

Abstract
Ischemic preconditioning (IPC) protects tissues such as the heart from prolonged ischemia-reperfusion (IR) injury. We previously showed that the lysine deacetylase SIRT1 is required for acute IPC, and has numerous metabolic targets. While it is known that metabolism is altered during IPC, the underlying metabolic regulatory mechanisms are unknown, including the relative importance of SIRT1. Thus, we sought to test the hypothesis that some of the metabolic adaptations that occur in IPC may require SIRT1 as a regulatory mediator. Using both ex-vivo-perfused and in-vivo mouse hearts, LC-MS/MS based metabolomics and 13C-labeled substrate tracing, we found that acute IPC altered several metabolic pathways including: (i) stimulation of glycolysis, (ii) increased synthesis of glycogen and several amino acids, (iii) increased reduced glutathione levels, (iv) elevation in the oncometabolite 2-hydroxyglutarate, and (v) inhibition of fatty-acid dependent respiration. The majority (83%) of metabolic alterations induced by IPC were ablated when SIRT1 was acutely inhibited with splitomicin, and a principle component analysis revealed that metabolic changes in response to IPC were fundamentally different in nature when SIRT1 was inhibited. Furthermore, the protective benefit of IPC was abrogated by eliminating glucose from perfusion media while sustaining normal cardiac function by burning fat, thus indicating that glucose dependency is required for acute IPC. Together, these data suggest that SIRT1 signaling is required for rapid cardioprotective metabolic adaptation in acute IPC.
Keywords: Ischemia, Glucose, Fatty Acids, Reperfusion, Sirtuin, Preconditioning
1. Introduction
Two hallmarks of cardiac metabolism under normal conditions are a preference for mitochondrial fatty acid oxidation (FAO) as an energy source, and a degree of metabolic plasticity, i.e., the ability to adapt to different substrate availabilities [1]. Under pathologic conditions (e.g., following ischemia-reperfusion (IR) injury or pathologic hypertrophy) metabolic maladaptation occurs, accompanying the development of heart failure. A down-regulation of FAO and up-regulation of glycolysis are characteristic of heart failure [1–5]. Although such metabolic remodeling is often detrimental, it may also serve a beneficial adaptive role in early stage heart failure [6–8].
In addition to long-term adaptations, cardiac metabolism is also regulated on a rapid time-scale in response to acute energy demand. In the context of IR injury, metabolic interventions such as the stimulation of glucose metabolism [9], inhibition of mitochondrial FAO [10,11], mild mitochondrial uncoupling [12], and transient respiratory inhibition (see [13] for review), have all been shown to elicit cardioprotection. Furthermore, several of these metabolic events are documented to occur naturally in acute ischemic preconditioning (IPC), the phenomenon in which several short IR cycles can protect against subsequent prolonged IR injury [13–17].
The signals regulating cardiac metabolism on a rapid timescale (e.g., during IPC) are not extensively studied, and a detailed metabolomics analysis of acute IPC has not been performed. While some metabolic phenomena in IPC have been linked to p38 MAP kinase [16], the signals that drive the vast majority of rapid metabolic changes in acute IPC are currently unclear.
Among the plethora of known metabolic regulators, the sirtuin family of lysine deacylases are thought to be key orchestrators of several metabolic pathways [18], with SIRT1 emerging as an important regulator of both glycolysis [19] and FAO [20]. Moreover, although cardioprotective signaling is both complicated and redundant [21], we recently showed that SIRT1 is both up-regulated in acute IPC, and is necessary for cardioprotection in this paradigm [22,23]. Specifically, we demonstrated that either genetic ablation or acute pharmacologic inhibition of SIRT1 abolished the cardioprotective effects of “first window” IPC. Furthermore, endogenous cardioprotection observed in transgenic SIRT1 over-expressing mice was blocked by acute treatment with the SIRT1 inhibitor splitomicin. These studies establish that SIRT1 mediated cardioprotection requires activity of the enzyme during the immediate time frame of ischemia itself.
Therefore, we hypothesized that some of metabolic adaptations that occur in IPC may require SIRT1 as a regulatory mediator. To test this hypothesis, we conducted metabolomics analysis on acutely preconditioned myocardium, while modulating SIRT1 activity using splitomicin. Such a pharmacologic approach avoids the confounding issue of long-term metabolic adaptations that occur in SIRT1 knockout or transgenic animals. Our results suggest that SIRT1 is an important player in the orchestra of signals that regulate rapid metabolic alterations in the setting of acute IPC.
2. Methods (see also supplement)
For in-vivo studies, 2 mo. old C57BL/6J mice were anesthetized and instrumented as previously described [23]. For in-vitro perfused heart studies, hearts were perfused ex-vivo as described previously [23] with minor modifications. Krebs-Henseleit (KH) buffer was supplemented with 5 mM glucose and/or 100 μM palmitate-BSA. A membrane oxygenator was used to saturate KH with 95% O2, 5% CO2. Cardiac function was monitored throughout via a left ventricular balloon pressure transducer. A schematic describing all perfusion protocols is shown in Fig. S1. Briefly, experiments were stratified into those studying IR injury (Fig. S1B), and those in which hearts were sampled for metabolomics analysis (Fig. S1A). IR injury comprised 25 min. global ischemia followed by 60 min. reperfusion. At the end of reperfusion infarct size was measured by tetrazolium chloride staining and planimetry. IPC comprised 3 cycles of 5 min. ischemia plus 5 min. reperfusion. The SIRT inhibitor splitomicin (10 μM final) was infused for 20 min. where indicated (Fig. S1).
In separate experiments samples were harvested at the end of either control perfusion or IPC to measure relative levels of metabolites (“steady state”). In addition, for 13C labeling studies, perfusion began with unlabeled (12C) substrates for control or IPC protocols. Then, at the end of perfusion protocols one substrate (either glucose or palmitate) was replaced with its U-13C labeled equivalent for 5 min., with continued presence of the other, unlabeled, substrate (Fig. S1).
