

Abstract
Previous studies demonstrate that intrastriatal injections of fibrillar alpha-synuclein (α-syn) into mice induce Parkinson’s disease (PD)-like Lewy body (LB) pathology formed by aggregated α-syn in anatomically interconnected regions and significant nigrostriatal degeneration. The aim of the current study was to evaluate whether exogenous mouse α-syn pre-formed fibrils (PFF) injected into the striatum of rats would result in accumulation of LB-like intracellular inclusions and nigrostriatal degeneration. Sprague Dawley rats received unilateral intrastriatal injections of either non-fibrillized recombinant α-syn or PFF mouse α-syn in 1- or 2- sites and were euthanized at 30, 60 or 180 days post-injection (pi). Both non-fibrillized recombinant α-syn and PFF α-syn injections resulted in phosphorylated α-syn intraneuronal accumulations (i.e., diffuse Lewy neurite (LN)- and LB-like inclusions) with significantly greater accumulations following PFF injection. LB-like inclusions were observed in several areas that innervate the striatum, most prominently the frontal and insular cortices, the amygdala, and the substantia nigra pars compacta (SNpc). α-Syn accumulations co-localized with ubiquitin, p62, and were thioflavin-S-positive and proteinase-k resistant, suggesting PFF-induced pathology exhibits properties similar to human LBs. Although α-syn inclusions within the SNpc remained ipsilateral to striatal injection, we observed bilateral reductions in nigral dopamine neurons at the 180-day time point in both the 1- and 2-site PFF injection paradigms. PFF injected rats exhibited bilateral reductions in striatal dopaminergic innervation at 60 and 180 days and bilateral decreases in homovanillic acid; however, dopamine reduction was observed only in the striatum ipsilateral to PFF injection. Although the level of dopamine asymmetry in PFF injected rats at 180 days was insufficient to elicit motor deficits in amphetamine-induced rotations or forelimb use in the cylinder task, significant disruption of ultrasonic vocalizations was observed. Taken together, our findings demonstrate that α-syn PFF are sufficient to seed the pathological conversion and propagation of endogenous α-syn to induce a progressive, neurodegenerative model of α-synucleinopathy in rats.
Keywords: alpha-synuclein, Parkinson’s disease, rat model, pre-formed fibrils, neurodegeneration, propagation, rats
Introduction
Parkinson’s disease (PD) pathology is characterized by the formation of intraneuronal Lewy body (LB) and Lewy neurite (LN) inclusions which are comprised primarily of misfolded, fibrillar α-synuclein (α-syn) [1]. α-syn is a 140-amino acid protein that adopts a α-helical structure when bound to membranes; however, it can easily fold into a β-sheet-rich structure that polymerizes into fibrils and aggregates [2]. Duplication, triplication or genetic mutations in the α-syn gene (A53T, A30P, E46K, G51D, etc.) are linked to autosomal dominant PD; thus implicating its role in the pathogenesis of PD [3–6]. Moreover, these mutations affect α-syn amyloid fibril formation in vitro, by either accelerating the formation of fibrils [7] or by making them unstable and easier to propagate than wild-type (WT) fibrils [8]. Taken together, these data suggest the rate of fibril formation and propagation (gradual, systematic spread) is an important factor involved in the disease process.
Mounting evidence suggests pathological forms of α-syn self-propagate and spread progressively throughout interconnected brain regions, thereby correlating to the staging of clinical parkinsonian symptoms [9, 10]. This concept evolved from a hypothetical caudo-rostral progression pattern of pathological α-syn accumulations in post-mortem tissue from select cases of PD both prior to and after the onset of motor deficits [9, 11–13]. Until recently, this hypothesis was widely criticized since the transmission of α-syn had not been demonstrated. However, the discovery of LB pathology in fetal grafts from PD patients at autopsy strongly suggests host-to-graft transmission occurs within the human brain [14, 15]; an observation that was reproduced in grafting and cell culture models [16, 17]. The mechanism whereby pathological α-syn is taken up and transported to adjacent cells is unknown; yet, based on the current evidence it is hypothesized that pathological α-syn species spread from cell-to-cell as a result of the templated misfolding and/or aggregation of nascent or properly configured α-syn, which is subsequently transferred to neighboring cells [18–20]. Further evidence in support of this transcellular spread of pathological α-syn comes from the initial observation that intrastriatal injection of synthetic α-syn preformed fibrils (PFF) into WT mice induces α-syn pathology that propagates throughout anatomically interconnected regions [21]. Moreover, endogenous α-syn is required for pathological transmissibility, as fibril injection into α-syn knockout mice produces no LB/LN pathology. Several other groups have also demonstrated in mice and non-human primates that pathological α-syn can be transferred to the central nervous system (CNS) and induce a similar pattern of PD-like α-syn pathology [19, 22–24].
