

Abstract
Reactivation of telomerase, the chromosome end-replicating enzyme, drives human cell immortality and cancer. Point mutations in the telomerase reverse transcriptase (TERT) gene promoter occur at high frequency in multiple cancers, including urothelial cancer (UC), but their impact on telomerase function has been unclear. In a study of 23 human UC cell lines, we show that these promoter mutations correlate with higher levels of TERT mRNA, TERT protein, telomerase enzymatic activity and telomere length. While previous studies found no relationship between TERT promoter mutations and UC patient outcome, we find that elevated TERT mRNA expression strongly correlates with reduced disease-specific survival (DSS) in two independent UC patient cohorts (n = 35; n = 87). These results suggest that high telomerase activity may be a better marker of aggressive UC tumors than TERT promoter mutations alone.
Telomerase activity is high in embryonic and stem cells but nearly undetectable in most somatic cells due primarily to transcriptional down-regulation of TERT, the catalytic subunit of the ribonucleoprotein particle (RNP) (1, 2). Telomerase activity is upregulated in 85% – 90% of cancers (3), and the recent identification of two highly recurrent point mutations in the TERT promoter in multiple cancer types suggests one likely mechanism for TERT reactivation. These mutations were first reported in melanoma (4, 5), then quickly found in many other cancers such as UC, the fifth most common cancer in the western world (6). For some cancers, such as UC, these mutations occur more frequently than any other mutation, including TP53, and a recent report suggests they may be the most prevalent of all non-coding mutations in cancer (6, 7). The two mutations reside 124 and 146 base pairs (bp) upstream from the ATG translation start site (Fig. 1A) and are proposed to augment transcription by recruiting E-twenty-six (ETS) transcription factors to newly generated GGA(A/T) motifs (4, 5).
Fig. 1. TERT gene copy number and promoter mutations in UC cell lines.
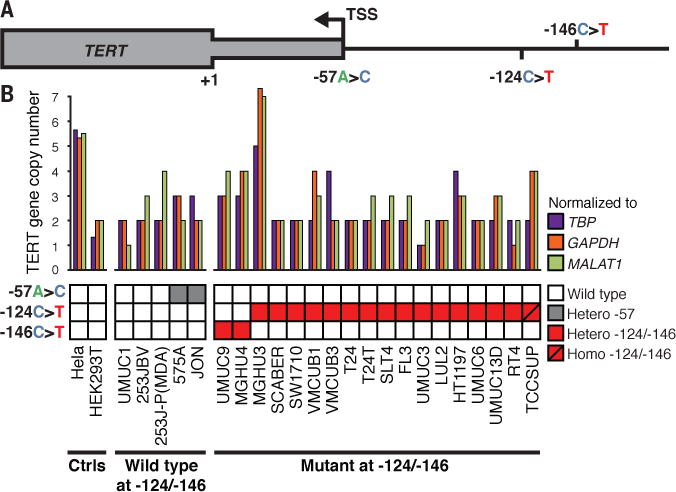
(A) Map of the proximal TERT promoter. Relevant mutations, the major annotated TSS and the translational start codon (+1) are indicated. (B) TERT CNV (top) and TERT promoter genotype (bottom) in the UC23 and in reference cell lines (Ctrls). The genes TATA Binding Protein (TBP), Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1) were used for normalization.