For metabolomics sampling in-vivo or in-vitro, the heart was clamped in pre-cooled Wollenberger tongs and plunged into liquid N2, then ground in a cooled pestle & mortar. Immediately, metabolites were serially extracted from tissue powder in 80% MeOH. For amino acid detection samples were derivatized with triethylamine and benzyl chloroformate.
Metabolites were separated by reverse phase HPLC (Shimadzu) and identified by single reaction monitoring on a triple-quadrupole mass spectrometer (Thermo Quantum Ultra). Data were analyzed using publicly available MzRock machine learning tool kit (http://code.google.com/p/mzrock/), which automated metabolite identification based on retention time, whole molecule mass, collision energy, and fragment mass. Selected metabolites were also analyzed using XCalibur Qual Browser (Thermo Scientific). Data for each run were median-normalized, and missing values (5.4%) and outliers (2.4%) imputed as weighted medians [24]. Fractional carbon saturation (F-SAT) was corrected for natural 13C abundance [25]. F-SAT values and isotopologue analysis for glycolytic and TCA cycle metabolites are presented in Figs. 3, 4, S3, S4 and S5.
Figure 3. Fractional carbon saturation (F-SAT) of glycolytic intermediates originating from U13C-labeled substrates; effect of IPC ±SIRT1 inhibition.

Following control perfusion or IPC, ± splitomicin (Sp), hearts were perfused for 5 min. with U13C-labeled glucose or palmitate (see methods and Fig. S1A), with the corresponding non-labeled partner substrates (glucose or palmitate) also present. Graphs show F-SAT, i.e., the fraction of a metabolite that became occupied by 13C from the labeled substrate within 5 min. (A): Schematic presentation of glycolysis and associated pathways. (B): F-SAT of metabolites originating from U13C-glucose (blue) or U13C-palmitate (red), after control perfusion (pale colors) or IPC (dark colors). (C): F-SAT of metabolites originating from U13C-glucose (green) or U13C-palmitate (brown) after control perfusion (pale colors) or IPC (dark colors), all in the presence of the SIRT1 inhibitor splitomicin (Sp). All data are means ± SEM, n=4–6, *p<0.05 between Ctrl. and IPC groups (ANOVA). ND = not detectable. Full data set in Table S2.
Figure 4. Fractional carbon saturation (F-SAT) of TCA cycle intermediates originating from U13C-labeled substrates; effect of IPC ±SIRT1 inhibition.

Following control perfusion or IPC, ± splitomicin (Sp), hearts were perfused for 5 min. with U13C-labeled glucose or palmitate (see methods), with the corresponding non-labeled partner substrates (glucose or palmitate) also present. Graphs show F-SAT, i.e., the fraction of a metabolite that became occupied by 13C from the labeled substrate within 5 min. (A): Schematic presentation of TCA cycle and related pathways. ETFQOR = electron transfer flavoprotein quinone oxido-reductase of FAO. (B): F-SAT of metabolites originating from U13C-glucose (blue) or U13C-palmitate (red), after control perfusion (pale colors) or IPC (dark colors). (C): F-SAT of metabolites originating from U13C-glucose (green) or U13C-palmitate (brown) under control perfusion (pale colors) or IPC (dark colors), all in the presence of the SIRT1 inhibitor splitomicin (Sp). All data are means ± SEM, n=6, *p<0.05 between Ctrl. and IPC groups (ANOVA). ND = not detectable. Full data set in Table S2.
Adult mouse cardiomyocytes (Ca2+-tolerant, ~6×105 cells per heart, >85% viability) were isolated as previously described [26]. Cells were plated on laminin-coated V7-PS plates (Seahorse Bioscience), and experiments performed in media containing 5 mM glucose plus 100 μM palmitate-BSA. Cells were exposed to simulated IPC (sIPC), comprising a single cycle of 20 min. ischemia, 60 min. reperfusion, inside the Seahorse XF24 apparatus as previously outlined [26]. In separate experiments cells were subjected to simulated IR injury (60 min. ischemia, 60 min. reperfusion, with or without prior sIPC, and cell death was assayed by LDH release (schematic, Fig. 5A).
Figure 5. Simulated cardiomyocyte IR injury (sIR) and simulated IPC (sIPC) in seahorse XF.

(A): Schematic showing treatment and measurement protocol for adult mouse cardiomyocytes. OCR = time point at which oxygen consumption rate was measured. (B): Cell death (LDH release) following sIR with or without prior sIPC. Data are means ± SEM, n=4, *p<0.05 vs. sIR (paired t-test). (C): Oxygen consumption rate (OCR) in cardiomyocytes at baseline and after sIPC. Cells were incubated with either glucose, palmitate or both substrates. Data are means ± SEM, n=6, *p<0.05 vs. glucose (ANOVA), #p<0.05 vs. corresponding baseline (ANOVA).
Six to eight independent experiments were performed. Significance between two groups was determined using paired Student’s t-test. ANOVA was used for comparisons among multiple groups with Tukey’s multiple comparison test (Prism 6.0 for Windows; GraphPad). Pathway impact analysis and principal component analysis for steady state metabolomics data were performed using publicly available MetaboAnalyst software [27].
3. Results & Discussion
3.1 Model validation (in-vivo vs. ex-vivo metabolomics)
To validate that the perfused heart adequately reflects in-vivo metabolism, we first performed steady-state metabolomics analysis on rapidly sampled in-vivo hearts, and hearts perfused with a physiologically relevant substrate mix (5 mM glucose plus 100 μM palmitate-BSA). These data (Fig. 1) show a strong correlation (r2=0.80) between metabolite profiles of in-vivo and ex-vivo hearts. One outlier was di-P-glycerate, a regulator of hemoglobin O2 affinity enriched in red blood cells, which was present in in-vivo samples at a level ~750 fold higher than in perfused hearts, suggesting that rapidly sampled in-vivo hearts were contaminated with blood.
