and Dll4 inhibitors was in agreement with previous observations on Dll4 and Notch inhibitors in various tumor models (19, 20). The reduction in vascular permeability and the increase in blood perfusion caused by DAPT and Dll4 blockade in JE-3 and BeWo tumors indicate that PlGF could reverse the Notch inhibitor–induced nonproductive tumor vasculatures to become functional microvascular networks that support tumor growth. Therefore, PlGF displays a robustly antagonistic effect against Dll4-Notch inhibition–triggered angiogenesis, vascular tortuosity, leakage, and poor perfusion in tumors.
PlGF modulates cell proliferation and apoptosis by alteration of hypoxia in anti-Dll4-Notch–treated human tumors
Modulation of blood perfusion and vascular leakage by PlGF in DAPT- and Dll4-treated human tumors might lead to substantial changes in the tumor microenvironment, such as tumor hypoxia. Vehicle-treated control JE-3 and BeWo tumors exhibited high degrees of tissue hypoxia as measured by the pimonidazole probe (Fig. 3, A and B). Intriguingly, DAPT and Dll4 blockade treatments markedly improved tumor hypoxia in these PlGF-expressing tumors (Fig. 3, A and B). In contrast, DAPT and Dll4 blockade induced severe tumor hypoxia in PlGF-negative MDA-MB-231 and Hep3B tumors (Fig. 3C and fig. S2, B and G). These findings show that PlGF opposes DAPT- and Dll4 blockade–induced tumor hypoxia.
Fig. 3. Hypoxia, proliferation, and apoptosis of Dll4-Notch inhibition–treated human tumors.

(A to C) Left: Representative images of tissue hypoxia in vehicle (VT)–, DAPT-, and anti-Dll4 antibody–treated human JE-3, BeWo, and MDA-MB-231 tumors. Green, pimonidazole-positive signals; blue, 4′,6-diamidino-2-phenylindole (DAPI). Scale bars, 100 μm. Right: Quantification of tumor hypoxia (n = 4 to 6 random fields per group); data are presented as means ± SEM. FITC, fluorescein isothiocyanate. (D to F) Left: Representative images of tumor cell proliferation and apoptosis in vehicle-, DAPT-, and anti-Dll4 antibody–treated human JE-3, BeWo, and MDA-MB-231 tumors. Red, Ki67+ proliferating or cleaved caspase 3+ apoptotic cells; blue, DAPI. Scale bars, 100 μm. Right: Quantification of tumor hypoxia (n = 4 to 6 random fields per group); data are presented as means ± SEM. PA index was calculated using the formula (% apoptotic cells/total cells)/(% Ki67-positive cells/total cells). Arrowheads indicate apoptotic cells. Scale bars, 50 μm. *P < 0.05; **P < 0.01; ***P < 0.001.
The increase in blood perfusion and the improvement of tissue hypoxia by PlGF in DAPT- and Dll4 blockade–treated JE-3 and BeWo tumors suggested that alteration of the tumor microenvironment might affect tumor cell growth rate and cellular apoptosis. Indeed, treatments with DAPT and Dll4 blockade led to significant increases of the Ki67+ proliferating tumor cell population in JE-3 and BeWo tumors, whereas cellular apoptosis in these treated tumors was markedly decreased (Fig. 3, D and E). Both terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining and measurements of the activated or cleaved caspase 3 showed reductions of apoptotic cells in Dll4-Notch inhibitor–treated JE-3 and BeWo tumors (Fig. 3, D and E, and fig. S3, A, B, E, and F). Consequently, proliferation/apoptosis (PA) indexes showed a drift toward a proliferative phenotype. The increase in cell proliferation and the decrease in cellular apoptosis in Dll4-Notch inhibitor–treated JE-3 and BeWo tumors support our notion that these treated tumors grew at higher rates than did vehicle-treated control tumors (Fig. 1). In contrast, treatment of PlGF-negative MDA-MB-231 tumors with DAPT and Dll4 blockade showed completely opposite effects on cell proliferation and apoptosis, tipping the balance toward an apoptotic phenotype (Fig. 3F). To further validate these findings, we used another PlGF-negative human HCC cell line. As shown in figs. S2 and S3, treatment of Hep3B with DAPT and Dll4 blockade also markedly decreased cell proliferation and increased apoptosis (figs. S2, B and H to J, and S3, D and H). These results provide independent evidence that the inhibition of Dll4-Notch signaling in PlGF-negative tumors suppresses tumor growth by tipping the PA indexes toward cell death.
PlGF confers resistance to Dll4-Notch inhibition on mouse tumors
To further validate our findings in human tumor models, we performed gain-of-function experiments in mouse tumors. High expression levels of PlGF protein in Lewis lung carcinoma (LLC) and T241 tumors were confirmed by quantitative enzyme-linked immunosorbent assay (ELISA) (fig. S1B). As described in previous findings (13–15, 17, 26, 27), genetically stable expression of PlGF in mouse LLC and T241 fibrosarcomas led to significant inhibition of tumor growth (Fig. 4, A and B, and fig. S4, A and B). Consistent with inhibition of primary tumor growth, the microvessel densities of PlGF-LLC and PlGF-T241 tumors were significantly reduced as compared with those of vector-transfected LLC and vector-T241 tumors (Fig. 4, C and D, and fig. S4, C and D). These findings support the general view that PlGF acts as a negative regulator of tumor growth and angiogenesis. In PlGF-negative vector-LLC and vector-T241 control tumors, inhibition of Dll4-Notch signaling by DAPT and Dll4 blockade markedly inhibited tumor growth (Fig. 4, A and B, and fig. S4, A and B). Surprisingly, treatments of PlGF-LLC tumor–bearing mice with DAPT and Dll4 blockade did not exhibit any antitumor activity, but rather augmented tumor growth to the rate of nontreated control vector-LLC tumors (Fig. 4, A and B). Similar findings were also obtained with an independent PlGF-T241 fibrosarcoma model that exhibited nearly identical effects of PlGF-triggered resistance to DAPT- and Dll4 blockade–mediated tumor inhibition (fig. S4, A and B). These data show that PlGF completely neutralizes the antitumor effects of DAPT and Dll4 blockade and thus confers resistance to these anticancer drugs.
Fig. 4. Dll4-Notch inhibition–altered tumor growth and microvasculatures in mouse PlGF– and PlGF+ tumors.
