

Abstract
Background
Low serum levels of the anti-inflammatory club cell secretory protein (CC16) have been associated with an accelerated FEV1 decline in COPD. Whether low circulating CC16 precedes lung function deficits and incidence of COPD in the general population is unknown.
Methods
We used longitudinal data from adults who were COPD-free at baseline from the population-based TESAOD (N=960, mean follow-up: 14yrs), ECRHS-Sp (N=514, 11yrs) and SAPALDIA (N=167, 8yrs) studies. CC16 was measured in serum from baseline and associated with subsequent FEV1 decline and incidence of airflow limitation. To evaluate early life CC16 effects, we also measured circulating CC16 in samples from ages 4-6yrs to predict subsequent lung function in childhood in the CRS (N=427), MAAS (N=481), and BAMSE (N=231) birth cohorts.
Findings
In adults – after adjustment for sex, age, height, smoking status/intensity, pack-years, asthma, and initial FEV1 levels – baseline CC16 was inversely associated with subsequent decline of FEV1 in TESAOD (p=0.0014), ECRHS-Sp (p=0.023), and a similar trend was found in SAPALDIA (p=0.052). Low CC16 at baseline also predicted an increased risk for incident stage 2 airflow limitation (i.e., FEV1/FVC<70% plus FEV1 % predicted < 80%) in TESAOD and ECRHS-Sp. In children, the lowest tertile of CC16 at age 4–6yrs was associated with subsequent FEV1 deficits up to age 16yrs (meta-analyzed estimate from adjusted models on birth cohorts: −68ml, p=0.0001). Results were confirmed among subjects who never smoked by age 16yrs (−71ml, p<0.0001).
Interpretation
Low serum CC16 is associated with subsequent slower growth and accelerated decline of lung function, and increased risk of developing stage 2 airflow limitation.
Funding
US National Heart, Lung, and Blood Institute and EU Seventh Framework Programme. For a complete list of other funding agencies, please refer to the acknowledgements section of the paper.
Introduction
Chronic obstructive pulmonary disease (COPD) – one of the chronic diseases with the highest morbidity and mortality burden across the world1 – may be related to an accelerated decline of lung function during adult life, an impaired lung function growth during childhood, or a combination thereof2. Although smoking has been long known to be the main risk factor for an accelerated decline of lung function and the inception of COPD3, only a proportion of smokers will go on to develop the disease and a significant proportion of COPD cases occurs among never smokers. Apart from alpha1-antitrypsin deficiency, which only accounts for about 1% of all COPD cases4, there are no established biomarkers to identify adults at risk for COPD before the onset of the disease or children who could be predisposed to lung function deficits in adult life.
Club (formerly Clara) cell secretory protein (CC16, also known as CC10 and CCSP) is a homodimeric pneumoprotein that is mainly produced by club cells in the distal airways, but can be measured in circulation5–7. Recurrent noxious environmental exposures, such as cigarette smoking, result in chronically decreased numbers of club cells and levels of serum CC168,9.
Growing evidence indicates that CC16 has anti-inflammatory and anti-toxicant properties in the lung5–7 and may protect against obstructive lung diseases9. In most clinical studies10–15, lower levels of CC16 in blood and airways have been associated with prevalence and severity of COPD. In addition, low serum CC16 was associated with a faster subsequent decline of forced expiratory volume in one second (FEV1) among patients with COPD in the ECLIPSE16 and Lung Health Study17. However, no study to date has evaluated whether serum CC16 is a predictor of lung function trajectories and development of COPD in the general population.
The goals of our study were to determine 1) whether baseline levels of circulating CC16 are associated with subsequent decline of lung function and incidence of airflow limitation in adults and, if so, 2) whether the relation of low CC16 to subsequent lung function deficits is already established in childhood before the effects of active smoking have taken place.
Methods
For studies on adults, we used the Tucson Epidemiological Study of Airway Obstructive Disease (TESAOD) as the main cohort and results were tested for replication in three Spanish centers of the European Community Respiratory Health Survey (ECRHS-Sp) and in the Swiss Cohort Study on Air Pollution and Lung Diseases in Adults (SAPALDIA). For studies on children, we used data from the birth cohorts of the Tucson Children’s Respiratory Study (CRS), the UK Manchester Asthma and Allergy Study (MAAS), and the Swedish Barn/children, Allergy, Milieu, Stockholm, Epidemiological survey (BAMSE).
Adult cohorts
TESAOD is a population-based prospective cohort study of non-Hispanic White households in Tucson, Arizona18. At baseline and in up to 11 subsequent surveys performed over 24 years, participants completed standardized respiratory questionnaires and lung function tests. For the present study, we used data from 960 participants who at enrollment were 21 to 70 years old, had a ratio between FEV1 and forced vital capacity (FVC) ≥ 70%, had available serum samples, and completed lung function tests in at least one follow-up survey.
ECRHS19 enrollment included a random sample of individuals aged 20–44 years and an enrichment sample of subjects who reported taking asthma medication or having had asthma attacks or shortness of breath at night during the last year. Participants completed a detailed questionnaire and lung function tests at baseline and in two follow-up surveys taken about 9 and 20 years later. For the present study, we used data from 514 ECRHS-Sp participants who had FEV1/FVC ≥ 70% and sufficient serum samples from survey 2 (no serum samples were available from survey 1), and who also completed lung function tests in survey 3.
SAPALDIA20 enrollment included a random sample of adults aged 18 to 62 years recruited using population registries from eight areas in Switzerland. Standardized questionnaires and spirometric lung function tests were completed in two follow-up surveys taken approximately 11 and 19 years later. For the present study, we used data from 167 SAPALDIA participants who were selected as controls for a nested COPD study because they had normal lung function (FEV1/FVC ≥ 70% and FVC % predicted ≥ 80%) and sufficient serum samples from survey 2 (no serum samples were available from survey 1), and completed lung function tests in survey 3.
Birth cohorts
CRS is a longitudinal cohort that recruited 1246 healthy infants at birth21. Baseline and multiple follow-up questionnaires were completed up to adult life, including specific questions on active smoking from age 16. Blood was collected at the YR6 survey (mean age, SD: 6.1, 0.8 years) and spirometric lung function tests were performed at YR11 (10.9, 0.7) and YR16 (16.7, 0.6). For the present study, we used data from 438 participants who had YR6 serum (N=360) or plasma (N=78) samples available for CC16 measurements and completed lung function tests at YR11 and/or YR16.