Figure 1. In-vivo vs. ex-vivo metabolomics.
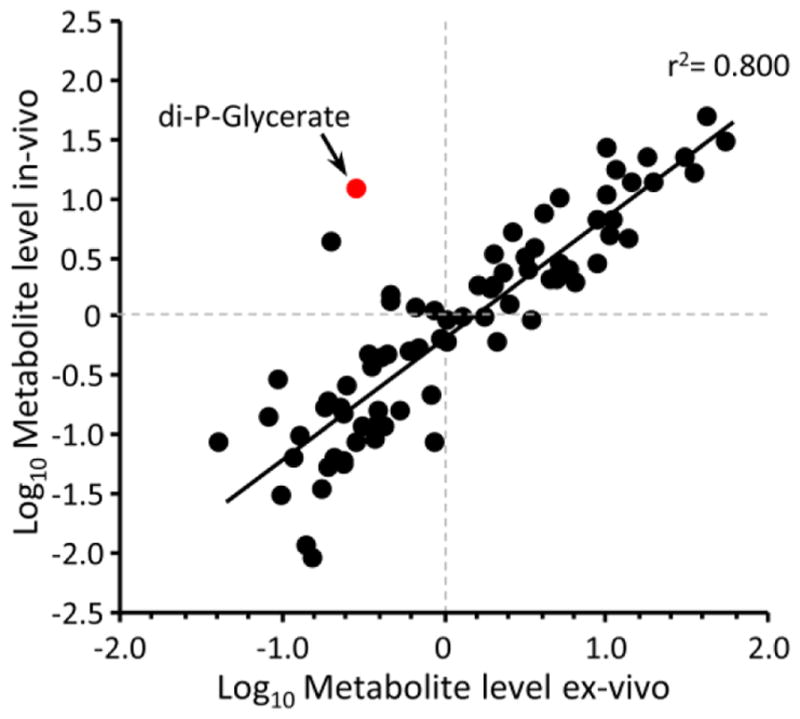
Whole hearts were rapidly sampled for metabolomic analysis as described in the methods, with starting material comprising either in-vivo hearts, or isolated hearts perfused with Krebs Henseleit buffer containing 5 mM glucose and 100 μM palmitate-BSA. Graph shows log-transformed steady-state metabolite levels, with each point representing a single metabolite. Data are means of 7 (in-vivo) or 8 (ex-vivo) independent experiments. The highlighted red point is di-P-glycerate, a contaminant from blood (see text). All data excluding this point were fitted with a linear regression shown on the graph (r2 for all data including di-P-glycerate was 0.745).
Since the heart is primarily a fat-burning organ, it was also important to validate that IPC could elicit cardioprotection, and this protection could be blocked by inhibiting SIRT1, in hearts perfused with a physiologically relevant substrate mix. The data in Figs. S1C/D confirm that IPC indeed yielded cardioprotection (improved post-IR functional recovery and lowered infarct size) in hearts perfused with glucose and fat, similar in magnitude to that previously observed with glucose alone [22,23], and this was blocked by the SIRT1 inhibitor splitomicin. Thus, we utilized this perfused heart system for the remainder of the study.
3.2 The metabolomic profile of IPC
Next we investigated how acute IPC altered cardiac metabolism, by sampling myocardium after control perfusion or immediately after the 3rd cycle of IPC, before index ischemia (arrow, Fig. S1A). Eighty one metabolites were reliably detected by LC-MS/MS with single reaction monitoring (Table S1). Based on a pathway impact analysis (Fig. S2), numerous metabolic pathways were adequately represented. To simplify the analysis, metabolites were parsed into functional pathways and their levels in IPC normalized to those in control perfused hearts (Fig. 2).
Figure 2. Steady-state metabolomics of IPC.

Schematics of perfusion protocols for steady-state metabolomics studies are presented in Fig. S1A. Metabolites are binned by pathway. Levels of each metabolite in the IPC condition are normalized to the same metabolite in the control condition. Ctrl = 1 (horizontal dotted line). Data are means ± SEM, n=8, *p<0.05 vs. Ctrl (paired t-test). Original data are in Table S1.
3.2.1 Glycolysis in IPC
Steady-state levels of most 6-carbon glycolytic intermediates (hexose-P, glucose-6-P, glucose-1-P, and fructose-bis-P) were significantly elevated in IPC (Fig. 2A), which is consistent with a reported stimulation of cardiac glucose uptake in IPC [16]. This is likely to be a functionally-relevant effect, since preventing glucose uptake by knock-down of cardiac GLUT4 is known to abolish cardioprotection by IPC [28].
Within glycolysis a key control point is phosphofructokinase (PFK), wherein PFK2 makes fructose-2,6-bis-P, an allosteric activator of PFK1 which makes fructose-1,6-bis-P. Although we observed accumulation of fructose-bis-P in IPC (Fig. 2A), our methodology could not discern fructose 1,6- or 2,6-bis-P. Nevertheless, it has been reported that stimulating PFK1 or exogenous delivery of fructose-1,6-bis-P can elicit cardioprotection [29,30], warranting further investigation of this metabolite in IPC.
In contrast to 6-carbon glycolytic intermediates, with the exception of di-P-glycerate most 3-carbon intermediates were unchanged or slightly lower in IPC. While together these observations (more C6, same C3) might suggest inhibition in the lower half of glycolysis, conclusions about enzymatic rates cannot be drawn based solely on steady-state metabolite pool sizes. Thus, we next conducted fractional saturation (F-SAT) analysis using 13C-labeled substrates. While working with whole hearts prevents extensive kinetic sampling, comparisons of pool sizes and labeling abundance at a single empirically-determined early non-saturated time point (F-SAT ~0.5) enables qualitative assessment of the relative progression of 13C label to that metabolite, a proxy for metabolic flux.