While the frequency of the TERT promoter mutations suggests their importance for telomerase reactivation in cancer, fundamental questions remain. First, only modest increases in gene expression were seen upon introducing these mutations into heterologous luciferase reporter constructs; ~1.5- to ~4-fold increases depending upon the particular cell line used (4, 5, 8). Measurements of TERT mRNA levels in tumor tissues of many diverse cancer types have yielded similarly small differences with the promoter mutation (9–12), such as a ~1.4-fold increase for UC (13). One report of ~40-fold increased mRNA expression in cirrhotic preneoplastic lesions that harbor promoter mutations, however, allows for the possibility of larger effects early in oncogenesis (10). Second, without evidence that these mutations have any consequence for telomerase activity and telomere length, their biological contribution to tumorigenesis is unclear. It is conceivable, for example, that TERT upregulation promotes tumorigenesis primarily by telomerase-independent mechanisms, such as by perturbation of the c-MYC or WNT signaling pathways [(14); however, see ref. (15)]. A third confounding observation is that roughly equal frequencies of these mutations are found across all stages and grades of UC and other cancers (8, 12, 13, 16), which might suggest that telomerase upregulation is not particularly important for tumor progression in these cases. One early study did find an association of telomerase activity level with pathological grade and clinical stage of bladder tumors (17); these authors were appropriately circumspect about their conclusions because they relied on a PCR-based assay to measure telomerase activity instead of the more reliable direct enzymatic activity assay. Also, TERT promoter mutations have been associated with reduced survival of patients with glioblastoma multiforme (6) and with larger tumors and lymph node metastasis in the case of conventional papillary thyroid carcinomas (12).
To explore the impact of the TERT promoter mutations on telomerase activity in UC, we studied a panel of 23 UC cell lines (UC23) derived from tumors of a wide range of stages and grades, including both muscle-invasive and non-invasive tumors (tables S1 to S3). We genotyped the TERT promoter in each of the UC23 and found frequent incidence of the −124 C>T mutation, and less frequent incidence of the −146 C>T mutation (Fig. 1B and fig. S1) (16). Two instances of a −57 A>C mutation, previously identified in a family prone to melanoma but otherwise not frequently observed across cancer types (7, 18), and several single nucleotide polymorphisms (SNPs) were also observed (fig. S1).
Because amplification of the TERT gene has also been reported to be a mechanism of TERT reactivation in some cancers (11, 18, 19), we measured TERT copy number variation (CNV) in the UC23. Some variation was observed; the MGHU3 cell line was estimated to have ~6 copies, the MGHU4 cell line was estimated to have ~3–4 copies and the HT1197, TCCSUP and UMUC9 cell lines were estimated to have ~3 copies each (Fig. 1B). As controls, our estimates of ~5 TERT copies per HeLa genome and ~2 per HEK293T genome agreed with previous reports (20, 21).
We then examined TERT expression and telomerase activity in each of the UC23. We compared cell lines harboring either the −124 or −146 mutation to those without. Although the −57 mutation also generates a GGA(A/T) motif, it lies exactly at the major annotated transcriptional start site (TSS) for TERT, and thus would probably affect transcription initiation by means other than simple upregulation (22, 23); indeed, the −57 mutation resulted in the smallest of all changes observed in expression of a luciferase reporter (4). We hypothesized that all of the UC23 would show more or less similar levels of telomerase activity since virtually all cancers reactivate telomerase whether by one mechanism (e.g., −124/−146 promoter mutation) or another (e.g., TERT CNV, upregulation of ETS or c-MYC transcription factors or downregulation of repressive chromatin modifications) and since most reports had found no association between the promoter mutations and severity of disease in UC (1–3, 8, 19, 24).
Contrary to our hypothesis, we found that TERT mRNA levels were dramatically increased in those cancer cell lines harboring the −124 or −146 promoter mutations relative to the others, with an 18-fold increase in median value as measured by qRT-PCR (Fig. 2A, fig. S2; p = 0.0067; all p-values were obtained using the Wilcoxon rank sum test). However, the majority of TERT transcripts are alternatively spliced variants that are not translated into a functional reverse transcriptase (RT) (1, 15). We therefore measured the levels of TERT protein (Fig. 2B,C) and telomerase enzymatic activity (Fig. 2D,E) in each of the UC23, using immunopurification (IP) of TERT followed by immunoblot analysis and direct enzymatic activity assays, respectively (25, 26). Both protein and telomerase activity levels were higher in cell lines harboring promoter mutations, although the 2-fold increases in median values were much more modest than the 18-fold increase in mRNA levels (Fig. 2B,D). These more modest changes may reflect additional, post-transcriptional regulation of TERT expression. It is also possible that the mild lysis conditions used for IP, necessary to preserve integrity and activity of the telomerase RNP, were not sufficient to completely solubilize some of the UC23. This would result in reduced efficiency of TERT recovery in some cell lines and subsequently an underestimation of protein and activity levels. Measurements of TERT mRNA levels and telomere lengths on the other hand, for which harsher methods of cell disruption were used, were significantly different when comparing wild type and mutant groups (Fig. 2A and 3B). Still, both TERT mRNA and protein levels correlated strongly with telomerase activity in the UC23 (Fig. 2F,G and fig. S3), while levels of the telomerase RNA (TR) subunit did not (Fig. 2H).