MAAS is a population-based birth cohort, whose participants were recruited prenatally and followed prospectively until age 16 years22. Follow-up visits at YR5, YR8, YR11, and YR16 included both standardized questionnaires and lung function tests. For the present study, we used data from 481 participants who had serum samples available from YR5, completed lung function tests both at YR5 and in at least one of the subsequent surveys YR8, YR11, and YR16.
BAMSE is a population-based birth cohort, whose participants completed standardized questionnaires at baseline and multiple follow-up surveys up to age 1623. Lung function tests were completed at YR8 and YR16. For the present study, we used data from 231 participants who were selected for nested molecular studies based on a random plus asthma-enriched sampling strategy, had available plasma samples from YR4, and completed lung function tests at YR8 and/or YR16.
Lung function
Complete information on lung function tests is provided in the supplementary appendix.
In the present study, based on guidelines from the Global Initiative for Chronic Obstructive Lung Disease (GOLD)1 airflow limitation was defined as FEV1/FVC < 70% and stage 2 airflow limitation as FEV1/FVC < 70% plus FEV1 % predicted < 80%. Sensitivity analyses were also completed using lower limit of normal (LLN) cut-off values (see supplementary appendix). Because bronchodilator response was not tested in most surveys, definitions were based on pre-bronchodilator values and we used the terms “airflow limitation” to define our incident outcome throughout this manuscript.
CC16 measurements
Circulating CC16 was measured in stored samples from “baseline” (survey 1 in TESAOD, survey 2 in ECRHS-Sp and SAPALDIA, YR6 in CRS, YR5 in MAAS, and YR4 in BAMSE) using a commercially available ELISA kit (BioVendor, with branches in Asheville, NC and Modrice, Czech Republic). In addition, in TESAOD CC16 levels were also measured in serum samples from follow-up surveys in the subset of 601 participants for whom they were available. Serum samples were used for all cohorts, with the exception of BAMSE (plasma) and of 78 plasma samples from CRS (see supplementary appendix and Figure E3 for adjustments made for comparability). Because the distribution of CC16 values was skewed, long tail to the right, values were log-transformed and inverse standardized levels were generated24 to test effects related to 1-SD decrease in baseline CC16 levels. Tertiles of CC16 were also used to determine the risk associated with having low CC16 (i.e., lowest tertile) for accelerated FEV1 decline and incident airflow limitation in adults and for deficits in FEV1 growth in children.
Statistical analyses
Complete statistical methods are provided in the supplementary appendix.
In the adult cohorts, we tested the effects of baseline CC16 on subsequent FEV1 decline using multivariate linear regression models that included the rate of FEV1 decline (i.e., the change of FEV1 per year) as the dependent variable and a list of potential predictors (including baseline CC16 and baseline FEV1) as the independent variables. To avoid effects of observations with short follow-up periods, only participants with ≥ 5 years follow-up were included in these analyses in TESAOD (all ECRHS-Sp and SAPALDIA participants had ≥ 5 years follow-up). Results were also confirmed in TESAOD in analyses that included all participants. Because multiple observations for each participant were available in TESAOD, in this cohort we also used random coefficients models25, which adjusted for the intra-household cluster correlation and intra-subject serial correlation of repeated observations, to assess the effects of CC16 at baseline on FEV1 levels during the study follow-up. These models included covariates and an interaction term between CC16 tertiles and years of follow-up to test whether FEV1 decline differed across CC16 tertiles.
Incident airflow limitation was studied in TESAOD and ECRHS-Sp. In TESAOD, the relation of baseline CC16 to the risk of incident airflow limitation was tested in Cox proportional hazards models. Because in ECRHS-Sp only one follow-up survey was available after baseline CC16 measurements, CC16 associations with incident airflow limitation were tested in logistic regression models. Model discrimination was assessed by the Harrell’s C statistics in Cox models and by the area under the curve (AUC) in logistic regression models. Because a household-based recruitment strategy was used in TESAOD, household-clustered sandwich estimators of standard errors were used in regression and Cox models for this cohort.
Among the 960 TESAOD participants included in this study, 601 (63%) had serum samples available during the follow-up, which were used for a prospective CC16 measurement. The mean number of years between the baseline and prospective measurement was 6.5 years with a range of 1 to 11 years. Prospective CC16 data in TESAOD were analyzed by generating two variables based on: 1) the quartiles of the rate of change of serum CC16 between baseline and the prospective measurement as previously done24; and 2) the combination of CC16 at baseline and at the prospective measurement categorized into three groups (“persistently low CC16”: i.e. being in the lowest CC16 tertile at both surveys; “inconsistently low CC16”: being in the lowest tertile in one but not both surveys; and “persistently high CC16”: not being in the lowest tertile at either survey). These variables were then used as independent variables in regression and Cox models as described above. In these analyses, the follow-up for Cox models started at the time of the prospective measurement.
In analyses on birth cohorts, random effects models25 were used to assess the effects of low CC16 on lung function growth in childhood. In order to remove potential effects by active smoking, in sensitivity analyses the same models were also tested after removing observations of children who smoked by age 16 in CRS and BAMSE. Because in MAAS information on active smoking was not available from YR16, in these analyses only YR8 and YR11 observations were included for this cohort.
Role of the funding source
The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. All authors had final responsibility for the decision to submit the paper for publication.
Results
Tables E1–3 summarize the baseline characteristics of participants from the main adult cohort of TESAOD and the two replication adult cohorts of ECRHS-Sp and SAPALDIA. In TESAOD, the study population included 59% females, 58% ever smokers, and 9% asthmatics (Table E1). At baseline, participants had a mean age of 45 years and a mean FEV1 % predicted of 98%. Similar distributions by demographic and clinical characteristics were found in the replication cohorts, although ECRHS-Sp had less females and SAPALDIA less smokers and asthmatics. Table E4 compares the characteristics of TESAOD participants who were included and excluded from the present study. As compared with participants excluded from the study, those included were slightly more likely to be female and older and less likely to be underweight. None of the other characteristics differed between the two groups.
The median and inter-quartile serum CC16 levels were 8.0 (5.7 – 10.9) ng/ml. Table 1 shows univariate associations of baseline characteristics of TESAOD participants with serum CC16 and Table E5 the independent effects of sex, age, BMI, and smoking on serum CC16 in TESAOD. CC16 levels were higher in males than females, had a U-shaped distribution across age categories, and were strongly and inversely associated with current smoking and pack-years. No association was found with asthma, but serum CC16 was directly associated with FEV1 % predicted, FVC % predicted, and the FEV1/FVC ratio.