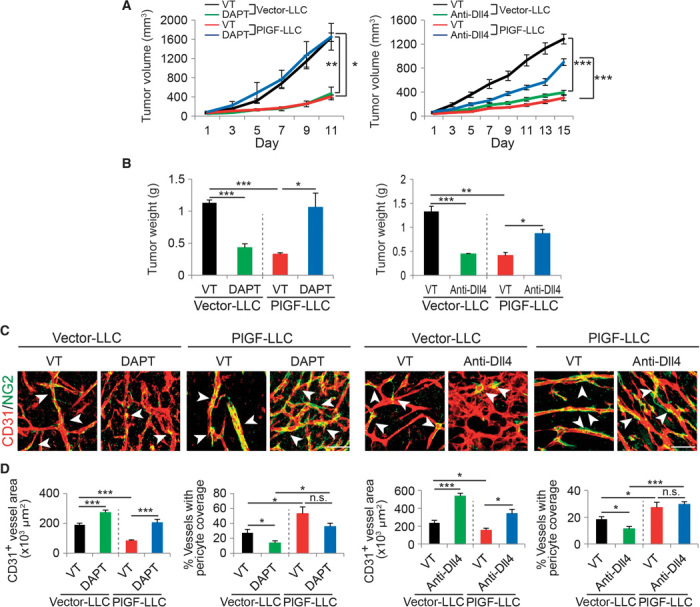
(A and B) Tumor growth rates (A) and weights (B) of vehicle (VT)–, DAPT-, and anti-Dll4 antibody–treated vector-LLC and PlGF-LLC tumors (four to six mice per group). *P < 0.05; **P < 0.01; ***P < 0.001. (C) Representative images of CD31+ tumor vessels in vehicle-, DAPT-, and anti-Dll4 antibody–treated vector-LLC and PlGF-LLC tumors. Red, CD31+ signals; green, NG2+ signals. Arrowheads point to pericyte coverage in tumor vessels. Scale bar, 50 μm. (D) Quantification of microvessel density and pericyte coverage 1 in vehicle-, DAPT-, and anti-Dll4 antibody–treated vector-LLC and PlGF-LLC tumors (5 to 10 random fields per group). Data are presented as means ± SEM.
PlGF mediates opposing vascular effects in nontreated and Dll4-Notch inhibitor–treated mouse tumors
Consistent with inhibition of tumor growth, expression of PlGF in LLC and T241 tumors also resulted in inhibition of tumor angiogenesis (Fig. 4, C and D, and fig. S4). The vasculatures of PlGF-expressing LLC and T241 tumors generally lacked excessive sprouts and branches as compared with those of vector-LLC and vector-T241 control tumors (Fig. 4C and fig. S4). However, remaining microvessels in PlGF-LLC and PlGF-T241 tumors were dilated with a significant increase in NG2+ pericyte coverage (Fig. 4, C and D, and fig. S4, C and D). Treatment of PlGF-LLC and PlGF-T241 tumor–bearing mice with DAPT markedly increased the total number of tumor microvessels without affecting pericyte coverage (Fig. 4, C and D, and fig. S4, C and D). Similarly, DAPT treatment also increased tumor vessel density in vector-LLC and vector-T241 control tumors. DAPT treatment of these PlGF-negative control tumors significantly decreased pericyte coverage (Fig. 4, C and D, and fig. S4, C and D). Likewise, treatment of PlGF-LLC and vector-LLC and PlGF-T241 and vector-T241 tumors with Dll4 blockade produced similar effects on tumor angiogenesis, vascular remodeling, and pericyte coverage as seen with DAPT (Fig. 4, C and D, and fig. S4, C and D).
We next correlated these vascular changes with their functions. In PlGF-negative control tumors, DAPT and Dll4 blockade treatments markedly increased the tortuosity, disorganization, and leakiness of tumor vasculatures as measured by extravasation of 70-kD dextran (Fig. 5, A and B, and fig. S5). In contrast, DAPT and Dll4 blockade had virtually no additional effects on vascular leakiness in PlGF-LLC and PlGF-T241 tumors (Fig. 5, A and B, and fig. S5, A and B). Whereas DAPT and Dll4 blockade treatments significantly reduced blood perfusion in vector-LLC and vector-T241 control tumors, these same drugs markedly improved blood perfusion in PlGF-LLC and PlGF-T241 tumor vessels (Fig. 5, A and C, and fig. S5, A and C). These findings provide compelling evidence that PlGF antagonizes Dll4 and Notch inhibition–modulated vascular structural and functional changes.
Fig. 5. Dll4-Notch inhibition–altered microvascular functions, tumor cell proliferation, and apoptosis in mouse PlGF– and PlGF+ tumors.

(A) Representative images of 70- and 2000-kD LRD, pimonidazole, Ki67, and cleaved caspase 3 in vehicle (VT)–, DAPT-, and anti-Dll4 antibody–treated vector-LLC and PlGF-LLC tumors. Arrowheads in the upper two rows of panels point to extravasated 70-kD (green) or perfused 2000-kD LRD (yellow). Tumor vessels were stained with CD31 (red). Tumor hypoxia in different groups was detected with pimonidazole probe (green in the middle rows of panels). Proliferating tumor cells were detected by Ki67 staining (red), and apoptotic tumor cells were detected by cleaved caspase 3 (red). Arrowheads in the bottom rows indicate apoptotic tumor cells. Scale bars, 50 μm. (B to G) Quantification of 70-kD LRD, 2000-kD LRD, and pimonidazole+, Ki67+, and cleaved caspase 3+ signals in vehicle (VT)–, DAPT-, and anti-Dll4 antibody–treated vector-LLC and PlGF-LLC tumors (n = 4 to 6 per group). Data are presented as means ± SEM. PA index was calculated using the formula (% apoptotic cells/total cells)/(% Ki67-positive cells/total cells). *P < 0.05; **P < 0.01; ***P < 0.001.