Fig. 2. TERT promoter mutations are associated with increased TERT mRNA and protein and telomerase activity.
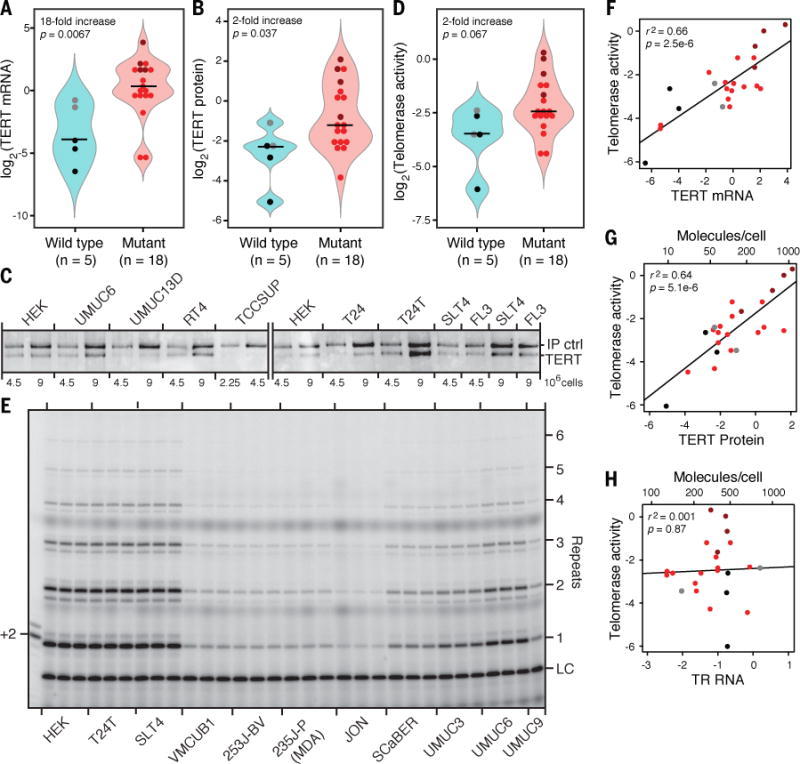
(A) Quantification of TERT mRNA in cell lines with wild-type versus mutant TERT promoters by qRT-PCR amplification of exon 14 normalized to 18S rRNA or GAPDH mRNA. (Throughout this work, “mRNA” refers to transcripts containing TERT exon 14 or exon 3, which will include alternatively spliced variants that do not yield a functional RT.) Two cell lines that are wild-type at −124/−146 have the −57 A>C mutation (gray dots). T24T is a more tumorigenic variant of T24, and SLT4 and FL3 were derived from tumors that had metastasized in mice injected with T24T cells (these cell lines are indicated with magenta dots; also see tables S1–S3). (B) Quantification of TERT protein by IP followed by immunoblot. (C) Representative immunoblots showing TERT protein levels in several of the UC23. IP ctrl, see Supplemental Methods. The efficiency of IP was found to be similar from lysates prepared from each cell line (fig. S4). (D) Quantification of telomerase activities in UC23 cell extracts, measured by extension of a telomeric oligonucleotide in the presence of [α-32P]dGTP. (E) Representative autoradiogram of telomeric oligonucleotides extended by telomerase following TERT IP. LC, DNA loading control. (F) Log2(telomerase activity) versus log2(TERT mRNA) for each of the UC23. Cell lines are color-coded as in panel A. (G) Log2(telomerase activity) versus log2(TERT protein). (H) Log2(telomerase activity) versus log2(TR RNA). In G and H, molecules/cell calculated as in (25).