Table 1.
Associations of baseline characteristics with baseline CC16 values in TESAOD.
| Serum CC16 levels | |
|---|---|
|
Geometric Means of serum CC16 in ng/ml P value |
|
|
| |
| Sex | |
| Females (N=570) | 7.38 |
| Males (N=390) | 8.08 P = 0.014 |
|
| |
| Age categories | |
| 21 ≤ years < 30 (N=245) | 8.29 |
| 30 ≤ years < 45 (N=212) | 6.67 |
| 45 ≤ years < 60 (N=278) | 7.21 |
| 60 ≤ years ≤ 70 (N=225) | 8.61 PANOVA < 0.0001; PTREND = 0.378 |
|
| |
| BMI categories | |
| Underweight (N=14) | 5.63 |
| Normal weight (N=538) | 7.77 |
| Overweight (N=308) | 7.61 |
| Obese (N=67) | 7.04 PANOVA = 0.106; PTREND = 0.536 |
|
| |
| Smoking status | |
| Never (N=406) | 8.81 |
| Former (N=220) | 8.16 |
| Current (N=333) | 6.21 PANOVA < 0.0001 |
|
| |
| Ever physician-confirmed asthma: | |
| No (N=870) | 7.72 |
| Yes (N=89) | 7.15 P = 0.222 |
|
| |
|
Spearman corr coeff with CC16 levels P value |
|
|
| |
| Pack-years (all subjects, N=959) | −0.249 P < 0.0001 |
|
| |
| Pack-years (only smokers, N=553) | −0.178 P < 0.0001 |
|
| |
| FEV1 % predicted (N=960) | 0.121 P = 0.0002 |
|
| |
| FVC % predicted (N=960) | 0.066 P = 0.041 |
|
| |
| FEV1/FVC ratio (N=960) | 0.073 P = 0.023 |
The TESAOD participants included in this study completed on average 6.8 lung function tests (SD: 3; range: 2 to 12) over a mean follow-up of 13.7 years (SD: 7, range: 1 to 23 years) for a total of 6,549 lung function observations. Younger age, normal weight, never smoking, and a slightly better lung function at baseline were related to having a longer follow-up. However, no relation of baseline levels of serum CC16 to length of follow-up was found (Table E6).
In TESAOD – after adjusting for sex, age, height, smoking status and intensity, pack-years, asthma, and initial FEV1 levels – baseline serum CC16 levels predicted subsequent decline of FEV1 during the study follow-up, with 1-SD decrease in baseline CC16 being associated with a 4.4 ml/yr faster FEV1 decline (Table 2, p = 0.0014). The inverse association between baseline CC16 and subsequent FEV1 decline was replicated in ECRHS-Sp and a similar trend was found in SAPALDIA (Table 2). These results were confirmed after removing subjects with FEV1/FVC below LLN at baseline (TESAOD p=0.0033, ECRHS-Sp p=0.023, SAPALDIA p=0.052) and after further adjustment for BMI categories (TESAOD p=0.0008, ECRHS-Sp p=0.012, SAPALDIA p=0.057). When the rate of FEV1 decline was compared across CC16 tertiles in adjusted random coefficients models in TESAOD (Figure 1), males and females with low CC16 at baseline had an FEV1 decline during the study follow-up that was on average 9.7 ml/yr (p=0.0043) and 6.3 ml/yr (p=0.0034) faster, respectively, than that of their peers with high CC16.
Table 2.
Association between baseline serum CC16 and subsequent decline of FEV1 in TESAOD, ECRHS-Sp, and SAPALDIA.
| Increase in FEV1 decline associated with 1-SD decrease in baseline CC16 Beta coefficient* (95% CI) p value |
|
|---|---|
|
TESAOD N subjects = 800**; N obs = 6114 Mean yrs follow-up = 16.0 |
−4.4 (−7.1, −1.7) ml/yr p = 0.0014 |
|
ECRHS-Sp N subjects = 495ˆ; N obs = 990 Mean yrs follow-up = 11.5 |
−2.4 (−4.5, −0.3) ml/yr p = 0.023 |
|
SAPALDIA N subjects = 164ˆˆ; N obs = 328 Mean yrs follow-up = 8.3 |
−4.5 (−9.0, 0.0) ml/yr p = 0.052 |
| Meta-analyzed estimate | −3.3 (−4.8, −1.8) ml/yr p < 0.0001 |
Adjusted for sex, age, height, smoking status and intensity, pack-years, asthma, and FEV1 levels at baseline. These covariates were included because they are known to affect FEV1 levels and/or decline. ECRHS-Sp models also included center and sample type (random versus enriched) and SAPALDIA models study area. The effects of CC16 on FEV1 decline were also tested for interaction with smoking and with age (see Tables E7 and E8).
TESAOD analyses included participants with ≥ 5 years follow-up.
Of the 514 ECRHS-Sp participants, 19 had missing smoking status and/or intensity information and were excluded from analyses.
Of the 167 SAPALDIA participants, 3 had missing smoking intensity information and were excluded from analyses.
Figure 1.
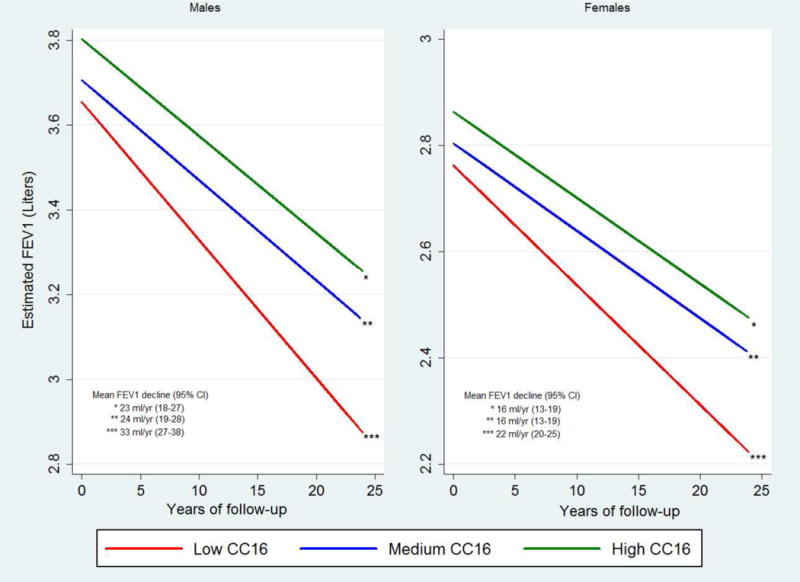
FEV1 levels at baseline and decline during the study follow-up as estimated from fully adjusted random coefficients models for TESAOD participants across the three tertiles of serum CC16 at baseline (models were run separately for males and females). Depicted values represent predicted values for a 175-cms tall male and a 170-cms tall female who were 45 years old at baseline.