Through observations from clinical and post-mortem studies and numerous in vitro and in vivo investigations, significant strides have been made toward understanding both the etiology and pathogenesis of PD. Despite the advancement of the field, major gaps still exist regarding our understanding of its molecular and cellular underpinnings. As a consequence, researchers rely heavily on experimental models to gain greater insight into the cause and pathogenesis of PD. While genetic studies have lead to the development of multiple transgenic PD models [25]; none exhibit the typical degeneration of substantia nigra (SN) dopaminergic neurons that is inherent to the disease process. In fact, a majority of traditional models do not recapitulate many of the characteristic features of the human condition. For example, the commonly utilized toxin models (6-OHDA, MPTP, etc.) are excellent for examining symptomatic therapies or studying the end stages of PD; however, these models exhibit acute neuronal loss and lack LB/LN pathology. To better understand the role α-syn plays in PD, viral vector-mediated overexpression of WT or mutant human α-syn has been targeted to the nigrostriatal system of naïve mice or rats. While this form of α-syn overexpression results in aggregation and nigral degeneration, these events occur at an accelerated pace (1–2 months) and neuropathology is limited to the nigrostriatal circuitry [26, 27] and therefore does not model the widespread pathological α-syn accumulation observed in the brains of PD subjects. More recently, viral vectors were used to express human α-syn in the rat vagus nerve, resulting in more widespread caudo-rostral pathology, but not neurodegeneration [12]. However, the α-syn PFF mouse model [21] offers a novel platform in which the time-dependent accumulation and propagation of widespread LB-like α-syn pathology and subsequent death of nigrostriatal dopamine neurons more closely models the human condition.
Using the rat as a model system offers distinct advantages over mice. Rats have more complex motor behaviors than mice and exhibit fine motor coordination, behaviors that have been extensively characterized [28]. Additionally, rats are more similar to humans in their genetics and pharmacokinetics than mice [29, 30], suggesting that results of therapeutics tested in rats would provide greater predictive validity than testing in mice. The greater synaptic complexity observed in the rat brain indicates that neural plasticity may be better modeled in the rat rather than mice [28]. On a practical level, the larger brain and body size of the rat allows researchers to collect more tissue and fluid samples, enables neurosurgical interventions and in vivo electrophysiology, facilitates intrathecal drug delivery and can allow for neuroimaging procedures that prove more difficult in mice. With these advantages in mind, the aim of the current study was to determine whether exogenous PFF formed by recombinant WT full length mouse α-syn injected into the striatum of naive rats results in the widespread α-syn pathology and nigrostriatal degeneration previously observed following PFF injections into WT mice [21]. Further, we evaluated the effects of intrastriatal α-syn PFF injections on ultrasonic vocalizations (USVs), which we previously found to be affected in response to viral vector induced nigrostriatal α-syn overexpression [27].
Materials and Methods
Animals
Adult male Sprague Dawley rats (200–225g; Harlan Laboratory, Indianapolis, IN) were utilized in all experiments. Studies were conducted at Michigan State University (MSU). Rats at MSU were housed in the Van Andel Research Institute vivarium. The animal facility is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care and complied with all Federal animal care and use guidelines. The Institutional Animal Care and Use Committee approved all protocols.
Experimental Design
Experiment 1
Three-month-old male Sprague Dawley rats were randomized and divided into four groups [1-site recombinant α-syn (α-syn, n=12), 1-site fibrillized α-syn (PFF, n=12), 2-site recombinant α-syn (n=6), 2-site fibrillized α-syn (n=6); Fig. 1)]. Rats received either a 1-site or 2-site intrastriatal injection of the same dose of recombinant mouse α-syn (8ug total) or sonicated pre-formed α-syn fibrils (PFF; 8ug total). To determine the time course for pathological α-syn propagation and accumulation, select cohorts of animals from the 1-site group were euthanized at 30 days (30d; n=3 recombinant α-syn; n=3 PFF) and 60 days (60d; n=3 recombinant α-syn; n=3 PFF) post-injection (pi). Remaining animals (n=6) were followed for 180 days (180d) and assessed for motor deficits at varying intervals throughout the study. Additionally, the vocal ability of rats was assessed at the conclusion of the study, which correlated to 180d post-injection (pi). Immunocytochemical analyses of the striatum were conducted on 1-site fibrillized α-syn PFF and recombinant α-syn injected rats whereas striatal neurochemical analyses were conducted on 2-site fibrillized α-syn PFF and recombinant α-syn injected rats due to the methodological incompatibility between these two techniques. A group of naïve age-matched rats (n=8) were utilized as controls. These animals were euthanized and processed for immunohistochemistry (IHC) (n=4) or high performance liquid chromatography (HPLC) (n=4) for morphological and biochemical comparisons. This overall design for Experiment 1 is illustrated in Figure 1.
Figure 1. Experimental timeline.
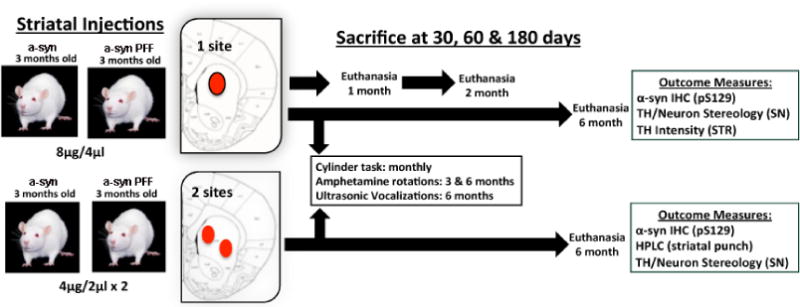
Three-month-old male Sprague Dawley rats received either a 1-site or 2-site intrastriatal injection of recombinant mouse α-syn or α-syn pre-formed fibrils (PFF). Select cohorts of animals from the 1-site group were sacrificed at early time-points pi (1 month: n=3 per group; 2 month: n=3 per group) and paraformaldehyde fixed tissue sections (1:6) were processed for IHC to assess the rate of phosphorylated α-syn accumulation relative to nigral dopamine neuron loss. Remaining animals from the 1-site (n=6) and 2-site (n=6) groups were followed for a 6-month duration and assessed for motor or vocalization deficits prior to euthanasia. HPLC analysis was conducted in the 2-site group to assess striatal dopamine levels 6-months post-PFF injection. Abbreviations: α-syn=alpha-synuclein; PFF=pre-formed alpha synuclein fibrils; IHC=immunohistochemistry; SN=substantia nigra; HPLC=high performance liquid chromatography; STR=striatum; μl=microliter; μg=microgram, pS129=phosphorylation of α-syn at serine 129 site; TH=tyrosine hydroxylase.