Fig. 3. TERT promoter mutations are associated with long telomeres in the UC23, as determined by telomeric restriction fragment (TRF) analysis.
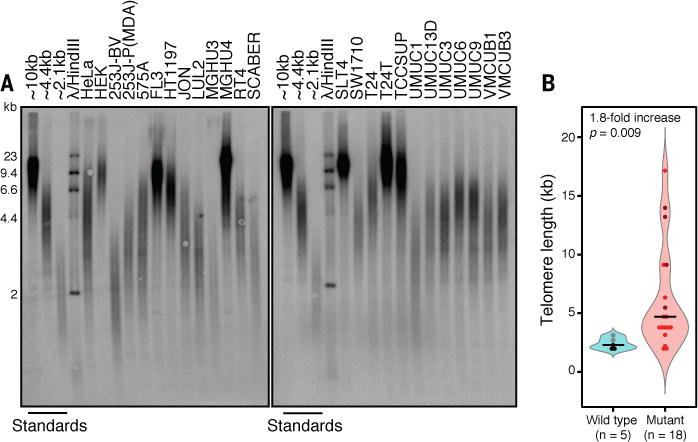
(A) Southern blot with a telomeric DNA probe was performed as in (34). Standards, TRFs from HeLa cell lines previously characterized as having long, intermediate, or short telomeres (34). (B) Quantification of TRFs in wild-type versus promoter-mutant cell lines; dots colored as in Fig. 2.
The eight cell lines with the greatest levels of TERT protein expression and the nine cell lines with the greatest levels of telomerase activity all harbored the −124 or −146 mutation. These results support the assertion that, in UC, these mutations often result in higher levels of TERT reactivation than that achieved by other reactivating mechanisms and confirm that TERT, rather than TR or some other factor, is the limiting component for formation of the telomerase RNP in the UC23 (one striking exception is the SW1710 cell line, which was measured to have ~850 TERT protein molecules but only ~140 TR molecules per cell).
Whether a cell enters replicative senescence or becomes immortal is not thought to depend upon telomerase activity per se, but rather upon telomere maintenance. We therefore measured telomere lengths for each of the UC23 and found longer average lengths in promoter-mutant cell lines (Fig. 3 and fig. S5). Overall, telomerase activity correlated strongly with telomere length (fig. S6). Interestingly, no correlation was seen between increased TERT gene copy number and TERT mRNA or protein levels, telomerase activity or telomere lengths (fig. S7). These data, alongside recent observations that there is no significant TERT CNV in UC, suggest that gene amplification is not an important mechanism for telomerase reactivation in this type of cancer (19).
To determine the clinical relevance of these observations, we evaluated whether TERT mRNA expression was predictive of DSS of UC patients. To make consistent comparisons, we focused our analysis on publically available gene expression profiles of patients for whom DSS endpoint data were available and who had undergone radical cystectomy as definitive treatment. This eliminated the confounding factor of patients who had undergone chemotherapy, since these treatment regimens varied amongst these individuals and can impact DSS (27). A subset of patients from two cohorts met these selection criteria, one from the Chungbuk National University Hospital (CNUH; n = 35) (28) and the other from the Memorial Sloan-Kettering Cancer Center (MSKCC; n = 87) (29). We found that increased expression of TERT mRNA in the tumors of these patients correlated with reduced DSS in both of these cohorts (p = 0.0042 and 0.034, respectively) (Fig. 4). Considering our finding that increased TERT mRNA levels serve as a reliable indicator for increased telomerase activity in the UC23 (Fig. 2F), these results suggest that robust telomerase reactivation is indeed important for UC tumor progression. In contrast, previous studies found no correlation between the presence of promoter mutations and severity of UC (12, 13, 16). This apparent discrepancy may be explained by the fact that, although these mutations do correlate with a more robust reactivation of the enzyme in general, other genetic or epigenetic differences also contribute. For example, the T24 and T24T cell lines have the same number of TERT copies and −124 promoter mutations, but the more tumorigenic T24T cells (30, 31) have ~5-fold higher levels of TERT protein and ~4-fold higher levels of telomerase activity. Taken together, these data suggest that the better marker for patient prognosis is not promoter mutation per se, but TERT expression levels, which directly determine telomerase activity levels.