* Results come from random coefficients models adjusted for age, smoking intensity, and physician-confirmed asthma (all variables at baseline), and for height, smoking status, pack-years, and years of follow-up (time-dependent variables). The model for males included 389 participants for a total of 2546 observations. The model for females included 569 participants for a total of 3864 observations.
Although in TESAOD CC16 associations with FEV1 decline appeared stronger in smokers than never smokers, we did not find evidence of such interaction in either ECRHS-Sp or SAPALDIA (Table E7). No statistical evidence of an interaction with age was found either, even though in TESAOD CC16 associations tended to be stronger in subjects ≥ 45 years than in those < 45 years old at baseline (Table E8).
Consistent with results on FEV1 decline, in Cox models TESAOD participants in the lowest CC16 tertile were found to be at increased risk of incident airflow limitation and incident stage 2 airflow limitation during the study follow-up, although only the latter association held true after full adjustment for covariates including initial levels of FEV1/FVC (Table 3a). In fully adjusted models, 1-SD decrease in baseline CC16 was associated with a 27% increased risk for incident stage 2 airflow limitation (adjHR: 1.03–1.56) (Table 3a). The inverse association between serum CC16 and incidence of stage 2 airflow limitation was replicated in the ECRHS-Sp study (Table 3b), although it was significant only when CC16 was used on a continuous scale.
TABLE 3.
Crude and adjusted HRs and ORs associated with baseline serum CC16 for incident airflow limitation in TESAOD (Table 3a) and in the replication population of ECRHS-Sp (Table 3b). The highest CC16 tertile is the reference group.
| a | Hazard Ratios for incident airflow limitation N subjects who developed airflow limitation/total N subjects (251/958)ˆ | Hazard Ratios for incident stage 2 airflow limitation N subjects who developed stage 2 airflow limitation/total N subjects (106/958)ˆ | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Crude HR (95% CI) P value |
Adj* HR (95% CI) P value |
Adj** HR (95% CI) P value |
Crude HR (95% CI) P value |
Adj* HR (95% CI) P value |
Adj** HR (95% CI) P value |
|
| Effect associated with baseline CC16 tertiles | ||||||
| HIGH | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) |
| MEDIUM | 1.09 (0.79 – 1.49) p = 0.613 |
1.17 (0.86 – 1.60) p = 0.325 |
1.15 (0.84 – 1.57) p = 0.376 |
1.53 (0.89 – 2.60) p = 0.121 |
1.65 (0.97 – 2.82) p = 0.067 |
1.71 (1.00 – 2.93) p = 0.051 |
| LOW | 1.45 (1.07 – 1.96) p = 0.017 |
1.15 (0.84 – 1.58) p = 0.387 |
1.10 (0.79 – 1.51) p = 0.573 |
2.36 (1.44 – 3.88) p = 0.0007 |
1.82 (1.08 – 3.07) p = 0.024 |
1.81 (1.06 – 3.09) p = 0.029 |
| P value for trend | 0.017 | 0.403 | 0.596 | 0.0005 | 0.026 | 0.032 |
|
| ||||||
| Effect associated with 1-SD decrease in CC16 | 1.17 (1.02 – 1.33) p = 0.022 |
1.09 (0.93 – 1.27) p = 0.284 |
1.06 (0.91 – 1.23) p = 0.458 |
1.37 (1.16 – 1.61) p = 0.0002 |
1.29 (1.04 – 1.59) p = 0.019 |
1.27 (1.03 – 1.56) p = 0.026 |
|
| ||||||
| Harrell’s C statistics (model with CC16 tertiles) | 0.55 | 0.73 | 0.81 | 0.60 | 0.79 | 0.83 |
| b | Odds Ratios for incident airflow limitation N subjects who developed airflow limitation/total N subjects (70/495)ˆ | Odds Ratios for incident stage 2 airflow limitation N subjects who developed stage 2 airflow limitation/total N subjects (28/495)ˆ | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Crude OR (95% CI) P value |
Adj* OR (95% CI) P value |
Adj** OR (95% CI) P value |
Crude OR (95% CI) P value |
Adj* OR (95% CI) P value |
Adj** OR (95% CI) P value |
|
| Effect associated with baseline CC16 tertiles | ||||||
| HIGH | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) | 1.00 (ref) |
| MEDIUM | 1.53 (0.76 – 3.06) p = 0.232 |
1.47 (0.72 – 3.02) p = 0.289 |
1.37 (0.56 – 3.36) p = 0.488 |
2.04 (0.60 – 6.91) p = 0.253 |
1.89 (0.53 – 6.66) p = 0.324 |
1.80 (0.46 – 7.02) p = 0.397 |
| LOW | 2.41 (1.25 – 4.64) p = 0.0083 |
2.14 (1.06 – 4.30) p = 0.033 |
2.12 (0.89 – 5.05) p = 0.091 |
4.18 (1.37 – 12.8) p = 0.012 |
3.35 (1.02 – 10.9) p = 0.045 |
3.06 (0.84 – 11.2) p = 0.090 |
| P value for trend | 0.0070 | 0.032 | 0.085 | 0.0072 | 0.036 | 0.079 |
|
| ||||||
| Effect associated with 1-SD decrease in CC16 | 1.47 (1.17 – 1.85) p = 0.0011 |
1.39 (1.08 – 1.80) p = 0.012 |
1.38 (1.00 – 1.92) p = 0.051 |
1.75 (1.27 – 2.40) p = 0.0005 |
1.64 (1.13 – 2.40) p = 0.010 |
1.53 (1.00 – 2.34) p = 0.049 |
|
| ||||||
| AUC (model with CC16 tertiles) | 0.60 | 0.70 | 0.92 | 0.65 | 0.81 | 0.91 |
adjusted for sex, age, smoking status and intensity, pack-years, and asthma at baseline
adjusted for sex, age, smoking status and intensity, pack-years, asthma, and FEV1/FVC at baseline. These covariates were included because they are known to affect the risk for COPD.