Experiment 2
In a follow-up study, three-month-old male Sprague Dawley rats were divided into two groups and received 1-site intrastriatal injection of either sonicated pre-formed α-syn fibrils (PFF; 8ug total, n=10) or an equal volume of saline (n=7). Accumulation of pathological α-syn was verified and the time course of nigrostriatal degeneration was evaluated. PFF or saline injected rats were euthanized at 60 days (2 months, PFF n=3, saline n=2), 120 days (4 months, PFF n=2, saline n=2) and 180 days (6 months; PFF n=5, saline n=3) post-injection (pi).
Intrastriatal Recombinant α-syn and PFF Injections
Purification of recombinant, full-length mouse α-syn protein and in vitro fibril assembly was performed as previously described [21, 31]. To evaluate the composition of recombinant α-syn and PFF used for injections, each solution was diluted in sample buffer and analyzed via Western blot (Fig. 2h). As expected, blots show an absence of high molecular weight species in the recombinant α-syn solution and show several high bands in the PFF solution. Immediately prior to injection, PFF were thawed, diluted in sterile saline, and sonicated at room temperature using an ultrasonic homogenizer (300VT; Biologics, Inc., Manassas, VA) with pulser at 20%, power output at 30% for 60 pulses at one second each. Recombinant α-syn solution was thawed on ice, diluted in ice-cold sterile saline, and kept on ice throughout the surgical session. Anesthetized rats (0.3 mg/kg, equithesin, i.p.) were placed into a stereotaxic device and either PFF, recombinant α-syn solution or sterile saline was injected into the left striatum at one site (4 ul/8 ug protein; AP +1.6, ML +2.4, DV −4.2 from skull) or two sites (2 ul/4 ug protein per site, 4 ul/8 ug protein total; AP +1.6, ML +2.4, DV −4.2; AP −1.4, ML +0.2, DV −2.8 from skull) at a rate of 0.5 μl per minute (see experimental timeline, Figure 1). After each injection, the needle (pulled glass needle attached to a Hamilton syringe) was left in place for 2 minutes and then slowly withdrawn. Animals were monitored weekly following surgery and sacrificed at various pre-determined time points (30, 60 & 180 days).
Figure 2. Intrastriatal injection of exogenous pre-formed alpha-synuclein fibrils initiate the formation of Lewy-body-like inclusions in regions that innervate the striatum.
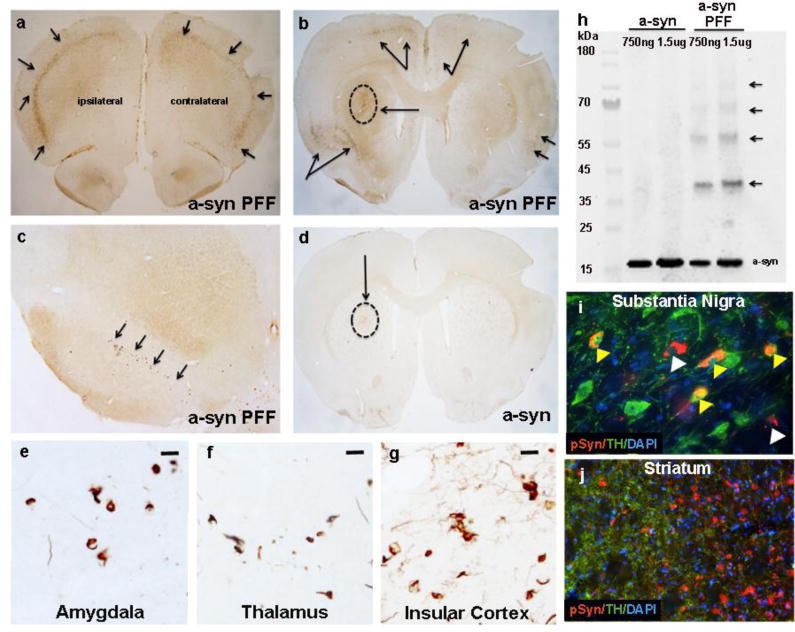
All rats received a unilateral intrastriatal injection of either pre-formed fibril (PFF) or recombinant α-syn (8μg). One month post injection (pi), α-syn PFF injected rats exhibited phosphorylated α-syn intraneuronal accumulations [depicted by phosphorylation at serine129 (pSyn) staining] in several areas that project to the striatum, most prominently in the pre-frontal (a) and insular (b) cortices, and the ipsilateral substantia nigra pars compacta (SNpc; c). Arrows indicate areas marked by the presence of pSyn labeled accumulations. α-syn pathology developed bilaterally within the cortical regions, but remained ipsilateral to the injection site in the substantia nigra. Circle and arrow in b indicates site of intrastriatal α-syn PFF injection. (d). Rats that received injection of non-fibrillized recombinant α-syn did not exhibit α-syn accumulation at this time-point. Circle and arrow indicates site of non-fibrillized recombinant α-syn injection. Other regions such as the amygdala (e), thalamus (medial dorsal nucleus) (f) and insular cortex (g) also developed α-syn pathology post-PFF injection (e-g; six-month time-point shown). (h). Western blot detecting α-syn in diluted non-fibrillized recombinant α-syn solution versus the PFF α-syn solutions used for intrastriatal injections. Note the absence of high molecular weight bands (arrows) in the recombinant α-syn conditions. (i). Immunofluorescence revealing co-localization of phosphorylated α-syn (pSyn, red) inclusions within dopamine neurons (TH+, green) in the SNpc (i; yellow arrows). It is notable that multiple neurons with pSyn accumulations exhibited reduced or absent TH staining (white arrows), suggesting α-syn accumulation reduces the TH phenotype and may induce neuronal toxicity. (j). Co-localization of TH and pSyn was not observed in striatum (6-month time-point). Scale bar in e, f and g=10μm. Abbreviations: α-syn=alpha-synuclein; PFF=preformed fibrils; TH=tyrosine hydroxylase; pSyn=phosphorylation of α-syn; μg=micrograms; ng=nanograms; kDa=kilo Daltons.