Fig. 4. Tumor expression level of TERT mRNA is inversely associated with disease-specific survival of patients with UC.
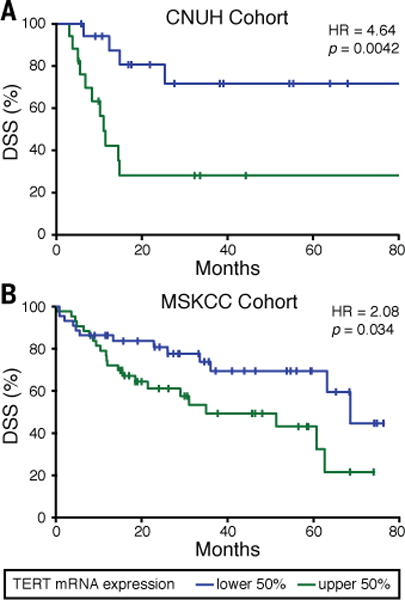
The prognostic value of TERT mRNA expression for DSS in patients who underwent radical cystectomy and who did not receive chemotherapy or additional treatments before or after cystectomy in the (A) CNUH and (B) MSKCC cohorts. Kaplan-Meier curves for patients with low and high expression levels of TERT mRNA were generated (Supplementary Materials) and the corresponding log-rank p-values and hazard ratios (HR) are as indicated. CNUH data include only patients with muscle-invasive tumors, while MSKCC data include patients both with muscle-invasive tumors and non-invasive tumors. A similar analysis could not be performed using the The Cancer Genome Atlas since this dataset is not limited to DSS.
Reactivation of some mechanism for telomere maintenance is considered to be essential for oncogenesis. Because UC does not typically employ the Alternative Lengthening of Telomeres mechanism (32), we originally hypothesized that all of the UC23 would have a similar level of telomerase upregulation; i.e., the promoter mutations would represent only one of several equally effective mechanisms for TERT reactivation (5, 12, 13, 16)(33). Instead, we observed that: (i) the UC cell lines varied widely (~100-fold) in the level of TERT protein and telomerase activity (Fig. 2G), which correlated with their respective telomere lengths (Fig. 3) and (ii) although the mere presence of a promoter mutation did not necessarily lead to high telomerase activity, those cell lines with the highest levels of activity did indeed harbor such mutations (Fig. 2D). These results suggest that TERT promoter mutations provide a particularly effective mechanism for high-level telomerase reactivation in UC. Future studies will be needed to test whether UC tumors harboring high levels of telomerase activity are particularly susceptible to treatment with targeted telomerase inhibition.
Supplementary Material
Acknowledgments
We thank C. Owens for providing the UC23, as well as detailed information on their origins and growth conditions. This work was supported by NIH grants CA075115 and CA104106 to D.T. and R01 GM099705 to T.R.C. S.B.C. is supported by Cancer Council NSW (Australia). T.R.C. is an investigator of the Howard Hughes Medical Institute.