Of the 960 TESAOD participants, 2 had missing smoking or asthma information and were excluded from analyses
adjusted for center, sample type (random versus enriched), sex, age, smoking status and intensity, pack-years, and asthma at baseline
adjusted for center, sample type (random versus enriched), sex, age, smoking status and intensity, pack-years, asthma, and FEV1/FVC at baseline
Of the 514 ECRHS-Sp participants, 19 had missing smoking status and/or intensity information and were excluded from analyses
Results from analyses on the subset of 601 TESAOD participants with available CC16 measurements from baseline and follow-up are shown in the appendix. After full adjustment, we found a trend between a steeper CC16 decrease from the baseline to the follow-up survey and a higher risk of incident stage 2 airflow limitation. In addition, as compared with participants who never had low CC16 those with persistently low CC16 had a 9.0 (4.3 – 13.7; p=0.0004) ml/yr faster FEV1 decline over the follow-up.
To determine whether the association of CC16 levels with subsequent lung function originates early in life and also affects lung growth, we used data from the CRS, MAAS, and BAMSE birth cohorts, in which circulating CC16 was measured in samples collected at age 6, 5, and 4 years, respectively. The baseline characteristics of the children included in these analyses are summarized in Tables E9–11. Univariate and multivariate analyses on the associations of circulating CC16 levels at age 6 years with baseline characteristics of CRS participants are shown in Table E12. Figure 2 shows that in adjusted random effects models on the three cohorts low levels of early CC16 at age 4–6 years predicted subsequent FEV1 deficits in childhood. The lowest tertile of early CC16 was associated with a meta-analyzed 68-ml deficit in FEV1 up to age 16 years (p=0.0001), which was equivalent to 2–3% of expected FEV1 levels across the three cohorts. Estimates were not substantially reduced by further adjustment for asthma (Figure E4) and associations were confirmed after restricting analyses to subjects who never smoked up to age 16 years (Figure E5).
Figure 2.
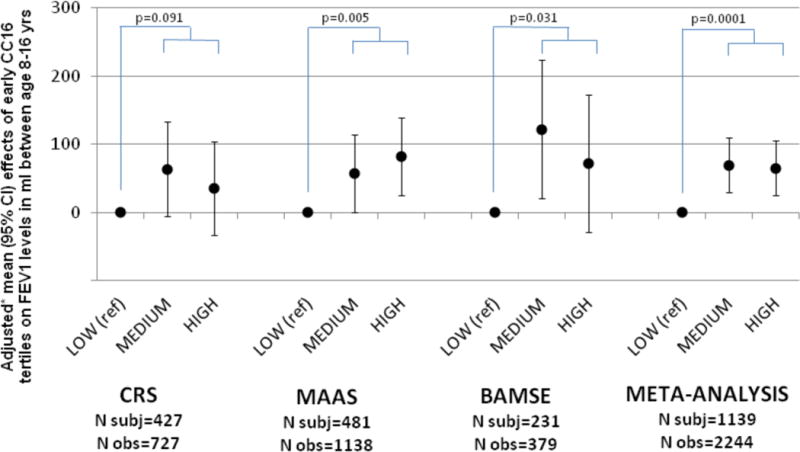
Relation of the medium and high tertile of early circulating CC16 (as compared with the lowest tertile) to subsequent FEV1 levels achieved during childhood up to age 16 years in the CRS, MAAS, and BAMSE birth cohorts.
* Results come from random effects models adjusted for sex, age, height, survey, maternal smoking, ethnicity (CRS and MAAS), and baseline FEV1 (MAAS). These covariates were included because of their possible effects on early CC16 and/or lung function growth. The dependent variable of the models was FEV1 at ages 11 and 16 yrs in CRS, FEV1 at ages 8, 11, and 16 yrs in MAAS, and FEV1 at ages 8 and 16 yrs in BAMSE.
Early CC16 was measured at age 6 years (mean 6.1 yrs; SD 0.8 yrs) in CRS, 5 years (5.0, 0.1) in MAAS, and 4 years (4.0, 0.1) in BAMSE.
Reported p values refer to the comparison of the lowest early CC16 tertile versus the other two tertiles (medium and high) combined.
Discussion
This is the first prospective study to show that decreased circulating CC16 levels are a risk factor for reduced growth of lung function in childhood, accelerated lung function decline in adulthood, and for the development of moderate airflow limitation in the general adult population. These findings are consistent with previous reports showing that lower baseline CC16 was associated with steeper FEV1 decline among patients with COPD16,17. Here, we show that this association holds true in subjects without COPD at the time of measurement, suggesting that the relationship between CC16 and FEV1 decline may be established before the inception of the disease. In support of this contention, we found that low circulating CC16 early in life predicted subsequent lung function deficits by adolescence in three birth cohorts. Thus, low CC16 was a risk factor for lung function deficits across the life span.
Among the factors that may affect CC16 levels in early childhood, in our study we found sex, age, BMI, maternal smoking, and active wheezing to be independent predictors of circulating CC16 (Table E12). However, after taking these factors into account only a small proportion (~6%) of the variability in CC16 levels was explained, supporting that other genetic, physiological, environmental, and developmental factors are involved, as also suggested by previous research26–29.
Whether the relation of CC16 to growth and decline of lung function and development of COPD is causal or is confounded by other unmeasured factors cannot be conclusively determined from our data. Nevertheless, the results from our birth cohorts support the conclusion that CC16 has effects on lung function that are at least partly independent of cigarette smoking. Interestingly, narrowing and disappearance of small airways on multidetector computed tomography has been shown to be present in early stages of COPD30, a finding that may be relevant to the CC16 deficits seen in this disease.
The molecular mechanisms by which CC16 could protect against development of COPD are not known. Consistent with possible anti-inflammatory and anti-toxicant activities of this molecule in the lung5–7, CC16-deficient mice31–33 have been shown to have increased lung epithelial injury, airway inflammation, and susceptibility to oxidative stress and infectious agents. Most animal studies also indicate an increased susceptibility of knockout mice to COPD-related phenotypes in response to cigarette smoking34,35, although another study failed to find an increased risk for emphysema17. Results from in vitro studies indicate that CC16 may affect inflammatory cell function36,37 and these effects could be linked to inhibition of PLA2 activity38, prostaglandins39, chemotaxis36,40, and cytokine production37,41.