Behavioral Evaluations
Behavioral tests to assess motor function were conducted at varying time points during the course of the study (Fig. 1). Assessment of motor function was determined by measurements of forelimb use in the cylinder task (conducted bi-monthly) and amphetamine-induced rotations (conducted on day 90 and 180). Ultrasonic vocalization testing was conducted during the final two weeks of the experiment (~180 days). Only animals in the 180 d cohorts (one or two site injections) were subjected to behavioral testing.
Cylinder task
Assessment of forelimb use asymmetry during spontaneous exploration of the walls of a cylindrical enclosure is a common test of motor function after unilateral 6-OHDA lesions [32] with the extent of striatal denervation significantly correlating with contralateral forelimb use [27]. Rats were tested bi-monthly for the duration of the study. During the dark cycle, rats were placed in a clear, 16.5 cm tall plexiglass cylinder and videotaped for five minutes. The number of weight-bearing forepaw placements on the wall of the cylinder (left, right, or both paws) was recorded (up to 20) in a given trial. Data are reported as the percentage of contralateral forelimb use: [(contra + 1/2 both) divided by (ipsi + contra + both)] × 100 as described previously [32].
Amphetamine-induced rotational asymmetry
To assess the extent of unilateral striatal dopamine depletion, rats were assessed for ipsilateral rotations at 90 and 180 days after α-syn injection (PFF or recombinant α-syn) as described previously [33]. Rats received an i.p. injection of 3.0 mg/kg amphetamine in 0.9% saline and were placed in cylindrical bowls within which rotational behavior was quantified using the TSE LabMaster (Germany) rotometer system. Immediately after the amphetamine injection, animals were acclimated to the cylindrical bowls for five minutes and then the number and direction of rotations were recorded for a total duration of 90 minutes. Data are expressed as the average number of ipsilateral rotations per minute.
Ultrasonic Vocalizations (USVs)
To determine the effect of α-syn accumulation on a rat’s vocal ability, USVs from male rats were evoked using a sexual motivation paradigm [34, 35]. In this paradigm, each male was paired with a receptive female rat (in estrous) for several days until reliable sexual interest (sniffing, chasing, and mounting) was achieved. USVs were examined 180d pi in rats treated with recombinant α-syn solution (n=12), PFF (n=12) and naïve age matched control rats (n=9). To record USVs, a sensitive condenser microphone (CM16 + CMPA, Avisoft, Germany) with a flat frequency response to 150 kHz and a working frequency response range of 10–180 kHz was utilized. The sampling rate was 214,174 Hz, 16-bits. On the day of testing, the microphone was secured 15cm above the test cage and was attached to an ultrasound recording interface (UltraSound Gate 116 Hb; Avisoft Bioacoustics, Germany). To elicit calls from each male, a receptive female rat was placed in the home cage until the male showed signs of sexual interest. The female was then removed and USVs from the male were recorded until at least 50 calls were recorded (~2–6 mins). A variety of acoustic parameters were analyzed including: duration (ms), bandwidth (Hz), intensity (dB), peak frequency (Hz), complexity, call rate, and latency to call [36].
Ultrasonic vocalization data analysis
Avisoft-SASlab Pro software (Avisoft Bioacoustics, Germany) was utilized to analyze acoustic parameters as previously described [34]. Briefly, spectrograms were generated under a 512 FFT (fast fourier transform) length and 75% overlap frame setup. Calls (50-kHz) were separated based on their complexity and divided into two general types of categories: simple and complex. The simple category included both simple and simple compound calls while complex included frequency modulated calls [34]. Due to the fact that there were so few complex calls made within this cohort, only simple calls were analyzed for the aforementioned acoustic parameters. Although acoustic parameters were measured automatically by the SASlab Pro software, categorization of each call was determined by visual and auditory inspection from a rater blinded to treatment groups. During inspection of calls, all extraneous sounds were removed so as to not interfere with USV measurements. For each animal, the maximum, average, and top 10 mean values were calculated for duration, bandwidth, intensity, and peak frequency. Call rate was determined over the first 60 seconds of recording and call latency was scored as the time when the first call was made after the female was removed from the cage and recording commenced.