PlGF controls hypoxia, tumor cell proliferation, and apoptosis in Dll4-Notch inhibitor–treated mouse tumors
Tumor vasculatures are essential for tumor growth. PlGF-modulated vascular functions in relation to Dll4-Notch inhibition might affect malignant cell proliferation and apoptosis. In PlGF-negative vector-LLC and vector-T241 control tumors, DAPT and Dll4 blockade treatments significantly increased tumor hypoxia (Fig. 5, A and D, and fig. S5, A and D). These findings were consistent with the reduced blood perfusion and elevated leakiness in these tumor vessels (Fig. 5A and fig. S5A). In sharp contrast, DAPT and Dll4 blockade treatments of PlGF-LLC and PlGF-T241 tumors markedly improved the hypoxic microenvironment, leading to virtually nondetectable levels of the pimonidazole probe in these PlGF-positive tumors (Fig. 5, A and D, and fig. S5, A and D). Improvement of tumor hypoxia in DAPT- and Dll4 blockade–treated PlGF-LLC and PlGF-T241 tumors also increased tumor cell proliferation as measured by Ki67 positivity (Fig. 5, A and E, and fig. S5, A and E). In contrast, tumor cell apoptosis in DAPT- and Dll4 blockade–treated PlGF-LLC and PlGF-T241 tumors was considerably decreased (Fig. 5, A and F, and figs. S3, H and I, and S5, A and F). As a result, PA indexes in DAPT- and Dll4 blockade–treated PlGF-LLC and PlGF-T241 tumors were significantly increased (Fig. 5G and fig. S5G), favoring tumor growth. In contrast to PlGF-LLC and PlGF-T241 tumors, DAPT and Dll4 blockade treatments of vector-LLC and vector-T241 tumors produced totally opposite effects on tumor hypoxia, proliferation, and apoptosis, leading to decreased PA indexes in these tumors. Together, the PlGF-modulated opposing vascular effects in relation to Dll4-Notch inhibition alter tumor growth by changing the microenvironment, tumor cell proliferation, and apoptosis.
Loss of PlGF function by Plgf short hairpin RNA ablates PlGF-potentiated tumor growth and vascular functions in anti–Dll4-Notch–treated human JE-3 tumors
To further validate our findings and define PlGF as the primary factor augmenting the growth of anti–Dll4-Notch–treated tumors, we performed a loss-of-function experiment using a short hairpin RNA (shRNA)–based knockdown approach (fig. S1C). Expectedly, specific knockdown of Plgf in JE-3 tumor cells retarded the anti-Dll4–induced tumor growth as compared with a scrambled control shRNA (Fig. 6A). Consistent with impaired tumor growth rates, the vasculatures of anti-Dll4–treated Plgf shRNA JE-3 tumors appeared to be disorganized, have less pericyte coverage, poorly perfused, and highly leaky as those seen in anti-Dll4–treated PlGF-negative tumors (Fig. 6, B to F). Consequently, tumor hypoxia was significantly increased, the Ki67+ proliferating tumor cell population was decreased, and the cleaved caspase 3+ and TUNEL+ apoptotic tumor cells were markedly increased (Fig. 6, B and G to I, and fig. S3, I and J). Thus, the PA index was decreased (Fig. 6J). These findings demonstrate that PlGF is primarily responsible for the augmented tumor growth of anti-Dll4–treated human JE-3 tumor cells.
Fig. 6. Tumor growth, microvessel density, pericyte coverage, blood perfusion, leakiness, hypoxia, tumor cell proliferation, and apoptosis of anti-Dll4–treated scrambled control shRNA and Plgf shRNA JE-3 tumors.

(A) Tumor weights of nonimmune immunoglobulin G (IgG)– and Dll4 blockade–treated scrambled control shRNA and Plgf shRNA tumors (n = 6 mice per group). **P < 0.01; ***P < 0.001. (B) Representative images of CD31+ tumor vessels, 70- and 2000-kD LRD, pimonidazole, Ki67, and cleaved caspase 3 in nonimmune IgG– and anti-Dll4 antibody–treated control shRNA and Plgf shRNA tumors. Arrowheads in the top row point to pericyte coverage in tumor vessels (yellow). Arrowheads in the second and third rows point to extravasated 70-kD (green) or perfused 2000-kD LRD (yellow). Tumor vessels were stained with CD31 (red). Scale bars, 50 μm. Tumor hypoxia in different groups was detected with pimonidazole probe (green in the fourth row; scale bar, 100 μm). Proliferating tumor cells were detected by Ki67 staining (red), and apoptotic tumor cells were detected by cleaved caspase 3 (red). Arrowheads in the lowest row indicate apoptotic tumor cells. Scale bars, 50 μm. (C to J) Quantification of microvessel density, pericyte coverage, 70-kD LRD extravasation, 2000-kD LRD perfusion, and pimonidazole+, Ki67+, and cleaved caspase 3+ signals in nonimmune IgG– and anti-Dll4 antibody–treated control shRNA and Plgf shRNA tumors (six random fields per group). Data are presented as means ± SEM. PA index was calculated using the formula (% Ki67-positive cells/total cells)/(% apoptotic cells/total cells). *P < 0.05; **P < 0.01; ***P < 0.001.
PlGF modulates angiogenesis and tumor growth of anti–Dll4-Notch–treated tumors through a VEGFR1-dependent mechanism
Because VEGFR1 is the key tyrosine kinase (TK) receptor for PlGF, we next studied the role of VEGFR1 in mediating the PlGF-regulated anti–Dll4-Notch effects on angiogenesis, vascular remodeling, and tumor growth. First, we found that Vegfr1 mRNA expression levels in DAPT-treated tumors were significantly down-regulated relative to those in vehicle-treated tumor tissues (Fig. 7A). In contrast, Vegfr2 mRNA levels remained unchanged in DAPT- and vehicle-treated tumors. Down-regulation of Vegfr1 mRNA occurred in endothelial cells because treatment of primary endothelial cell–derived LLC tumors with DAPT also significantly decreased the mRNA expression level of Vegfr1 but not that of Vegfr2 (Fig. 7B). Similarly, DAPT treatment of human umbilical vein endothelial cells (HUVECs) in vitro also led to a reduced expression level of VEGFR1 as compared with vehicle-treated cells (Fig. 7C). These findings agree with the general view that VEGFR1 plays a negative role in the regulation of tumor angiogenesis (10).
Fig. 7. VEGFR-1 mediates PlGF-modulated tumor and vascular responses to Dll4-DAPT inhibition.