Although this study supports the temporal relation of CC16 to subsequent lung health outcomes, the possible relevance of this biomarker in the clinical or public health setting remains to be determined. In most cohorts we analyzed data from a single CC16 measurement, although results from the TESAOD subset with prospective CC16 measurements supported the potential added value of temporal trajectories of CC16 in risk prediction. No information was available on other physiological predictors of systemic CC16 levels, such as glomerular filtration rate (which is inversely related to serum CC16)42 and diurnal variation43. Longitudinal studies that can control for such physiological factors and can model serial CC16 measurements will be required to establish conclusively the potential value of this molecule in informing prevention or treatment of COPD, either in the general population or in targeted subgroups. In TESAOD, CC16 effects appeared stronger in smokers than never smokers, but we found no evidence for such trend in the two replication cohorts. This may reflect no real interaction with smoking, true inter-population differences, or inter-cohort methodological variability.
Our analyses on lung function have some limitations. No bronchodilator response was tested in TESAOD and therefore we could not determine how CC16 effects relate to the risk of COPD defined using post-bronchodilator lung function1. Also, differences in the number of available tests, length of follow-up, and instruments may have reduced comparability of lung function measurements across the adult and birth cohorts. This may be the case also for CC16 measurements, based on inter-cohort differences in blood collection protocols and serum storage conditions, although CC16 levels have been shown to be quite stable in serum44. However, the use of standardized levels and tertiles of CC16 within each cohort and the implementation of a prospective study design with inclusion of participants with comparable follow-up periods both in the adult and pediatric cohorts are expected to have reduced the impact of such differences. In addition, while these differences may affect inter-cohort comparability they do not impact the internal validity of the results.
Among the strengths of our study are the long-term and population-based nature of our populations, the extensive longitudinal characterization of lung function, and the replication of findings in multiple independent cohorts that cover a significant part of lifespan.
In summary, we found low circulating CC16 levels to be an independent predictor for subsequent deficits in lung function growth in childhood and for accelerated lung function decline and incident COPD in adult life.
Supplementary Material
Panel: Research in Context.
Evidence before this study
Low circulating levels of CC16 have been linked to subsequent accelerated decline of FEV1 among patients with COPD. These observations are in line with experimental evidence supporting anti-inflammatory and anti-oxidative effects of this molecule in the lung. However, whether low circulating CC16 predicts subsequent lung function decline and incidence of COPD in the general population remains unknown. It also remains unknown whether the potential effects of CC16 on subsequent lung function are already present in childhood before the effects of cigarette smoking have taken place. We completed several PubMed searches using search terms such as “CC16”, “CC10”, “CCSP”, “uteroglobin”, “Clara cell”, “Club cell”, “COPD”, “emphysema”, “chronic bronchitis”, “asthma”, and “lung function” without language restrictions, which returned several cross-sectional studies linking circulating CC16 to asthma, COPD, and lung function and two clinical studies linking circulating CC16 to subsequent decline of FEV1 among patients with COPD. However, we found no previous prospective study that assessed circulating CC16 in relation to subsequent lung function growth/decline or in relation to subsequent incidence of COPD in the general population. The date of our last search was Nov 28, 2014.
Added value of this study
In six population-based cohorts, we found consistent relations of low circulating levels of CC16 to subsequent accelerated decline of lung function and incidence of COPD in adults and to subsequent lung function deficits in children up to age 16 years. These results held true after adjustment for other known risk factors and they cannot be simply explained by residual confounding by smoking. Our study provides novel evidence that implicates low CC16 as an independent risk factor for subsequent lung function deficits and COPD.
Implications of all the available evidence
Our results indicate the possible value of CC16 for risk stratification. Longitudinal studies designed to determine determinants of circulating CC16 and to model serial CC16 measurements are warranted to establish conclusively the potential value of this molecule in informing prevention or treatment of COPD.
Acknowledgments
This study was supported by awards HL107188, HL095021, and HL056177 from the National Heart, Lung, and Blood Institute, US National Institutes of Health; FIS award PS09/01354 from the Spanish Instituto de Salud Carlos III; a post-doctoral fellowship to IL by the Environment and Health Fund, Israel; grant 33CSCO-134276 from the Swiss National Science Foundation; grant awards by the Swedish Research Council, the Swedish Heart-Lung Foundation, and the Stockholm County Council (ALF); grants G0601361 and MR/K002449/1 by the Medical Research Council, UK; a grant award by the JP Moulton Charitable Foundation; and grant agreement number 261357 (Mechanisms of the Development of ALLergy – MeDALL) by the EU Seventh Framework Programme.
We are grateful to Dr Duane Sherrill for the valuable advice provided for this study.
Conflict of Interest Statements: Stefano Guerra reports grants from NIH, during the conduct of the study.
Marilyn Halonen reports grants from National Institutes of Health, during the conduct of the study; grants from National Institutes of Health, outside the submitted work.
Monica Vasquez reports grants from NIH/NHLBI, during the conduct of the study.
Wayne Morgan reports grants from NIH, during the conduct of the study; personal fees from Genentech, personal fees from Cystic Fibrosis Foundation, grants from Cystic Fibrosis Foundation, grants from NIH, personal fees from American Thoracic Society, personal fees from American College of Chest Physicians, personal fees from American Academy of Allergy, Asthma, and Immunology, outside the submitted work.
Lluisa Tares reports grants from Instituto de Salud Carlos III (Spain), during the conduct of the study.
Jean Bousquet reports personal fees from Almirall, Meda, Merck, MSD, Novartis, Sanofi-Aventis, Takeda, Teva, Uriach, personal fees from Almirall, AstraZeneca, Chiesi, GSK, Meda, Menarini, Merck, MSD, Novartis, Sanofi-Aventis, Takeda, Teva, Uriach, personal fees from Stallergènes, outside the submitted work.
Danielle Belgrave reports other from GlaxoSmithKline, outside the submitted work.
Angela Simpson reports grants from Medical research council, grants from The JP Moulton Charitable Foundation, grants from North West Lung Centre Charity, grants from National Institute of Health Research, during the conduct of the study.