Immunohistochemistry
Animals were euthanized via pentobarbital overdose (60 mg/kg) and intracardially perfused with saline followed by cold 4% paraformaldehyde (PFA) in 0.1 M PO4 buffer. Brains were extracted and post-fixed in 4% PFA for 24 hours and sunk in 30% sucrose. Brains were frozen on a microtome platform and cut to generate 40 μm thick sections. A 1:6 series of free-floating coronal sections was stained for either tyrosine hydroxylase (TH) or phosphorylated α-syn (pSyn). Tissue was incubated in 0.3% H202 for 45 minutes, rinsed and blocked in 10% normal goat serum (1 hour) then incubated in either primary mouse anti-pSyn (81a-1:10,000;[21]), mouse-anti-TH (1:8000; Immunostar, Hudson, WI), mouse-anti-NeuN (1:1,000, Millipore, Temecula, CA) or rabbit-anti-DARRP32 (1:10,000, Cell Signaling Technology, Danvers, MA) antibodies overnight at 4°C. Then, sections were incubated in biotinylated secondary antisera against either mouse (1:400, Millipore, Temecula, CA) or rabbit IgG (1:400, Millipore, Temecula, CA) followed by Vector ABC detection kit (Vector Labs, Burlingame, CA). Antibody labeling was visualized by exposure to 0.5 mg/ml 3,3′ diaminobenzidine (DAB), 2.5mg/ml nickel ammonium sulfate and 0.03% H2O2 in Tris buffer followed by incubation with the NovaRed kit (Vector Labs, Burlingame, CA). Sections were mounted on subbed slides, dehydrated to xylene and coverslipped with Cytoseal (Richard-Allan Scientific, Waltham, MA).
Immunofluorescence
For all immunofluorescence a 1:12 series of free-floating coronal sections was stained. Tissue was rinsed and blocked in 10% new goat serum in TBS (1 hours) then incubated in primary solution 10% bovine serum albumin in TBS with appropriate primary antibody overnight at 4°C. Then, sections were incubated with corresponding Alexa Fluor antibodies in 10% new goat serum (1 hour). Double labeling was done simultaneously with primaries from two different host species followed by incubation with appropriate secondary antibodies. For DAPI staining, an additional three-minute incubation in Tris buffer with DAPI (1:500; Invitrogen, Carlsbad, CA) was performed. Primary antibodies used include rabbit anti-TH (1:4000; Millipore, Temecula, CA), mouse anti-pSyn (81a-1:15,000), rabbit anti-α-syn (1:5000; Cell Signaling Technology, Danvers, MA), mouse anti-ubiquitin (1:5000; Millipore, Temecula, CA), rabbit anti-SQSTM1 (p62) (1:5000; Cell Signaling Technology, Danvers, MA), rabbit anti-βIII-tubulin (1:2000; Cell Signaling Technology, Danvers, MA), rabbit anti-CNPase (2′,3′-cyclic nucleotide 3′-phosphodiesterase) (1:2000; Cell Signaling Technology, Danvers, MA), and rabbit anti-Glial Fibrillary Acidic Protein (GFAP) (1:10,000; Millipore, Temecula, CA). Secondary antibodies used include Alexa Fluor 568 goat anti-mouse IgG (1:500; Invitrogen, Carlsbad, CA) and Alexa Fluor 488 goat anti-rabbit IgG (1:500; Invitrogen, Carlsbad, CA).
TH and pS129 immunofluorescence for densitrometry analysis
Free-floating tissue sections from animals in the 1-site injection paradigm were blocked in Odyssey blocking buffer (LI-COR, Lincoln, NE) for 60 min at room temperature prior to primary antibody incubation for mouse anti-pSyn (81a-1:10,000) or rabbit-anti-TH (1:8000, Immunostar, Hudson, WI; in Odyssey blocking buffer with 0.2% Triton-X) overnight at room temperature. Following primary incubation, tissue was incubated in secondary antisera for 2 hours at room temperature (LI-COR IRDye 800-Goat anti-mouse, 1:250 in Odyssey blocking buffer). Sections were then rinsed in 0.1 M Tris-buffered saline and immediately mounted onto subbed slides, dehydrated, and coverslipped with Cytoseal. Slides were then imaged using the Odyssey infrared image system (LI-COR, Lincoln, NE) (800/700 nm channel, 42 μm resolution) to examine TH/pSyn expression in the nigrostriatal system (see Densitometry).
Densitometry
Striatal TH positive (TH+) innervations
We utilized densitometry to determine the extent of striatal dopamine denervation in the 1-site PFF injected animals compared to naïve control animals. Serial sections were fluorescently labeled for TH and pS129 and slides were viewed on a LI-COR Odyssey near-infrared scanner (LI-COR, Hudson, WI) and analyzed as described previously [27, 37]. Specifically, to determine unilateral loss of TH immunoreactivity, integrated signal intensities were collected from a contour drawn around the injected (or contralateral) striatal hemisphere. An investigator blinded to experimental conditions drew the contours and every sixth section throughout the entire striatum was included in the analysis. Slides were normalized by analyzing and subtracting background staining intensity from the contralateral cortex for each animal. The average of the raw integrated intensity values (arbitrary units) was calculated for each animal to normalize for disparities in number of sampling sites between animals.