Hearts were perfused with buffer containing both glucose and fat, and subjected to IPC as above. Five minutes before sampling, either glucose or palmitate in the perfusion media were replaced with their U13C-labeled analogs (with continued presence of the other unlabeled substrate). Preliminary experiments examining 0–20 min. 13C substrate infusion showed that 5 min. was the optimal time for 13C substrate delivery, to obtain a dynamic range of labeling (40–70%) without full saturation (i.e. steady state equilibration of the label) in key metabolites (Figs. S3 & S4).
As shown in Fig. 3B, F-SAT of both the C6 and C3 metabolites of glycolysis was elevated in IPC (vs. Ctrl). Notably, both steady-state levels (pool sizes, Fig. 2A) and F-SAT values (Fig. 3B) were elevated for hexose-P, glucose-1-P, fructose-bis-P, and 1,3-di-P-glycerate, suggesting that progression of 13C label through the upper half of glycolysis was elevated in IPC, resulting in accumulation of these intermediates.
In contrast for the lower half of glycolysis, although F-SAT values were elevated in IPC (Fig. 3B), suggesting enhanced progression of 13C label down the glycolytic pathway, the steady-state pool sizes of these metabolites were not elevated in the same manner as the upper half of glycolysis (Fig. 2A). One possible explanation for this could be a rapid exit of carbon from the lower half of glycolysis. Notably, the steady state levels of both alanine (from pyruvate) and serine (from 3-P-glycerate) were elevated in IPC (Fig. 2D), while the pool size of acetyl-CoA was unchanged and lactate was slightly lower (Fig. 2A). Alanine accumulation in ischemia is thought to be due to inhibition of pyruvate dehydrogenase [31], and it has been proposed this may protect the heart by diverting carbon away from lactate [32]. While this would be consistent with the protective nature of IPC, the possibility cannot be excluded that these altered amino acid pools may originate at the level of proteolysis. Overall, the data suggest that IPC enhances progression of carbon through glycolysis, with possible diversion of carbon toward serine and alanine rather than Acetyl-CoA and lactate.
3.2.2. Glycolysis-linked pathways in IPC (gluconeogenesis, glycogen, lipogenesis, pentose phosphate)
Due to the lack of a glyoxylate cycle the mammalian heart cannot use carbon from fat to make glucose. Thus as expected, there was zero-to-minimal F-SAT of glycolytic intermediates with either 5 min. (Fig. 3B) or 20 min. (Fig. S4A) infusion of 13C-palmitate in the presence of unlabeled glucose. Isotopologue analysis of P-enol-pyruvate, pyruvate and lactate (Fig. S5A) indicated that with 13C-glucose as substrate, only the fully-labeled species were present, indicating their sole origin from glycolysis and not the anaplerotic TCA cycle (i.e., P-enol-pyruvate carboxykinase). In addition, 13C-palmitate yielded virtually no isotopologues for these 3 species (Fig. S5B) confirming no gluconeogenesis from fat. This profile did not change with IPC (Fig. 3).
Accumulation of glucose-1-P in IPC (Fig. 2A) might suggest an increase in glycogenolysis. However, the greater F-SAT of this metabolite in IPC (Fig. 3B) indicates that its source is exogenous 13C-glucose and not stored glycogen. Furthermore, the F-SAT of UDP-glucose was also elevated in IPC (Fig. 3B), with no change in its steady-state accumulation (Fig. 2F), suggesting an elevation in glycogen synthesis in IPC. Consistent with this, pre-ischemic glycogen accumulation has been shown to protect the heart against IR injury [33].
Neither steady-state levels nor F-SAT of glycerol-3-P were altered in IPC (Figs. 2A, 3B), suggesting no change in the exit of carbon from glycolysis toward triglyceride synthesis. Regarding the pentose phosphate pathway (PPP), F-SAT values were not available, but steady state data (Fig. 2G) showed that PPP intermediate pool sizes were unchanged in IPC. Consistent with this, steady-state levels of most nucleotide phosphates (Fig. 2C) and redox-active pyrimidine nucleotides (Fig. 2E) were also unchanged in IPC, together suggesting that PPP activity was unaltered.
3.2.3. TCA cycle in IPC
In the heart, the mitochondrial TCA cycle is the major carbon sink for acetyl-CoA originating from either glycolysis or FAO (Fig. 4A). Thus we examined both steady state pool sizes and F-SAT for TCA cycle intermediates under control and IPC conditions. As shown in Fig. 4B, 5 min. infusion of control hearts with 13C-glucose in the presence of unlabeled palmitate did not elicit substantial labeling of any TCA cycle intermediate (note scale 0–0.3). This situation was the same with 20 min. 13C-glucose infusion (Fig. S3B), but removal of palmitate did permit 13C-glucose entry to the TCA cycle (Fig. S3C). These data indicate that under these perfusion conditions (i.e. glucose plus fat, no lactate, pyruvate or insulin), glycolysis is not a major carbon source for the TCA cycle when fat is present, directly confirming the classical Randle effect in the heart, wherein glucose oxidation is inhibited in the presence of fatty acids [34].
Confirming the dogma that fat is the heart’s preferred fuel, 5 min. infusion of control hearts with 13C-palmitate in the presence of unlabeled glucose yielded 50–70% F-SAT of numerous TCA cycle intermediates (Fig. 4B). There was no change in F-SAT for TCA cycle intermediates from either 13C-glucose or 13C-palmitate in IPC (Fig. 4B), and with a few exceptions to be discussed later, the steady state levels of most TCA cycle metabolites were unchanged in IPC (Fig. 2B). Moreover IPC did not change isotopologue distributions for the TCA cycle intermediates (Fig. S5C), suggesting that entry or exit of carbon from the TCA cycle (i.e., anaplerosis) was not modulated by IPC.
3.2.4. Bioenergetics in IPC
Steady state ATP levels were slightly but significantly elevated in IPC (Fig. 2C), while energy charge (ATP+½ADP/ATP+ADP+AMP) was no different (0.95±0.02 in IPC vs. 0.93±0.02 in control), thereby suggesting that ATP accumulation in IPC may be a consequence of lower energy demand, as previously reported for preconditioned hearts [35], rather than stimulation of mitochondrial function.