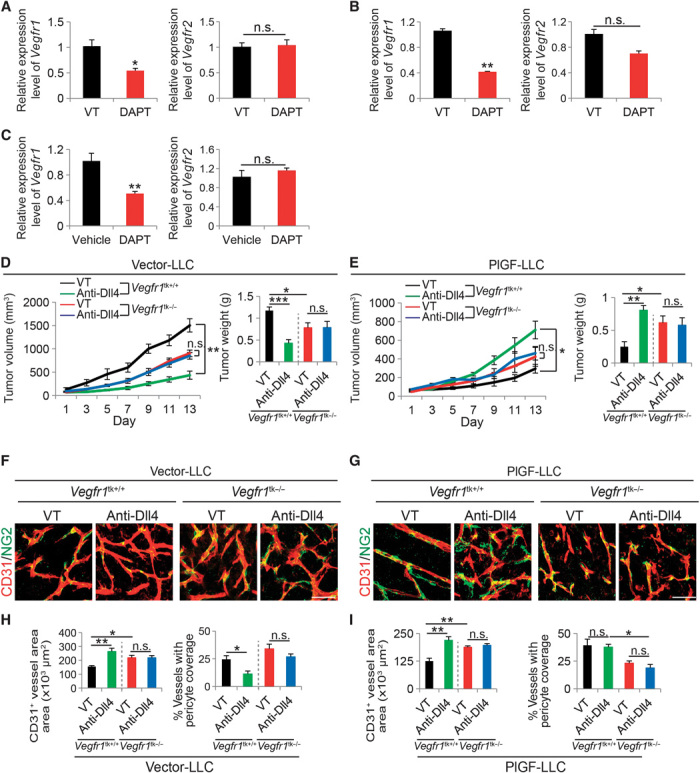
(A to C) qPCR quantification of Vegfr1 and Vegfr2 mRNA expression in vehicle (VT)– and DAPT-treated vector-LLC whole tumor tissues (A), isolated CD31+ endothelial cell fractions from vehicle- and DAPT-treated vector-LLC tumors (B), and vehicle- and DAPT-treated HUVECs (C). *P < 0.05; **P < 0.01. (D and E) Tumor growth rates and weights of anti-Dll4 antibody– and vehicle-treated vector-LLC and PlGF-LLC tumors grown in wild-type or Vegfr1tk–/– mice (six animals per group). *P < 0.05; **P < 0.01. (F to I) Tumor microvessels and pericyte coverage of anti-Dll4 antibody– and vehicle-treated vector-LLC and PlGF-LLC tumors grown in wild-type or Vegfr1tk–/– mice (four to six random fields per group). *P < 0.05; **P < 0.01; ***P < 0.001.
To understand the in-depth functional involvement of VEGFR1 in these experimental settings, we used a genetic Vegfr1-deficient mouse model that lacked the TK domain of VEGFR1 (Vegfr1tk−/−) (28). A decreased LLC tumor growth rate was observed in Vegfr1tk−/− mice as compared with wild-type mice (Fig. 7D). Surprisingly, the anti-Dll4 antibody–induced tumor suppression was completely abrogated in Vegfr1tk−/− mice (Fig. 7D), indicating that VEGFR1-transduced signaling is essential for anti-Dll4–induced tumor inhibition. Noteworthy, the anti-Dll4–stimulated growth of PlGF-LLC tumors grown in wild-type mice was completely attenuated in Vegfr1tk−/− mice (Fig. 7E). In concordance with the reduced tumor growth rate, the anti-Dll4–induced tumor vessel growth in vector-LLC tumors was completely ablated in Vegfr1tk−/− mice (Fig. 7, F and H). Similarly, anti-Dll4–induced pericyte changes were also neutralized in Vegfr1tk−/− mice (Fig. 7, F and H). Deletion of the TK domain of VEGFR1 significantly increased tumor microvessel density (Fig. 7, G and I), suggesting that VEGFR1 triggers a negative signal for tumor angiogenesis. Furthermore, both tumor vascular density and pericyte coverage remained unchanged in anti-Dll4– and vehicle-treated PlGF-LLC tumors in Vegfr1tk−/− mice (Fig. 7, G and I). Together, these findings demonstrate that VEGFR1 is the crucial receptor that mediates the PlGF-modulated antitumor effects of anti–Dll4-Notch inhibitors.
DISCUSSION
Targeting the Dll4-Notch signaling pathway provides an attractive approach in antiangiogenic cancer therapy (21). Unlike other antiangiogenic drugs, Dll4-Notch inhibitors increase, rather than decrease, the tumor microvessel density. Despite marked increases in tumor microvessel density, treatment of primary tumors with Dll4-Notch inhibitors in most cases delays tumor growth (19, 20). Dll4-Notch inhibition has been shown to promote the formation of disorganized tumor vessels that lack appropriate functions. On the basis of these preclinical findings, Dll4-Notch inhibition–induced formation of nonproductive tumor vessels has been proposed as a new approach in cancer treatment (29, 30).
The concept of stimulation of nonproductive vessels in cancer therapy opposes the conventional view of inhibition of tumor angiogenesis through reducing the number of tumor microvessels using endogenous inhibitors, generic inhibitors, and angiogenic factor antagonists (31). However, there are a few important issues that need in-depth mechanistic understanding of the functions of tumor microvessels and the complex interactions between various angiogenic signaling pathways in the tumor microenvironment: (i) do all tumors respond to Dll4-Notch inhibitors in a uniform fashion? According to published studies (19, 23, 24, 32), various tumors seem to respond differently to Dll4-Notch inhibition. These findings lead to another clinically related issue: (ii) are there any reliable biomarkers that could distinguish responders from nonresponders? For the nonresponders, would these Dll4-Notch inhibitors cause any potential harmful effects? As of this writing, these important issues remain unanswered. (iii) Do blood vessels in tumors simply supply oxygen and nutrients for tumor growth? Endothelial cells of tumor microvessels could potentially influence tumor behavior through paracrine mechanisms. Endothelial cells of tumor angiogenic vessels produce growth factors and cytokines that might potentially affect tumor growth and invasion (33). The high numbers of tumor microvessels associated with Dll4-Notch inhibition might potentiate tumor growth and invasion through a paracrine mechanism. Finally, (iv) the relation between the Dll4-Notch signaling and other angiogenic signaling pathways has not been well studied.