Adnan Custovic reports grants from Medical Research Council, grants from J P Moulton Charitable Foundation, grants from EU FP7 programme, personal fees from Thermo Fisher, personal fees from Novartis, personal fees from GlaxoSmithKline, personal fees from AstraZeneca, outside the submitted work.
Fernando Martinez reports grants from NHLBI/NIH, during the conduct of the study; grants from NIH, other from Abbott, outside the submitted work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributors
SG and FDM designed the study. SG, MMV, DAS, AEC, DK, JH, and DCMB analyzed the data. AS measured CC16 in the TESAOD, SAPALDIA, and CRS samples with supervision by MH. LT measured CC16 in the ECRHS samples with supervision by EB. IL measured CC16 in the BAMSE samples with supervision by CD. Leadership and coordination for the specific cohorts were provided by SG and MMV (TESAOD), JPZ, JMM, IU, and JS (ECRHS-Sp), NPH and MI (SAPALDIA), FDM, ALW, SG, and WJM (CRS), AC and AS (MAAS), and EM and MW (BAMSE). JMA and JB are the principle investigators of the MeDALL project. SG and FDM drafted the manuscript with input from all authors. All authors approved the final version of the manuscript.
The other authors declared no conflicts of interest.
References
- 1.Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–65. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 2.Guerra S, Martinez FD. Natural History. In: Barnes PJ, Drazen JM, Rennard SI, Thomson NC, editors. Asthma and COPD. 2nd. Academic Press; 2009. pp. 23–35. [Google Scholar]
- 3.Burrows B, Knudson RJ, Cline MG, Lebowitz MD. Quantitative relationships between cigarette smoking and ventilatory function. Am Rev Respir Dis. 1977;115(2):195–205. doi: 10.1164/arrd.1977.115.2.195. [DOI] [PubMed] [Google Scholar]
- 4.Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. The New England journal of medicine. 2009;360(26):2749–57. doi: 10.1056/NEJMcp0900449. [DOI] [PubMed] [Google Scholar]
- 5.Broeckaert F, Bernard A. Clara cell secretory protein (CC16): characteristics and perspectives as lung peripheral biomarker. Clin Exp Allergy. 2000;30(4):469–75. doi: 10.1046/j.1365-2222.2000.00760.x. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee AB, Zhang Z, Chilton BS. Uteroglobin: a steroid-inducible immunomodulatory protein that founded the Secretoglobin superfamily. Endocr Rev. 2007;28(7):707–25. doi: 10.1210/er.2007-0018. [DOI] [PubMed] [Google Scholar]
- 7.Lakind JS, Holgate ST, Ownby DR, et al. A critical review of the use of Clara cell secretory protein (CC16) as a biomarker of acute or chronic pulmonary effects. Biomarkers. 2007;12(5):445–67. doi: 10.1080/13547500701359327. [DOI] [PubMed] [Google Scholar]
- 8.Shijubo N, Itoh Y, Yamaguchi T, et al. Serum and BAL Clara cell 10 kDa protein (CC10) levels and CC10-positive bronchiolar cells are decreased in smokers. Eur Respir J. 1997;10(5):1108–14. doi: 10.1183/09031936.97.10051108. [DOI] [PubMed] [Google Scholar]
- 9.Rava M, Tares L, Lavi I, et al. Serum levels of Clara cell secretory protein, asthma, and lung function in the adult general population. J Allergy Clin Immunol. 2013;132(1):230–2. doi: 10.1016/j.jaci.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Lomas DA, Silverman EK, Edwards LD, Miller BE, Coxson HO, Tal-Singer R. Evaluation of serum CC-16 as a biomarker for COPD in the ECLIPSE cohort. Thorax. 2008;63(12):1058–63. doi: 10.1136/thx.2008.102574. [DOI] [PubMed] [Google Scholar]
- 11.Bernard A, Marchandise FX, Depelchin S, Lauwerys R, Sibille Y. Clara cell protein in serum and bronchoalveolar lavage. Eur Respir J. 1992;5(10):1231–8. [PubMed] [Google Scholar]
- 12.Pilette C, Godding V, Kiss R, et al. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(1):185–94. doi: 10.1164/ajrccm.163.1.9912137. [DOI] [PubMed] [Google Scholar]
- 13.Braido F, Riccio AM, Guerra L, et al. Clara cell 16 protein in COPD sputum: a marker of small airways damage? Respir Med. 2007;101(10):2119–24. doi: 10.1016/j.rmed.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 14.Ye Q, Fujita M, Ouchi H, et al. Serum CC-10 in inflammatory lung diseases. Respiration. 2004;71(5):505–10. doi: 10.1159/000080636. [DOI] [PubMed] [Google Scholar]
- 15.Sin DD, Leung R, Gan WQ, Man SP. Circulating surfactant protein D as a potential lung-specific biomarker of health outcomes in COPD: a pilot study. BMC Pulm Med. 2007;7:13. doi: 10.1186/1471-2466-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. The New England journal of medicine. 2011;365(13):1184–92. doi: 10.1056/NEJMoa1105482. [DOI] [PubMed] [Google Scholar]
- 17.Park HY, Churg A, Wright JL, et al. Club cell protein 16 and disease progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(12):1413–9. doi: 10.1164/rccm.201305-0892OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lebowitz MD, Knudson RJ, Burrows B. Tucson epidemiologic study of obstructive lung diseases. I: Methodology and prevalence of disease. Am J Epidemiol. 1975;102(2):137–52. doi: 10.1093/oxfordjournals.aje.a112141. [DOI] [PubMed] [Google Scholar]
- 19.The European Community Respiratory Health Survey, II. Eur Respir J. 2002;20(5):1071–9. doi: 10.1183/09031936.02.00046802. [DOI] [PubMed] [Google Scholar]
- 20.Ackermann-Liebrich U, Kuna-Dibbert B, Probst-Hensch NM, et al. Follow-up of the Swiss Cohort Study on Air Pollution and Lung Diseases in Adults (SAPALDIA 2) 1991–2003: methods and characterization of participants. Soz Praventivmed. 2005;50(4):245–63. doi: 10.1007/s00038-005-4075-5. [DOI] [PubMed] [Google Scholar]
- 21.Taussig LM, Wright AL, Morgan WJ, Harrison HR, Ray CG. The Tucson Children’s Respiratory Study. I. Design and implementation of a prospective study of acute and chronic respiratory illness in children. Am J Epidemiol. 1989;129(6):1219–31. doi: 10.1093/oxfordjournals.aje.a115242. [DOI] [PubMed] [Google Scholar]