To examine the effects of IPC on mitochondrial function under conditions of differential substrate choice, we used freshly isolated primary adult mouse cardiomyocytes and a Seahorse XF-24 analyzer to measure oxygen consumption (OCR) in a model of simulated IPC (sIPC, Fig. 5A). As shown in Fig. 5B, sIPC yielded a reduction in cell death following subsequent simulated IR injury (sIR).
In separate experiments, cells were incubated in the presence of different substrates, and their metabolic function was assayed. With palmitate alone or a mix of glucose plus palmitate, baseline OCR was faster than with glucose alone (Fig. 5C). This additional respiration may be due to augmentation of mitochondrial Ox-Phos by electrons from β-oxidation entering the respiratory chain at the level of the electron transfer flavoprotein quinone oxidoreductase (ETF-QOR, see Fig. 4A, top left).
When myocytes respiring on different substrates were subjected to sIPC alone, no effect on respiration was observed in glucose-burning cells, but the additional respiration seen with palmitate was abolished by sIPC (Fig. 5C). Although these data might suggest that IPC inhibits FAO, as mentioned above there was no difference in penetration of label from 13C-palmitate into the TCA cycle upon IPC (Fig. 4B). The cause of this discrepancy – i.e. inhibition of fatty-acid-linked respiration but no change in apparent 13C label progression through β-oxidation – is not clear.
3.3. The role of SIRT1 in rapid metabolic adaptation in IPC
SIRT1 is required for cardioprotection by acute IPC [22,23], and although SIRT1 can regulate several metabolic pathways [36], the role of SIRT1 in governing rapid metabolic adaptations in IPC is unknown. To investigate this, a pharmacologic approach was used (splitomicin), which capitalizes on the fact that SIRT1 is only important for protection within the acute time scale of IR injury [23], and avoids complications of knock-out animals (i.e., long-term metabolic adaptation). Splitomicin was originally discovered as an inhibitor of the archetypal sirtuin, Sir2p (yeast silent information regulator 2p), and mammalian SIRT1 is the isoform most closely related to Sir2p [37]. Splitomicin has been widely used as mammalian SIRT1 inhibitor in cardiac and vascular cells [38–42].
Of the 23 metabolites significantly altered in IPC (Fig. 2), 19 of these (i.e. 83%) showed either a blockade or reversal of the effects of IPC, in the presence of splitomicin (Fig. 6). In addition, all differences in F-SAT values in IPC (Figs. 3B & 4B), were abolished (i.e., rendered non-significant) in the presence of splitomicin (Figs. 3C & 4C). Full data sets for the IPC + splitomicin conditions, as well as for the effect of splitomicin alone (without IPC), are in Tables S1 and S2. The predominant metabolite class whose alterations in IPC were not affected by splitomicin, was the amino acids (Fig. 6) suggesting that such changes are modulated in IPC by signals other than SIRT1. This is consistent with the well-known complexity and redundancy of IPC signaling, involving numerous kinases and other signaling molecules (e.g. Akt, GSK-3β, eNOS, PKC) [21].
Figure 6. Steady state metabolomics of IPC with SIRT1 inhibition.

Hearts were subjected to control perfusion or IPC, in the presence of 10 μM splitomicin (Sp). The subset of metabolites that were significantly altered in IPC alone (Fig. 2) are shown as circles, using the same color scheme as Fig. 2 (expressed relative to control, dotted line = 1). N.B. symbols to indicate significance in this IPC alone group (circles) are removed for clarity, since all are significant. The effect of IPC on these metabolites in the presence of splitomicin is shown in corresponding triangles, relative to control perfusion with splitomicin alone (no IPC, dotted line = 1). Data are means ± SEM, n=8. *p<0.05 (paired t-test) for IPC with splitomicin, vs. control perfusion with splitomicin (i.e. triangles).
A principal component analysis (PCA, Fig. S6) showed that the effect of IPC on steady state metabolite profiles was fundamentally different in nature when splitomicin was present (i.e., the principal components did not overlap). Together, these data suggest that rapid metabolic adaptations in acute IPC require SIRT1, and that when SIRT1 is inhibited IPC still affects metabolism but in a different manner.
Regarding the mechanism by which SIRT1 elicits rapid metabolic changes in IPC, the most likely route is via deacetylation of metabolic enzymes or the signaling pathways that regulate them. In this regard, we previously identified a number of important substrates for SIRT1 deacetylation during IPC, including GAPDH, eNOS, p65, and ICDH [22,23]. This is in addition to an ever-growing catalog of known SIRT1 metabolic targets [36,43].
It is important to note that these results do not preclude the possibility that additional sirtuins (e.g. SIRT3) may be important for regulating metabolism in the setting of acute IPC. However, the lack of specific inhibitors for these isoforms, and possible long term metabolic adaptations in knockout animals, may cause difficulties in studying the role of other sirtuins in regulating acute IPC metabolism.
3.4. IPC cardioprotection is metabolism-dependent
Although the SIRT1 inhibitor splitomicin blocks protection by IPC (Fig. S1C/D), and blocks the metabolic alterations induced by IPC, it is not clear whether the blockage of protection stems from the blockage of metabolic alterations. In this regard, it is interesting to note that the effect of splitomicin alone on metabolism (Tables S1 and S2) is somewhat similar to the effect of IPC alone. However, despite these similar effects on metabolism, these interventions have opposite effects on protection: IPC protects but splitomicin does not.
One possible interpretation of these data, could be that in IPC hearts metabolism is further remodeled in a beneficial manner during subsequent pathological ischemia, whereas in splitomicin treated hearts (depending on the duration of drug action) metabolism may be refractory to further changes, thus preventing the beneficial effects of IPC from taking place during pathologic ischemia.