References and Notes
- 1.Kilian A, et al. Hum Mol Genet. 1997;6:2011–2019. doi: 10.1093/hmg/6.12.2011. [DOI] [PubMed] [Google Scholar]
- 2.Gunes C, Lichtsteiner S, Vasserot AP, Englert C. Cancer Res. 2000;60:2116–2121. [PubMed] [Google Scholar]
- 3.Shay JW, Bacchetti S. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 4.Horn S, et al. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 5.Huang FW, et al. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Killela PJ, et al. Proc Natl Acad Sci USA. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Nat Genet. 2014;46:1160–1165. doi: 10.1038/ng.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rachakonda PS, et al. Proc Natl Acad Sci USA. 2013;110:17426–17431. doi: 10.1073/pnas.1310522110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heidenreich B, et al. Nat Commun. 2014;5:3401. doi: 10.1038/ncomms4401. [DOI] [PubMed] [Google Scholar]
- 10.Nault JC, et al. Nat Commun. 2013;4:2218. doi: 10.1038/ncomms3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tallet A, et al. Oncogene. 2013;33:3748–3752. doi: 10.1038/onc.2013.351. [DOI] [PubMed] [Google Scholar]
- 12.Vinagre J, et al. Nat Commun. 2013;4:2185. doi: 10.1038/ncomms3185. [DOI] [PubMed] [Google Scholar]
- 13.Allory Y, et al. Eur Urol. 2014;65:360–366. doi: 10.1016/j.eururo.2013.08.052. [DOI] [PubMed] [Google Scholar]
- 14.Park JI, et al. Nature. 2009;460:66–72. doi: 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Listerman I, Gazzaniga FS, Blackburn EH. Mol Cell Biol. 2014;34:280–289. doi: 10.1128/MCB.00844-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hurst CD, Platt FM, Knowles MA. Eur Urol. 2014;65:367–369. doi: 10.1016/j.eururo.2013.08.057. [DOI] [PubMed] [Google Scholar]
- 17.Lin Y, et al. Clin Cancer Res. 1996;2:929–932. [PubMed] [Google Scholar]
- 18.Cao Y, Bryan TM, Reddel RR. Cancer Sci. 2008;99:1092–1099. doi: 10.1111/j.1349-7006.2008.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoadley KA, et al. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adey A, et al. Nature. 2013;500:207–211. doi: 10.1038/nature12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kjeldsen E, Tordrup D, Hubner GM, Knudsen BR, Andersen FF. Anticancer Res. 2010;30:3257–3265. [PubMed] [Google Scholar]
- 22.Meyerson M, et al. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 23.Li LH, Nerlov C, Prendergast G, MacGregor D, Ziff EB. EMBO J. 1994;13:4070–4079. doi: 10.1002/j.1460-2075.1994.tb06724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gui Y, et al. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xi L, Cech TR. Nucleic Acids Res. 2014;42:8565–8577. doi: 10.1093/nar/gku560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen SB, Reddel RR. Nat Methods. 2008;5:355–360. doi: 10.1038/nmeth.f.209. [DOI] [PubMed] [Google Scholar]
- 27.Grossman HB, et al. N Engl J Med. 2003;349:859–866. doi: 10.1056/NEJMoa022148. [DOI] [PubMed] [Google Scholar]
- 28.Kim WJ, et al. Mol Cancer. 2010;9:3. doi: 10.1186/1476-4598-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. J Clin Oncol. 2006;24:778–789. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 30.Gildea JJ, Golden WL, Harding MA, Theodorescu D. Genes Chromosomes Cancer. 2000;27:252–263. doi: 10.1002/(sici)1098-2264(200003)27:3<252::aid-gcc5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 31.Gildea JJ, et al. Cancer Res. 2002;62:6418–6423. [PubMed] [Google Scholar]
- 32.Heaphy CM, et al. Am J Pathol. 2011;179:1608–1615. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The use of microarray datasets to identify alternative mechanisms of TERT reactivation employed by those cell lines that were wild type at positions −124 and −146 were unsuccessful; average mRNA levels were roughly similar for the (i) ETS1 and ETS2 transcription factors (~1.1-fold higher in the wild type group), (ii) c-MYC transcription factor (~1.4-fold higher in the wild type group) and (iii) core components of the Polycomb Repressive Complex-2 (between ~0.91-fold and ~1.3-fold higher in the wild type group).
- 34.Nandakumar J, et al. Nature. 2012;492:285–289. doi: 10.1038/nature11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.