Although lacking appropriate functions owing to disorganized and excessive growth of endothelial cells in angiogenic vessels, the Dll4-Notch inhibition–induced nonproductive vessels may be potentially reorganized into functional vessels by other vascular remodeling factors. Our recent work shows that tumor-derived PlGF induces normalization of tumor vasculatures that consist of large-diameter vascular networks without excessive branches and sprouts. Noteworthy, the PlGF-normalized tumor vessels are highly coated with pericytes and perfused with blood, indicating that PlGF-remodeled tumor vessels are functional. Here, we provide evidence that PlGF markedly remodels the Dll4-Notch inhibition–induced disorganized and nonproductive tumor vasculatures toward functionally vascular networks. The PlGF-initiated functional improvement is dependent on its receptor, VEGFR1, which is involved in vascular reorganization (34). For example, genetic deletion of the Vegfr1 gene results in embryonic lethality owing to uncontrolled growth of endothelial cells and disorganization of developing vascular networks (34). The Vegfr1-null vascular phenotype is completely different from that of the Vegfr2-null mice, although deletion of both receptors leads to early embryonic lethality (34, 35). Whereas Vegfr1-null mouse embryos show disorganization of vasculatures that cause functionally impairment, Vegfr2-null mouse embryos completely lack detectable blood vessels (34, 35). On the basis of our present findings and published work, it is obvious that VEGFR1 mediates a similar vascular remodeling function in tumors as seen in mouse embryos. There seems to be a fundamental difference between PlGF and VEGF even though both factors bind to VEGFR1. VEGF promotes angiogenesis and vascular permeability through the VEGFR2-mediated signaling pathway (7), and its remodeling function may remain modest. In contrast, PlGF as a VEGFR1-exclusive binding ligand is unable to induce angiogenesis but is dedicated to vascular remodeling functions (10). In the presence of Dll4-Notch inhibitors, VEGF probably plays a critical role in guiding and driving vessel growth because VEGF-VEGFR2 signaling is critically involved in the tip cell formation at the leading edge of vascular growth (18). Conversely, PlGF does not play such a guiding role in vascular development and remodels tumor vessels toward a normalized phenotype by trimming excessive vascular branches and by increasing pericyte coverage.
For most targeted therapeutics, defining reliable biomarkers that distinguish responders from nonresponders and overcoming drug resistance are challenging issues related to clinical practice. With no exceptions for therapeutic agents targeting the Dll4-Notch signaling pathway, defining such reliable biomarkers not only ensures accurate selection of responders but also avoids drug-induced acceleration of tumor growth. In some cases, inadequate treatment of nonresponders with targeted drugs might accelerate tumor growth and invasion. Indeed, anti-VEGF and anti–platelet-derived growth factor receptor drugs have been reported to stimulate invasiveness in experimental tumor models (36–38). Would PlGF expression levels in tumors serve as such a reliable biomarker for Dll4-Notch inhibitors in human cancer patients? On the basis of our preclinical findings, this would most likely be the case. A number of human tumors express high levels of PlGF (12, 39), and thus, measurement of PlGF expression levels has profound implications for anti–Dll4-Notch therapy. Our recent work shows that tumor expression levels of PlGF might also serve as a biomarker to predict the therapeutic outcome of anti-VEGF drugs (27). Therefore, combination therapy with anti-VEGF and anti–Dll4-Notch would likely produce synergistic effects on PlGF-positive tumors. This interesting possibility warrants clinical validation. Of interest, high PlGF–expressing tumors such as choriocarcinoma also have hypermethylation of p16ink4a, which may be mediated by the reactive oxygen species–mediated pathway for regulation of PlGF expression (40–42).
Together, our data shed new mechanistic insights on the complex interactions between different signaling pathways that stimulate angiogenesis and remodel microvessels in the tumor microenvironment. On the basis of these findings, we propose that PlGF expression levels in tumors may serve as indicative markers for selection of anti–Dll4-Notch responders and for prediction of anti–Dll4-Notch resistance in human cancer patients.
MATERIALS AND METHODS
Cells, reagents, and antibodies
Human JE-3 choriocarcinoma and MDA-MB-231 breast cancer cell lines were purchased from the American Type Culture Collection (ATCC); human BeWo choriocarcinoma cells were provided by A. Barragan at the Department of Medicine, Karolinska Institute; the human Hep3B HCC cell line was obtained from T. Torimura at the Department of Medicine, Kurume University; and murine LLC and T241 fibrosarcoma cells stably expressing PlGF or an empty vector were established as previously described (13, 17, 26, 27). Both human and mouse tumor cell lines were grown and maintained in Dulbecco’s modified Eagle’s medium (HyClone) containing 20% (v/v) heat-inactivated fetal bovine serum (HyClone) with penicillin-streptomycin (100 U/ml; HyClone). HUVECs were purchased from ATCC and maintained in complete EGM-2 medium (Lonza). The DAPT γ-secretase inhibitor was obtained from AbMole BioScience. DAPT was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) and was diluted to a final concentration of 5 μM for the in vitro cell culture. A rabbit anti-human Dll4 neutralizing antibody was obtained from Genentech. Rabbit nonimmune IgG was obtained from Invitrogen. About 1 × 105 HUVECs were seeded on a 12-well plate and starved overnight before DAPT treatment. After starvation, the medium containing 5 μM DAPT or an equivalent amount of DMSO vehicle was replaced, treatment was continued for 24 hours, and RNA was collected from each group.
Animals
Male and female C57Bl/6 and severe combined immunodeficient (SCID) mice of ages 6 to 8 weeks were obtained from the breeding unit of the Department of Microbiology, Tumor and Cell Biology, Karolinska Institute, Stockholm, Sweden. Vegfr1-deficient mice that lacked a TK domain (28) were provided by M. Shibuya at the Institute of Medical Science, University of Tokyo, Tokyo, Japan, and genotyped using specific primers (table S1) and a Fast Tissue-to-PCR Kit (Fermentas).
Transfection of tumor cells with Plgf shRNA
Plasmids containing an shRNA specific for human Plgf and a lentiviral vector–based expression packaging kit were purchased from GeneCopoeia. The transfection procedure was executed according to the manufacturer’s protocol. To produce Plgf shRNA viral particles, a Plgf shRNA–containing plasmid and viral packaging vectors were cotransfected into human embryonic kidney 293T cells. Viral particles carrying Plgf shRNA were harvested from the conditioned medium and subsequently used to infect JE-3 choriocarcinoma cells. JE-3 choriocarcinoma cells containing stable Plgf knockdown were selected using puromycin (2 μg/ml). The knockdown efficiency was validated by the quantitative real-time polymerase chain reaction (qPCR) method using specific primers (table S1) as previously described (43).