- 22.Custovic A, Simpson BM, Murray CS, Lowe L, Woodcock A. The National Asthma Campaign Manchester Asthma and Allergy Study. Pediatr Allergy Immunol. 2002;13(Suppl 15):32–7. doi: 10.1034/j.1399-3038.13.s.15.3.x. [DOI] [PubMed] [Google Scholar]
- 23.Ekstrom S, Magnusson J, Kull I, et al. Maternal BMI in early pregnancy and offspring asthma, rhinitis and eczema up to 16 years of age. Clin Exp Allergy. 2014 doi: 10.1111/cea.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerra S, Vasquez MM, Spangenberg A, Halonen M, Martinez FD. Serum concentrations of club cell secretory protein (Clara) and cancer mortality in adults: a population-based, prospective cohort study. The Lancet Respiratory medicine. 2013;1(10):779–85. doi: 10.1016/S2213-2600(13)70220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown H, Prescott R. Applied mixed models in medicine. Chichester, UK: John Wiley & Sons, LTD; 2001. [Google Scholar]
- 26.Kim DK, Cho MH, Hersh CP, et al. Genome-wide association analysis of blood biomarkers in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(12):1238–47. doi: 10.1164/rccm.201206-1013OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernard A, Nickmilder M, Dumont X. Airway Epithelium Defects and Risks of Allergic Diseases: Multiple Associations Revealed by a Biomarker Study among Adolescents. Am J Respir Crit Care Med. 2015;191(6):714–7. doi: 10.1164/rccm.201409-1748LE. [DOI] [PubMed] [Google Scholar]
- 28.Broeckaert F, Arsalane K, Hermans C, et al. Serum clara cell protein: a sensitive biomarker of increased lung epithelium permeability caused by ambient ozone. Environmental health perspectives. 2000;108(6):533–7. doi: 10.1289/ehp.00108533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagerkvist BJ, Bernard A, Blomberg A, et al. Pulmonary epithelial integrity in children: relationship to ambient ozone exposure and swimming pool attendance. Environmental health perspectives. 2004;112(17):1768–71. doi: 10.1289/ehp.7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. The New England journal of medicine. 2011;365(17):1567–75. doi: 10.1056/NEJMoa1106955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Kundu GC, Yuan CJ, et al. Severe fibronectin-deposit renal glomerular disease in mice lacking uteroglobin. Science. 1997;276(5317):1408–12. doi: 10.1126/science.276.5317.1408. [DOI] [PubMed] [Google Scholar]
- 32.Stripp BR, Reynolds SD, Plopper CG, Boe IM, Lund J. Pulmonary phenotype of CCSP/UG deficient mice: a consequence of CCSP deficiency or altered Clara cell function? Ann N Y Acad Sci. 2000;923:202–9. doi: 10.1111/j.1749-6632.2000.tb05531.x. [DOI] [PubMed] [Google Scholar]
- 33.Snyder JC, Reynolds SD, Hollingsworth JW, Li Z, Kaminski N, Stripp BR. Clara cells attenuate the inflammatory response through regulation of macrophage behavior. Am J Respir Cell Mol Biol. 2010;42(2):161–71. doi: 10.1165/rcmb.2008-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laucho-Contreras ME, Polverino F, Gupta K, et al. Protective role for club cell secretory protein-16 (CC16) in the development of chronic obstructive pulmonary disease. Eur Respir J. 2015 doi: 10.1183/09031936.0134214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu L, Di PY, Wu R, Pinkerton KE, Chen Y. Repression of CC16 by Cigarette Smoke (CS) Exposure. PloS one. 2015;10(1):e0116159. doi: 10.1371/journal.pone.0116159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lesur O, Bernard A, Arsalane K, et al. Clara cell protein (CC-16) induces a phospholipase A2-mediated inhibition of fibroblast migration in vitro. Am J Respir Crit Care Med. 1995;152(1):290–7. doi: 10.1164/ajrccm.152.1.7541278. [DOI] [PubMed] [Google Scholar]
- 37.Dierynck I, Bernard A, Roels H, De Ley M. Potent inhibition of both human interferon-gamma production and biologic activity by the Clara cell protein CC16. Am J Respir Cell Mol Biol. 1995;12(2):205–10. doi: 10.1165/ajrcmb.12.2.7865218. [DOI] [PubMed] [Google Scholar]
- 38.Levin SW, Butler JD, Schumacher UK, Wightman PD, Mukherjee AB. Uteroglobin inhibits phospholipase A2 activity. Life sciences. 1986;38(20):1813–9. doi: 10.1016/0024-3205(86)90135-9. [DOI] [PubMed] [Google Scholar]
- 39.Mandal AK, Zhang Z, Ray R, et al. Uteroglobin represses allergen-induced inflammatory response by blocking PGD2 receptor-mediated functions. The Journal of experimental medicine. 2004;199(10):1317–30. doi: 10.1084/jem.20031666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasanthakumar G, Manjunath R, Mukherjee AB, Warabi H, Schiffmann E. Inhibition of phagocyte chemotaxis by uteroglobin, an inhibitor of blastocyst rejection. Biochemical pharmacology. 1988;37(3):389–94. doi: 10.1016/0006-2952(88)90204-3. [DOI] [PubMed] [Google Scholar]
- 41.Gamez AS, Gras D, Petit A, et al. Supplementing defect in Club Cell Secretory Protein attenuates airway inflammation in COPD. Chest. 2014 doi: 10.1378/chest.14-1174. [DOI] [PubMed] [Google Scholar]
- 42.Hermans C, Bernard A. Lung epithelium-specific proteins: characteristics and potential applications as markers. Am J Respir Crit Care Med. 1999;159(2):646–78. doi: 10.1164/ajrccm.159.2.9806064. [DOI] [PubMed] [Google Scholar]
- 43.Helleday R, Segerstedt B, Forsberg B, et al. Exploring the time dependence of serum clara cell protein as a biomarker of pulmonary injury in humans. Chest. 2006;130(3):672–5. doi: 10.1378/chest.130.3.672. [DOI] [PubMed] [Google Scholar]
- 44.Kenis G, Teunissen C, De Jongh R, Bosmans E, Steinbusch H, Maes M. Stability of interleukin 6, soluble interleukin 6 receptor, interleukin 10 and CC16 in human serum. Cytokine. 2002;19(5):228–35. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.