TH+ Dendrites in the SN pars reticulata (SNpr)
The density of TH+ dendrites (DAB visualized) within the SNpr of naïve and PFF-injected rats was determined using Nikon Elements imaging software. Three 10x fields immediately ventral to the SNpc were captured per hemisphere, the sections analyzed were those in closest proximity to the medial terminal nucleus of the accessory optic tract (approximately 3 sections per rat). All images were captured under identical exposure and light intensity parameters. Image capture and analysis was conducted by an experimenter blinded to treatment conditions. To measure density of TH immunoreactive dendrites the average pixel intensity was calculated per field and normalized to a TH negative region within the SNpr in the same section and hemisphere. Values represent the mean pixel intensity averaged across all sampling sites and sections per hemisphere.
Thioflavin-S Staining
A 1:12 series of free-floating coronal sections was stained to assess whether α-syn accumulations display cross-beta-sheet structures, which are a defining feature of insoluble amyloid fibrils [38]. Tissue was immersed in TBS (5 min) followed by an immersion in 0.05% KMnO4/TBS (20 min). After 5 min rinses (5X) in TBS, the tissue was de-stained in 0.2% K2S2O5/0.2% oxalic acid/TBS (3 min). Following de-staining, the slides were immersed in 0.0125% thioflavine-S/40%EtOH/60%TBS (3 min) and differentiated in 50% EtOH/50%TBS (15 min). After 5 min rinses (5X) in TBS and diH2O (3X), the slides were coverslipped with aqueous anti-fade medium (Vectashield; Vector Laboratories, Burlingame, CA).
Aggregated α-syn counts
MicroBrightfield stereological software (MBF Bioscience, Williston, VT) was used to assess the total number of aggregates (defined as dense, darkly stained cores of phosphorylated α-syn staining) within the SN at the 180d time-point. A contour was drawn around the SN using a 4X objective and then aggregates were systematically counted using the 20X objective. The total number of aggregates was recorded for every sixth section (1:6 series) throughout the rostro-caudal axis of the SN. The total number of aggregates for each section was compiled and reported as a total for each animal. Counts reflect actual numbers counted, not a population estimate derived from a sample within the SN.
Stereology
MicroBrightfield stereological software (MBF Bioscience, Williston, VT) was used to assess total population cell counts. Sections were counterstained with cresyl violet following TH immunolabeling in order to distinguish between loss of TH phenotype and neuronal loss. Glial nuclei were excluded from cresyl violet neuronal counts based on smaller size and homogeneous intense Nissl substance staining. Low magnification (1.25X) was used to outline the substantia nigra pars compacta (SNpc) and 20% of the designated area was sampled via a random series of counting frames (50 μm × 50 μm) systematically distributed across a grid (183 μm × 112 μm) placed over the SNpc. An investigator blinded to experimental conditions counted neurons using the optical fractionator probe (60X with oil). A marker was placed on each TH+ or cresyl-labeled neuron within the counting frame while focusing through a z-stack of images (1–2 μm). Between 50 and 300 objects were counted to generate the stereological estimates. The total number of stained neurons was calculated using optical fractionator estimations and the variability within animals was assessed via the Gundersen Coefficient of Error (< 0.1) [39].
Mapping of α-syn pathology
For cell mapping studies, coronal sections were stained for pSyn antibody and developed using the 3′-diaminobenzidine (DAB;Vector Laboratories) chromogen. Immunostained tissue was visualized on an Olympus BX60 microscope and images captured with a Nikon DXM1200 digital camera. Photoshop CS2 (Adobe Systems) was used to map pSyn occurrence/immunoreactivity throughout the rostrocaudal axis of the rat brain. A contour around each section was drawn at 10× magnification and then the visible immunoreactive inclusions/neurites were outlined and filled. Maps were developed using a representative sample from each time point (30, 60 and 180 days).
Western Blotting
To verify PFF did not form spontaneously in the recombinant α-syn solution during storage, solutions of PFF and recombinant α-syn were thawed and diluted in sample buffer, then analyzed via immunoblot. Both solutions (750ng and 1.5ug) were separated on 4–12% Bis-Tris gels (Invitrogen; Carlsbad, CA) and then transferred onto nitrocellulose membranes (Invitrogen, Carlsbad, CA). The blots were probed with the following antibodies: rabbit polyclonal α-syn (1:2,000; Santa Cruz, Dallas, TX) followed by goat anti-rabbit (1:10,000; Novus, Littleton, CO). Protein bands were detected and quantified with the OdysseyClx infrared scanning system (Li Cor, Lincoln, Nebraska).
High performance liquid chromatography (HPLC)
Frozen brains (stored at −80°C) from animals in the 2-site injection paradigm were equilibrated in a cryostat chamber at a temperature of −12 °C prior to dissection. A dorsolateral striatal tissue punch (2×2 mm) was taken from both hemispheres. Frozen punches were placed individually in vials supercooled on dry ice and stored at −80 °C until analysis. Tissue was homogenized and analyzed as described previously [40, 41]. The Pierce BCA Protein Kit (Rockford, IL) was utilized for protein determination. Samples were separated on a Microsorb MV C-18 column (5 Am, 4.6_250 mm, Varian, Palo Alto, CA) and simultaneously examined for norepinephrine, serotonin, DA, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA). Compounds were detected using a 12-channel coulometric array detector (CoulArray 5200, ESA, Chelmsford, MA) attached to a Waters 2695 Solvent Delivery System (Waters, Milford, MA) under the following conditions: flow rate of 1 ml/min; detection potentials of 50, 175, 350, 400 and 525 mV; and scrubbing potential of 650 mV. The mobile phase consisted of a 10% methanol solution in distilled H2O containing 21 g/l (0.1 M) citric acid, 10.65 g/l (0.075 M) Na2HPO4, 176 mg/l (0.8 M) heptanesulfonic acid and 36 mg/l (0.097 mM) EDTA at a pH of 4.1. Data are expressed as ng/mg protein.