Human and mouse tumor models
Human JE-3, BeWo, MDA-MB-231, and Hep3B cells at a density of 3 × 106 cells in 100 μl of phosphate-buffered saline (PBS) were subcutaneously injected into the mid-dorsal region of each SCID mouse. Murine LLC and T241 tumor cells at a density of 1 ×106 cells in 100 μl of PBS were subcutaneously implanted into each C57Bl/6 or Vegfr1tk−/− mouse. Tumor sizes were measured every other day, and tumor volume was calculated according to the following standard formula: length × width2 × 0.52 (44). Tumor-bearing mice were randomly divided into various experimental groups to receive treatment with DAPT or an anti-Dll4 antibody. Treatments were initiated when tumor sizes reached 100 to 200 mm3. Each mouse was intraperitoneally injected with DAPT (100 mg/kg, daily) and an anti-Dll4 antibody (5 mg/kg, every 5 days). Experiments were terminated, and the mice were sacrificed by exposure to a lethal dose of CO2. Fresh tumor tissues were immediately collected for RNA extraction, fixed in 4% paraformaldehyde (PFA) overnight, and washed with PBS before whole-mount staining. For detection of tumor hypoxia, each mouse was intraperitoneally injected with 1.5 mg of pimonidazole (Hypoxyprobe) and was sacrificed at 30 min after injection.
Whole-mount staining
Tissue whole-mount staining was performed according to our previously described method (17). A rat anti-mouse CD31 antibody (BD Pharmingen; 1:200) and a rabbit anti-mouse NG2 antibody (Millipore; 1:200) were used as primary antibodies. The secondary antibodies included a Cy3- or Cy5-conjugated goat anti-rat IgG antibody (Invitrogen; 1:400) and a donkey anti-rabbit Cy5 antibody (Invitrogen; 1:400). Stained tissues were mounted in a Vectashield mounting medium (Vector Laboratories). Images were captured with a confocal microscope (Nikon D-Eclipse C1, Nikon Corp.). Quantitative analyses were performed from at least 10 different random fields using the Adobe Photoshop CS software program.
Vascular permeability and perfusion
LRD at a molecular weight of 70 or 2000 (Invitrogen) was injected into the tail vein of each tumor-bearing mouse when the tumor size was about 0.8 cm3. At 5 min after injection with 2000-kD dextran and 15 min after injection with 70-kD dextran, the mice were sacrificed, and tumor tissues were immediately fixed in 4% PFA overnight and further analyzed by whole-mount staining.
Immunohistochemistry
Staining of paraffin-embedded tissues was performed according to our standard protocol (45). Briefly, paraffin-embedded tissues were sectioned at a thickness of 5 μm and baked at 60°C for 1 hour. Antigen retrieval was achieved with unmasking solution (Vector Laboratories, H3300). Samples were blocked with 4% horse serum at room temperature for 30 min. A rabbit anti-mouse Ki67 antibody (eBioscience; 1:500) and a rabbit anti-mouse cleaved caspase 3 (R&D Systems; 1:500) were used as primary antibodies. A Cy3-conjugated goat anti-rabbit IgG antibody (Chemicon; 1:400) was used as a secondary antibody. For detection of tumor hypoxia, a FITC-conjugated mouse anti-pimonidazole monoclonal antibody (clone 4.3.11.3, Hypoxyprobe) was used. Slides were mounted with a Vectashield mounting medium containing DAPI for nuclear staining (Vector Laboratories). Stained tissue samples were examined with a confocal microscope (Nikon D-Eclipse C1) using NIS-Elements D software (Nikon).
Real-time qPCR
Real-time qPCR was performed according to the standard protocol (46, 47) using commercially available kits. The collected tumor samples were extracted using a GeneJet RNA Purification Kit (Thermo Scientific), and complementary DNA (cDNA) synthesis was achieved using a RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific). Real-time qPCR was performed with a Power SYBR Green Master Mix Kit (Applied Biosystems) and the Step One Plus Real-Time PCR system (Applied Biosystems). The specific primers used for real-time qPCR are given in table S1.
Enzyme-linked immunosorbent assay
Human PlGF protein was quantitatively analyzed using a PlGF ELISA Kit (R&D Systems Inc.). The measurement procedures were performed according to the manufacturer’s instructions.
Isolation of CD31+ cells from tumor tissues by fluorescence-activated cell sorting
Subcutaneous vector LLC tumor tissues were prepared as a single-cell suspension using 0.15% collagenase I and II (Sigma). Cells were stained for 45 min on ice with a rat anti-mouse CD31 antibody (1:100; BD Pharmingen). After being washed with PBS, the cells were incubated with an Alexa Fluor 555–labeled goat anti-rat antibody (1:200; Invitrogen) for 30 min on ice. CD31+ cells were analyzed and sorted by fluorescence-activated cell sorting analysis as previously described (44, 45) (MoFlo XTD, Beckman Coulter), and RNA was extracted.
Statistical analysis
Data represent means ± SEM. P values were determined by unpaired Student’s t test or the Mann-Whitney test. Comparison of multiple groups was achieved using analysis of variance (ANOVA) with SPSS software. *P < 0.05 is considered significant, **P < 0.01 is considered highly significant, and ***P < 0.001 is considered extremely significant.
Supplementary Material
Acknowledgments
We thank M. Yan at the Genentech Inc. for providing the anti-Dll4 antibody. Funding: Y.C.’s laboratory is supported through research grants from the Swedish Research Council, the Swedish Cancer Foundation, the Karolinska Institute Foundation, the Karolinska Institute distinguished professor award, the Torsten Söderberg Foundation, the European Research Council (ERC) advanced grant ANGIOFAT (project no. 250021), the NOVO Nordisk Foundation, the Advanced grant from the NOVO Nordisk Foundation, and the Royal Alice Wallenberg Foundation. H.I. was supported in part by the International Research Fund for Subsidy of Kyushu University School of Medicine Alumni. Author contributions: Y.C. generated all ideas of this study, designed all experiments, designed all figures, analyzed the results, and wrote the manuscript. H.I. performed most experiments, analyzed all raw data, prepared all figures, and helped in writing the experimental procedures. Y.Z., X.Y., J.W., T.S., Y.Y., and M.N. helped perform some of the experiments. T.T. provided essential research materials and participated in discussion of the project. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/3/e1400244/DC1
Table S1. Sequences of the human and mouse primers used for genotyping and qPCR.
Fig. S1. PlGF protein and mRNA expression levels in human and mouse cancer cell lines.
Fig. S2. Tumor growth, microvessel density, pericyte coverage, blood perfusion and leakiness, hypoxia, tumor cell proliferation, and apoptosis of Dll4-Notch inhibitor–treated PlGF− tumors.
Fig. S3. TUNEL staining in each human and mouse tumor.