Another interpretation of this result is that the protective effect of IPC is perhaps unrelated to the metabolic changes that occur in IPC. To investigate the link between metabolism and protection, we performed experiments in which metabolic changes in acute IPC were prevented, by limiting substrate availability. As shown in Fig. 7, hearts perfused with palmitate as sole substrate were functionally normal and exhibited a similar degree of damage in response to IR injury (infarct size & functional recovery), compared to hearts perfused with glucose plus palmitate (Figs. S1C/D). However, the palmitate-only hearts were refractory to protection by IPC. These data suggest that indeed, there is a link between metabolism and protection in IPC, with the ability to up-regulate glucose utilization required for the cardioprotective benefit of acute IPC. Thus, further work is likely required to define the interplay between splitomicin effects on metabolism, and its blockage of IPC protection.
Figure 7. IR and IPC in the absence of glucose.

Isolated hearts were perfused with buffer supplemented with 100 μM palmitate-BSA alone (no glucose), and were subjected to IR injury as described in the methods. Optionally, hearts were also subjected to IPC prior to IR injury. Schematics of perfusion protocols for ischemia reperfusion injury and ischemic preconditioning are presented in Fig. S1B. (A): Cardiac function (rate pressure product) in IR and IPC+IR hearts. Data are means ± SEM. (B): Myocardial infarct, plotted as individual points (left) and means ± SEM (right). Images inset to the graph show representative TTC-stained cardiac cross-section slices (white = infarct, red = live myocardium). Data are means ± SEM, n=7. No significant differences were observed between IR and IPC groups.
Regarding the relationship between metabolism and protection in general, it is currently unclear how diverting metabolism toward glucose utilization confers a cardioprotective benefit. It is possible that simply facilitating anaerobic metabolism is beneficial under ischemic conditions, as previously proposed [44]. Alternatively, a detrimental role for succinate accumulation during ischemia, with its rapid consumption at reperfusion driving ROS generation, has recently been shown [45]. Although we did not observe any effect of acute IPC on succinate during the observed time-frame(Figs. 2B & 4B), the effects of IPC on succinate accumulation or disposal during subsequent IR injury remain unknown and may warrant further investigation.
3.5. Other metabolites in IPC
Within the TCA cycle, we found that 2-hydroxyglutarate (2-OHG) was significantly elevated during IPC (Fig. 2B). Typically this metabolite is associated with isocitrate dehydrogenase (ICDH) mutation in cancer, leading to its characterization as an oncometabolite [46]. Very recently it has been shown that 2-OHG accumulates in response to hypoxia [47,48], but the regulation of this phenomenon is unclear. In this regard, the elevation in 2-OHG levels seen in IPC was blocked by SIRT1 inhibition (Fig. 6) and it is also known that ICDH is a substrate for SIRT-mediated deacetylation [49]. These data raise the possibility that 2-OHG may be a normal alternate product of ICDH dependent on its acetylation status. As such, drugs under development to target this oncometabolite (e.g., NCT01915498, see [50] for review) may have unintended cardiovascular effects. Further work is required to elucidate the exact source of 2-OHG during IPC.
Several important redox metabolites were altered in IPC, including elevated levels of NAD+ (Fig. 2E) which facilitates the activity of several enzymes implicated in cardioprotection such as aldehyde dehydrogenase [51] and SIRTs. In addition, 5-P-ribosyl-1-pyro-P (Fig. 2C), an important intermediate in the resynthesis of purine nucleotides and NAD+ upon reperfusion [52], was elevated in IPC.
The observed increase in reduced glutathione (GSH) in IPC (Fig. 2E) could be due to greater reduction of oxidized glutathione (GSSG) by NADPH-dependent glutathione reductase. However, GSH/GSSG ratios in most tissues are 40:1, such that reduction of the entire GSSG pool can only elevate GSH levels slightly. The 3-fold increase in GSH, coupled with no change in GSSG, and no change in NADPH or the PPP (Fig. 2E & 2G), suggests that this increase in GSH in IPC may be due to de-novo glutathione synthesis.
In the same manner, another surprising observation was a large increase in the level of ADP-D-glucose (Fig. 2F), a synthetic precursor for disaccharides typically only found in invertebrates, where these molecules are thought to afford protection in stress conditions such as hypoxia [53]. Together these data suggest that the cell may generate more protective molecules in response to IPC.
The levels of free flavin mononucleotide (FMN) were also elevated in IPC (Fig. 2C). This free FMN may originate from complex I of the mitochondrial respiratory chain which loses FMN during ischemia [54].
In the hexosamine biosynthetic pathway which is linked to glycolysis, IPC significantly increased the level of UDP-N-acetylglucosamine (Fig. 2F), which serves as a substrate for O-GlcNac-ylation, an important protein post-translational modification which has been implicated in cardioprotection [55]. This change was also blocked by splitomicin, raising the possibility that O-GlcNac signaling may be regulated by SIRT1. Alternatively, UDP-N-acetylglucosamine may be used to make hyaluronic acid, which has recently been shown to serve a cardioprotective role [56].
4. Limitations of this study
The limitations of this study primarily concern the relevance of metabolomic analyses in perfused hearts to the in-vivo condition. The perfused heart model employed was not a working heart preparation (i.e., no afterload) but the hearts employed all exhibited excellent contractile function (rate x pressure product 48,500 ± 2,700) and rapid incorporation of 13C labeled substrates. Furthermore, although lactate and pyruvate were absent from the perfusion media, this was in order to measure these metabolites as part of our analysis.
While some of these limitations could be overcome by using the in-vivo mouse LAD occlusion model of cardiac ischemia, this model has several pitfalls which render it unsuitable for metabolomics studies. These include an inability to rapidly sample cardiac tissue without blood contamination, or to rapidly dissect ischemic vs. non-ischemic myocardium, without affecting metabolite pools during dissection. Furthermore, in-vivo models are not amenable to 13C labeled substrate delivery or tracing. Thus, overall despite its limitations, the Langendorff ex-vivo perfused mouse heart is the preferred model system for such studies.