Statistical Analysis
All statistical tests were completed using either the IBM SPSS statistics software (version 22.0, IBM, Armonk, NY) or GraphPad Prism software (version 6, GraphPad, La Jolla, CA). A two-tailed independent Student’s t-test was utilized to detect differences between the PFF and naïve age-matched controls NeuN counts and between the 1 and 2 site PFF groups for aggregate counts. Differences between three or more groups were analyzed using a one-way ANOVA and comparisons made across select time points were analyzed using a two-way ANOVA. Treatment effects between naïve, recombinant α-syn, 1-site PFF and 2-site PFF TH+ and cresyl+ neurons were analyzed using a two-way ANOVA with the ipsilateral and contralateral counts comprising the within subject variable. Behavioral measures were analyzed using a two-way ANOVA for repeated measures (except for the USVs which was only assessed at the 180d time-point). When appropriate, post-hoc comparisons were made between groups using the Student-Newman-Keuls or Bonferonni method. The level of significance was set at P ≤ 0.05.
Results
Unilateral, intrastriatal injection of synthetic mouse α-syn PFF results in widespread endogenous α-syn aggregation throughout the rat brain
To determine whether PFF assembled from recombinant mouse α-syn initiate the pathological conversion of endogenous α-syn when injected into rat, we administered a unilateral injection of either α-syn PFF or recombinant α-syn solution into the left dorsal striatum at one or two sites (Fig. 1). At the site of the striatal injection at 30d pi hyper-phosphorylated α-syn (detected by α-syn phosphorylated at serine129 (pSyn) immunohistochemistry) appeared as accumulations in neuritic processes (Fig. 2b, 3b). In contrast, phosphorylated α-syn intraneuronal accumulations were visible in several areas that project afferent innervation to the striatum by 30d pi, most prominently in the pre-frontal (Fig. 2a), insular, cingulate and motor cortical regions (layers IV & V) (Fig. 2b), and the substantia nigra pars compacta (SNpc; Fig. 2c). Despite the fact that PFF were injected unilaterally, after 30d the pSyn-positive intraneuronal and neuritic accumulations were present bilaterally in cortex, but remained ipsilateral to the injection site in the SNpc (Fig. 4a) Inclusions were not observed in astrocytes or oligodendrocytes (data not shown), suggesting inclusions specifically developed in neurons.
Figure 3. Time course of α-syn pathology throughout rat brain.
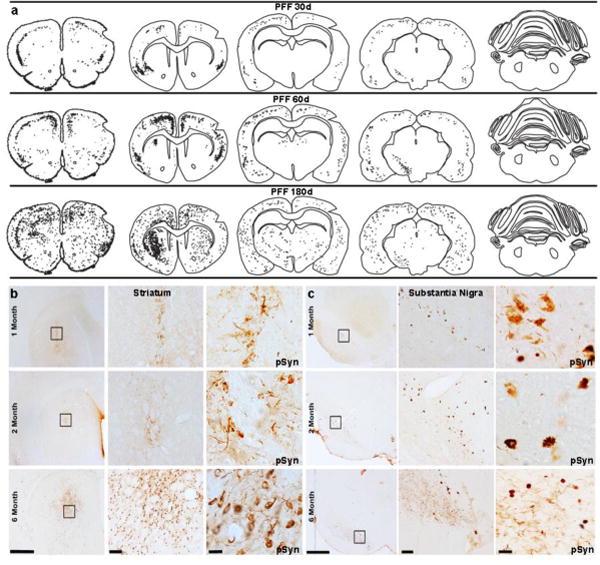
Post α-syn PFF injection, α-syn pathology developed bilaterally within the striatum and cortical regions, but remained ipsilateral to the injection site in the substantia nigra. (a). Representative illustrations detailing the spread and accumulation of α-syn (black dots and stipples, respectively) across both hemispheres of rats sacrificed at 30, 60, or 180 days post injection. (b, c). Representaive images of phosphorylated α-syn (pSyn) depicting α-syn aggregation/accumulation within the striatum (b) and substantia nigra pars compacta (SNpc, c) over time and at increasing magnifications. Scale bars from left to right within each structure panel=1000μm, 100μm and 10 μm. Abbreviations: α-syn=alpha-synuclein; PFF=preformed fibrils; d=days; pSyn=phosphorylation of α-syn
Figure 4. α-syn pathology in the substantia nigra remains ipsilateral to intrastriatal injection.