Fig. S4. Dll4-Notch inhibition–altered tumor growth and microvasculatures in mouse PlGF− and PlGF+ tumors.
Fig. S5. Dll4-Notch inhibition–altered microvascular functions, tumor cell proliferation, and apoptosis in mouse PlGF− and PlGF+ tumors.
REFERENCES AND NOTES
- 1.Cao Y., Tumor angiogenesis and molecular targets for therapy. Front. Biosci. (Landmark Ed.) 14, 3962–3973 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Jain R. K., Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 307, 58–62 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Folkman J., Angiogenesis: An organizing principle for drug discovery? Nat. Rev. Drug Discov. 6, 273–286 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Kerbel R. S., Tumor angiogenesis. N. Engl. J. Med. 358, 2039–2049 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao Y., Langer R., Optimizing the delivery of cancer drugs that block angiogenesis. Sci Transl Med 2, 15ps13 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Jubb A. M., Pham T. Q., Hanby A. M., Frantz G. D., Peale F. V., Wu T. D., Koeppen H. W., Hillan K. J., Expression of vascular endothelial growth factor, hypoxia inducible factor 1α, and carbonic anhydrase IX in human tumours. J. Clin. Pathol. 57, 504–512 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmes K., Roberts O. L., Thomas A. M., Cross M. J., Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 19, 2003–2012 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Shibuya M., Vascular endothelial growth factor (VEGF)-receptor2: Its biological functions, major signaling pathway, and specific ligand VEGF-E. Endothelium 13, 63–69 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Shibuya M., Ito N., Claesson-Welsh L., Structure and function of vascular endothelial growth factor receptor-1 and −2. Curr. Top. Microbiol. Immunol. 237, 59–83 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Cao Y., Positive and negative modulation of angiogenesis by VEGFR1 ligands. Sci. Signal. 2, re1 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Bais C., Wu X., Yao J., Yang S., Crawford Y., McCutcheon K., Tan C., Kolumam G., Vernes J. M., Eastham-Anderson J., Haughney P., Kowanetz M., Hagenbeek T., Kasman I., Reslan H. B., Ross J., Van Bruggen N., Carano R. A., Meng Y. J., Hongo J. A., Stephan J. P., Shibuya M., Ferrara N., PlGF blockade does not inhibit angiogenesis during primary tumor growth. Cell 141, 166–177 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Fischer C., Jonckx B., Mazzone M., Zacchigna S., Loges S., Pattarini L., Chorianopoulos E., Liesenborghs L., Koch M., De Mol M., Autiero M., Wyns S., Plaisance S., Moons L., van Rooijen N., Giacca M., Stassen J. M., Dewerchin M., Collen D., Carmeliet P., Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131, 463–475 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Eriksson A., Cao R., Pawliuk R., Berg S. M., Tsang M., Zhou D., Fleet C., Tritsaris K., Dissing S., Leboulch P., Cao Y., Placenta growth factor-1 antagonizes VEGF-induced angiogenesis and tumor growth by the formation of functionally inactive PlGF-1/VEGF heterodimers. Cancer Cell 1, 99–108 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Schomber T., Kopfstein L., Djonov V., Albrecht I., Baeriswyl V., Strittmatter K., Christofori G., Placental growth factor-1 attenuates vascular endothelial growth factor-A–dependent tumor angiogenesis during β cell carcinogenesis. Cancer Res. 67, 10840–10848 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Xu L., Cochran D. M., Tong R. T., Winkler F., Kashiwagi S., Jain R. K., Fukumura D., Placenta growth factor overexpression inhibits tumor growth, angiogenesis, and metastasis by depleting vascular endothelial growth factor homodimers in orthotopic mouse models. Cancer Res. 66, 3971–3977 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Lu D., Jimenez X., Zhang H., Wu Y., Bohlen P., Witte L., Zhu Z., Complete inhibition of vascular endothelial growth factor (VEGF) activities with a bifunctional diabody directed against both VEGF kinase receptors, fms-like tyrosine kinase receptor and kinase insert domain-containing receptor. Cancer Res. 61, 7002–7008 (2001). [PubMed] [Google Scholar]
- 17.Hedlund E. M., Hosaka K., Zhong Z., Cao R., Cao Y., Malignant cell-derived PlGF promotes normalization and remodeling of the tumor vasculature. Proc. Natl. Acad. Sci. U.S.A. 106, 17505–17510 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellström M., Phng L. K., Hofmann J. J., Wallgard E., Coultas L., Lindblom P., Alva J., Nilsson A. K., Karlsson L., Gaiano N., Yoon K., Rossant J., Iruela-Arispe M. L., Kalén M., Gerhardt H., Betsholtz C., Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445, 776–780 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Ridgway J., Zhang G., Wu Y., Stawicki S., Liang W. C., Chanthery Y., Kowalski J., Watts R. J., Callahan C., Kasman I., Singh M., Chien M., Tan C., Hongo J. A., de Sauvage F., Plowman G., Yan M., Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444, 1083–1087 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Noguera-Troise I., Daly C., Papadopoulos N. J., Coetzee S., Boland P., Gale N. W., Lin H. C., Yancopoulos G. D., Thurston G., Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444, 1032–1037 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Brzozowa M., Wojnicz R., Kowalczyk-Ziomek G., Helewski K., The Notch ligand Delta-like 4 (DLL4) as a target in angiogenesis-based cancer therapy? Contemp. Oncol. 17, 234–237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuhnert F., Kirshner J. R., Thurston G., Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc. Cell 3, 20 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J. L., Sainson R. C., Shi W., Leek R., Harrington L. S., Preusser M., Biswas S., Turley H., Heikamp E., Hainfellner J. A., Harris A. L., Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res. 67, 11244–11253 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Zhang J. P., Qin H. Y., Wang L., Liang L., Zhao X .C., Cai W. X., Wei Y. N., Wang C. M., Han H., Overexpression of Notch ligand Dll1 in B16 melanoma cells leads to reduced tumor growth due to attenuated vascularization. Cancer Lett. 309, 220–227 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Oon C. E., Harris A. L., New pathways and mechanisms regulating and responding to Delta-like ligand 4–Notch signalling in tumour angiogenesis. Biochem. Soc. Trans. 