5. Conclusions & Outlook
Overall, we conclude that acute IPC induces a unique set of rapid metabolic alterations including stimulation of glycolysis and glycogen synthesis, inhibition of fatty-acid dependent respiration, and several unique alterations in metabolites such as 2-OHG, UDP-N-acetylglucosamine, fructose-bis-P, NAD+, GSH, and 5-P-ribosyl-1-pyro-P. These alterations are largely (83%) blocked by the SIRT1 inhibitor splitomicin, suggesting a requirement for SIRT1 in this process, and consistent with the requirement for SIRT1 for cardioprotection by IPC. It should be emphasized that our data do not preclude the involvement or requirement of other signaling pathways (even other SIRTs) in IPC, either at the level of cardioprotection or in governing metabolic alterations.
Although numerous small molecule therapeutics are available that target cardiac metabolism [7–12], our data also suggest that molecules which modulate SIRT1 activity, including sirtuin activating compounds (STACs), and NAD+ precursors such as nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), may be therapeutically useful to alter cardiac metabolism and mimic a preconditioned-like state.
Supplementary Material
HIGHLIGHTS.
First metabolomic profile of the preconditioned state
Discovery that SIRT1 is critical for acute metabolic changes in IPC
Discovery that the fat-only perfused heart cannot be preconditioned
Acknowledgments
Funding
This work was funded by a grant from the US National Institutes of Health (RO1 HL-071158) to PSB and (R01 AI-081773) to JM.
Non-standard Abbreviations
- ETF-QOR
electron transfer flavoprotein quinone oxidoreductase
- FAO
fatty acid oxidation
- F-SAT
fractional saturation with 13C label
- IPC
ischemic preconditioning
- IR
ischemia-reperfusion
- LAD artery
left anterior descending artery
- LC-MS
liquid chromatography–mass spectrometry
- OCR
oxygen consumption rate
- PPP
pentose phosphate pathway
- SIRT1
silent information regulator two P homolog 1
Footnotes
Disclosures
The authors declare no conflicts of interest, financial or otherwise, regarding this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. Literature Cited
- 1.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 2.Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. 2014;7:634–42. doi: 10.1161/CIRCHEARTFAILURE.114.001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–45. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- 4.Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742–H750. doi: 10.1152/ajpheart.1994.267.2.H742. [DOI] [PubMed] [Google Scholar]
- 5.Pound KM, Sorokina N, Ballal K, Berkich DA, Fasano M, Lanoue KF, et al. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res. 2009;104:805–12. doi: 10.1161/CIRCRESAHA.108.189951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao R, Jain M, Cui L, D’Agostino J, Aiello F, Luptak I, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. 2002;106:2125–31. doi: 10.1161/01.cir.0000034049.61181.f3. [DOI] [PubMed] [Google Scholar]
- 7.Turcani M, Rupp H. Etomoxir improves left ventricular performance of pressure-overloaded rat heart. Circulation. 1997;96:3681–6. doi: 10.1161/01.cir.96.10.3681. [DOI] [PubMed] [Google Scholar]
- 8.Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112:3280–8. doi: 10.1161/CIRCULATIONAHA.105.551457. [DOI] [PubMed] [Google Scholar]
- 9.Taniguchi M, Wilson C, Hunter CA, Pehowich DJ, Clanachan AS, Lopaschuk GD. Dichloroacetate improves cardiac efficiency after ischemia independent of changes in mitochondrial proton leak. Am J Physiol Heart Circ Physiol. 2001;280:H1762–H1769. doi: 10.1152/ajpheart.2001.280.4.H1762. [DOI] [PubMed] [Google Scholar]
- 10.Lopaschuk GD, Wall SR, Olley PM, Davies NJ. Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res. 1988;63:1036–43. doi: 10.1161/01.res.63.6.1036. [DOI] [PubMed] [Google Scholar]
- 11.Dyck JR, Cheng JF, Stanley WC, Barr R, Chandler MP, Brown S, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. 2004;94:e78–e84. doi: 10.1161/01.RES.0000129255.19569.8f. [DOI] [PubMed] [Google Scholar]
- 12.Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res. 2006;72:313–21. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 13.Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut-down and gradual wake-up. J Mol Cell Cardiol. 2009;46:804–10. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 15.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395:611–8. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong H, Chen W, London RE, Murphy E, Steenbergen C. Preconditioning enhanced glucose uptake is mediated by p38 MAP kinase not by phosphatidylinositol 3-kinase. J Biol Chem. 2000;275:11981–6. doi: 10.1074/jbc.275.16.11981. [DOI] [PubMed] [Google Scholar]
- 17.Opie LH, Sack MN. Metabolic plasticity and the promotion of cardiac protection in ischemia and ischemic preconditioning. J Mol Cell Cardiol. 2002;34:1077–89. doi: 10.1006/jmcc.2002.2066. [DOI] [PubMed] [Google Scholar]
- 18.Boutant M, Canto C. SIRT1 metabolic actions: Integrating recent advances from mouse models. Mol Metab. 2014;3:5–18. doi: 10.1016/j.molmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallows WC, Yu W, Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J Biol Chem. 2012;287:3850–8. doi: 10.1074/jbc.M111.317404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–5. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674–99. doi: 10.1161/CIRCRESAHA.116.305348. [DOI] [PubMed] [Google Scholar]
- 22.Nadtochiy SM, Yao H, McBurney MW, Gu W, Guarente L, Rahman I, et al. SIRT1-mediated acute cardioprotection. Am J Physiol Heart Circ Physiol. 2011;301:H1506–H1512. doi: 10.1152/ajpheart.00587.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res. 2011;89:643–9. doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aittokallio T. Dealing with missing values in large-scale studies: microarray data imputation and beyond. Brief Bioinform. 2010;11:253–64. doi: 10.1093/bib/bbp059. [DOI] [PubMed] [Google Scholar]