39, 1612–1618 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Yang X., Zhang Y., Yang Y., Lim S., Cao Z., Rak J., Cao Y., Vascular endothelial growth factor-dependent spatiotemporal dual roles of placental growth factor in modulation of angiogenesis and tumor growth. Proc. Natl. Acad. Sci. U.S.A. 110, 13932–13937 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hedlund E. M., Yang X., Zhang Y., Yang Y., Shibuya M., Zhong W., Sun B., Liu Y., Hosaka K., Cao Y., Tumor cell-derived placental growth factor sensitizes antiangiogenic and antitumor effects of anti-VEGF drugs. Proc. Natl. Acad. Sci. U.S.A. 110, 654–659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiratsuka S., Minowa O., Kuno J., Noda T., Shibuya M., Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 95, 9349–9354 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noguera-Troise I., C. Daly, N. J. Papadopoulos, S. Coetzee, P. Boland, N. W. Gale, H. C. Lin, G. D. Yancopoulos, G. Thurston, Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Novartis Found. Symp. 283, 106–120; discussion 121–105, 238–141 (2007). [PubMed] [Google Scholar]
- 30.Thurston G., Noguera-Troise I., Yancopoulos G. D., The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat. Rev. Cancer 7, 327–331 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Folkman J., Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 285, 1182–1186 (1971). [DOI] [PubMed] [Google Scholar]
- 32.Segarra M., Williams C. K., Sierra Mde. L., Bernardo M., McCormick P. J., Maric D., Regino C., Choyke P., Tosato G., Dll4 activation of Notch signaling reduces tumor vascularity and inhibits tumor growth. Blood 112, 1904–1911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo P., Hu B., Gu W., Xu L., Wang D., Huang H. J., Cavenee W. K., Cheng S. Y., Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am. J. Pathol. 162, 1083–1093 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fong G. H., Rossant J., Gertsenstein M., Breitman M. L., Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376, 66–70 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Shalaby F., Rossant J., Yamaguchi T. P., Gertsenstein M., Wu X. F., Breitman M. L., Schuh A. C., Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66 (1995). [DOI] [PubMed] [Google Scholar]
- 36.Ebos J. M., Lee C. R., Cruz-Munoz W., Bjarnason G. A., Christensen J. G., Kerbel R. S., Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pàez-Ribes M., Allen E., Hudock J., Takeda T., Okuyama H., Viñals F., Inoue M., Bergers G., Hanahan D., Casanovas O., Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hosaka K., Yang Y., Seki T., Nakamura M., Andersson P., Rouhi P., Yang X., Jensen L., Lim S., Feng N., Xue Y., Li X., Larsson O., Ohhashi T., Cao Y., Tumour PDGF-BB expression levels determine dual effects of anti-PDGF drugs on vascular remodelling and metastasis. Nat. Commun. 4, 2129 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Cao Y. , H. Chen, L. Zhou, M. K. Chiang, B. Anand-Apte, J. A. Weatherbee, Y. Wang, F. Fang, J. G. Flanagan, M. L. Tsang, Heterodimers of placenta growth factor/vascular endothelial growth factor. Endothelial activity, tumor cell expression, and high affinity binding to Flk-1/KDR. J. Biol. Chem. 271, 3154–3162 (1996). [DOI] [PubMed] [Google Scholar]
- 40.Bhandarkar S. S., Jaconi M., Fried L. E., Bonner M. Y., Lefkove B., Govindarajan B., Perry B. N., Parhar R., Mackelfresh J., Sohn A., Stouffs M., Knaus U., Yancopoulos G., Reiss Y., Benest A. V., Augustin H. G., Arbiser J. L., Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J. Clin. Invest. 119, 2359–2365 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonner M. Y., Arbiser J. L., Targeting NADPH oxidases for the treatment of cancer and inflammation. Cell. Mol. Life Sci. 69, 2435–2442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue W. C., Chan K. Y., Feng H. C., Chiu P. M., Ngan H. Y., Tsao S. W., Cheung A. N., Promoter hypermethylation of multiple genes in hydatidiform mole and choriocarcinoma. J. Mol. Diagn. 6, 326–334 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji H., Cao R., Yang Y., Zhang Y., Iwamoto H., Lim S., Nakamura M., Andersson P., Wang J., Sun Y., Dissing S., He X., Yang X., Cao Y., TNFR1 mediates TNF-α-induced tumour lymphangiogenesis and metastasis by modulating VEGF-C-VEGFR3 signalling. Nat. Commun. 5, 4944 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Lim S., Y. Zhang, D. Zhang, F. Chen, K. Hosaka, N. Feng, T. Seki, P. Andersson, J. Li, J. Zang, B. Sun, Y. Cao, VEGFR2-mediated vascular dilation as a mechanism of VEGF-induced anemia and bone marrow cell mobilization. Cell Rep. 9, 569–580 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Xue Y., S. Lim, Y. Yang, Z. Wang, L. D. Jensen, E. M. Hedlund, P. Andersson, M. Sasahara, O. Larsson, D. Galter, R. Cao, K. Hosaka, Y. Cao, PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat. Med. 18, 100–110 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Dong M., Yang X., Lim S., Cao Z., Honek J., Lu H., Zhang C., Seki T., Hosaka K., Wahlberg E., Yang J., Zhang L., Länne T., Sun B., Li X., Liu Y., Zhang Y., Cao Y., Cold exposure promotes atherosclerotic plaque growth and instability via UCP1-dependent lipolysis. Cell Metab. 18, 118–129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jensen L. D., Cao Z., Nakamura M., Yang Y., Bräutigam L., Andersson P., Zhang Y., Wahlberg E., Länne T., Hosaka K., Cao Y., Opposing effects of circadian clock genes Bmal1 and Period2 in regulation of VEGF-dependent angiogenesis in developing zebrafish. Cell Rep. 2, 231–241 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/3/e1400244/DC1
Table S1. Sequences of the human and mouse primers used for genotyping and qPCR.
Fig. S1. PlGF protein and mRNA expression levels in human and mouse cancer cell lines.
Fig. S2. Tumor growth, microvessel density, pericyte coverage, blood perfusion and leakiness, hypoxia, tumor cell proliferation, and apoptosis of Dll4-Notch inhibitor–treated PlGF− tumors.
Fig. S3. TUNEL staining in each human and mouse tumor.
Fig. S4. Dll4-Notch inhibition–altered tumor growth and microvasculatures in mouse PlGF− and PlGF+ tumors.
Fig. S5. Dll4-Notch inhibition–altered microvascular functions, tumor cell proliferation, and apoptosis in mouse PlGF− and PlGF+ tumors.