- 25.Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc. 2008;3:1328–40. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nadtochiy SM, Madukwe J, Hagen F, Brookes PS. Mitochondrially targeted nitro-linoleate: a new tool for the study of cardioprotection. Br J Pharmacol. 2014;171:2091–8. doi: 10.1111/bph.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40:W127–W133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji L, Zhang X, Liu W, Huang Q, Yang W, Fu F, et al. AMPK-regulated and Akt-dependent enhancement of glucose uptake is essential in ischemic preconditioning-alleviated reperfusion injury. PLoS One. 2013;8:e69910. doi: 10.1371/journal.pone.0069910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Donthi RV, Wang J, Lange AJ, Watson LJ, Jones SP, et al. Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am J Physiol Heart Circ Physiol. 2008;294:H2889–H2897. doi: 10.1152/ajpheart.91501.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riedel BJ, Gal J, Ellis G, Marangos PJ, Fox AW, Royston D. Myocardial protection using fructose-1,6-diphosphate during coronary artery bypass graft surgery: a randomized, placebo-controlled clinical trial. Anesth Analg. 2004;98:20–9. doi: 10.1213/01.ANE.0000094336.97693.90. table. [DOI] [PubMed] [Google Scholar]
- 31.Lewandowski ED, White LT. Pyruvate dehydrogenase influences postischemic heart function. Circulation. 1995;91:2071–9. doi: 10.1161/01.cir.91.7.2071. [DOI] [PubMed] [Google Scholar]
- 32.Lewandowski ED, Johnston DL, Roberts R. Effects of inosine on glycolysis and contracture during myocardial ischemia. Circ Res. 1991;68:578–87. doi: 10.1161/01.res.68.2.578. [DOI] [PubMed] [Google Scholar]
- 33.Cross HR, Opie LH, Radda GK, Clarke K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ Res. 1996;78:482–91. doi: 10.1161/01.res.78.3.482. [DOI] [PubMed] [Google Scholar]
- 34.Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578–E591. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jennings RB, Murry CE, Reimer KA. Energy metabolism in preconditioned and control myocardium: effect of total ischemia. J Mol Cell Cardiol. 1991;23:1449–58. doi: 10.1016/0022-2828(91)90190-w. [DOI] [PubMed] [Google Scholar]
- 36.Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2014;25:138–45. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci U S A. 2001;98:15113–8. doi: 10.1073/pnas.261574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breitenstein A, Stein S, Holy EW, Camici GG, Lohmann C, Akhmedov A, et al. Sirt1 inhibition promotes in vivo arterial thrombosis and tissue factor expression in stimulated cells. Cardiovasc Res. 2011;89:464–72. doi: 10.1093/cvr/cvq339. [DOI] [PubMed] [Google Scholar]
- 39.Chapalamadugu KC, Panguluri SK, Bennett ES, Kolliputi N, Tipparaju SM. High level of oxygen treatment causes cardiotoxicity with arrhythmias and redox modulation. Toxicol Appl Pharmacol. 2015;282:100–7. doi: 10.1016/j.taap.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stavrou EX, Fang C, Merkulova A, Alhalabi O, Grobe N, Antoniak S, et al. Reduced thrombosis in Klkb1−/− mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood. 2015;125:710–9. doi: 10.1182/blood-2014-01-550285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stein S, Lohmann C, Schafer N, Hofmann J, Rohrer L, Besler C, et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur Heart J. 2010;31:2301–9. doi: 10.1093/eurheartj/ehq107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stein S, Schafer N, Breitenstein A, Besler C, Winnik S, Lohmann C, et al. SIRT1 reduces endothelial activation without affecting vascular function in ApoE−/− mice. Aging (Albany NY) 2010;2:353–60. doi: 10.18632/aging.100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foster DB, Liu T, Rucker J, O’Meally RN, Devine LR, Cole RN, et al. The cardiac acetyl-lysine proteome. PLoS One. 2013;8:e67513. doi: 10.1371/journal.pone.0067513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–55. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–5. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Megova M, Drabek J, Koudelakova V, Trojanec R, Kalita O, Hajduch M. Isocitrate dehydrogenase 1 and 2 mutations in gliomas. J Neurosci Res. 2014;92:1611–20. doi: 10.1002/jnr.23456. [DOI] [PubMed] [Google Scholar]
- 47.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015;22:304–11. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in l-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. 2012;287:14078–86. doi: 10.1074/jbc.M112.355206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomita A. Progress in molecularly targeted therapies for acute myeloid leukemia. Rinsho Ketsueki. 2015;56:130–8. doi: 10.11406/rinketsu.56.130. [DOI] [PubMed] [Google Scholar]
- 51.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–5. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dow JW, Bowditch J, Nigdikar SV, Brown AK. Salvage mechanisms for regeneration of adenosine triphosphate in rat cardiac myocytes. Cardiovasc Res. 1987;21:188–96. doi: 10.1093/cvr/21.3.188. [DOI] [PubMed] [Google Scholar]
- 53.Chen Q, Haddad GG. Role of trehalose phosphate synthase and trehalose during hypoxia: from flies to mammals. J Exp Biol. 2004;207:3125–9. doi: 10.1242/jeb.01133. [DOI] [PubMed] [Google Scholar]
- 54.Rouslin W, Ranganathan S. Impaired function of mitochondrial electron transfer complex I in canine myocardial ischemia: loss of flavin mononucleotide. J Mol Cell Cardiol. 1983;15:537–42. doi: 10.1016/0022-2828(83)90329-2. [DOI] [PubMed] [Google Scholar]
- 55.Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, et al. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172–82. doi: 10.1161/CIRCULATIONAHA.107.730515. [DOI] [PubMed] [Google Scholar]
- 56.Law CH, Li JM, Chou HC, Chen YH, Chan HL. Hyaluronic acid-dependent protection in H9C2 cardiomyocytes: a cell model of heart ischemia-reperfusion injury and treatment. Toxicology. 2013;303:54–71. doi: 10.1016/j.tox.2012.11.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.