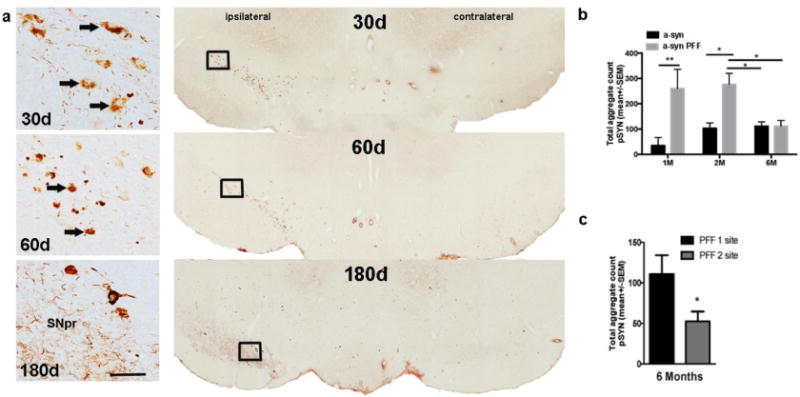
Rats were injected intrastriatally with equal amounts (8 μg) of recombinant α-syn or PFF at 1-site or 2-sites. Select cohorts were euthanized at 30, 60 or 180 days post injection (pi). Rats in both PFF groups developed substantial α-syn pathology (phosphorylated α-syn, pSyn) over time in the substantia nigra (SN) ipsilateral to injection. (a). Representative images of pSyn pathology (arrows) in the ipsilateral SN pars compacta (SNpc) illustrating the accumulation of pSyn α-syn aggregates at 30, 60 and 180 days post 1-site injection of α-syn PFF. At 30d pi, pSyn immunostaining appeared diffuse in the cellular cytoplasm, at 60d pi pSYN immunostaining appeared more compact and rounded, by 180d pi relatively few rounded or cellular accumulations are apparent the SNpc and diffuse neuritic pSyn immunostaining is evident in the SN pars reticulata (SNpr). Scale bar=20 um. (b). Graph depicting the time course of α-syn aggregation (pSYN) in the SN in rats that received a 1-site injection of either PFF or recombinant α-syn. *p<0.05, ** p<0.01 compared to recombinant α-syn at same time point; #p< 0.05 compared to 60d (2M) PFF groups. (c). Aggregate counts are significantly different between the 1-site and 2-site PFF injected rats at the 180d (6M) time-point. Data represent mean +/−SEM. Abbreviations: pSyn = phosphorylated α-syn at the serine 129 site; PFF = α-syn pre-formed fibril; PFF1=1 site injection group; PFF2=2 site injection group; contralateral=contralateral hemisphere (relative to injection site); ipsi=ipsilateral hemisphere (relative to injection site); d=days; α-syn=recombinant α-syn; SEM=standard error of the mean; pi=post injection, SNpr=substantia nigra pars reticulata.
Immunofluorescence was utilized to confirm localization of pSyn-positive inclusions in the SNpc. Results show co-localization of pSyn and TH in the SNpc, indicating inclusions accumulated within nigral dopamine neurons (Fig. 2i). pSyn-positive inclusions were also seen in SN neurons not labeled with TH, indicating α-syn aggregation/accumulation elicits a down regulation of the TH phenotype [42], but may also develop within other neuronal subtypes in the SNpc. Colocalization of pSyn and TH was not observed in striatum (Fig. 2j). Further analysis confirmed pSyn-positive staining co-localized with endogenous α-syn, p62, and ubiquitin indicating they share common features with LBs in the human condition (Supplemental Fig. 2). Furthermore, Thioflavin-S staining was evident in the SNpc and α-syn inclusions were proteinase-K resistant suggesting they develop into insoluble aggregates (Supplemental Fig. 2 k, l). Lastly, analysis of the non-fibrillized α-syn and PFF solutions revealed the presence of high molecular weight α-syn in the PFF condition and a relative absence of high molecular weight α-syn in the non-fibrillized α-syn solution (Fig. 2h).
Mapping of pSyn pathology throughout the rostrocaudal axis illustrates the time-dependent accumulation and propagation of LB-like α-syn pathology (Fig. 3a). In general, structures in which LB-like α-syn pathology was initially observed at the 30–60d time point demonstrated an increase in the abundance of α-syn pathology over time including the insular, cingulate and motor cortical regions, thalamus and amygdala (Fig. 2 e–g, Supplemental Figure 3), all regions with abundant innervation to the striatum. However, whereas α-syn pathology persisted in the SNpc at 180d, it was less abundant at 180d compared to 30d or 60d (Fig. 3a, 3c, Fig. 4). LB-like α-syn pathology also eventually developed in some regions that initially did not possess intraneuronal pSyn-positive inclusions (Fig. 3a). Most notably, α-syn accumulation develops more slowly in the striatum and extensive pSyn-positive intraneuronal accumulation/aggregation not evident until 180d (Fig. 3b). Within 180d pathology was not observed in regions not directly connected to the striatum such as the hippocampus, ventral tegmental area and cerebellum. It is notable that pSyn-positive accumulation/aggregation was not observed in other monoaminergic regions that have connections to the striatum at any of the time points examined (ie., locus coeruleus and dorsal raphe nucleus; data not shown).
Rats injected with the non-fibrillized recombinant α-syn solution did not exhibit any pSyn immunoreactivity at early time-points, but ultimately exhibited mild pathology characterized by pSyn-positive accumulation/aggregation within cortical areas and the SNpc at the 180-day time point (Supplemental Fig 1).
Unilateral PFF injection results in ipsilateral accumulation of α-syn aggregates in the SNpc
Rats that received unilateral intrastriatal injection of PFF developed substantial α-syn pathology over time in the ipsilateral SNpc. The progressive accumulation of α-syn aggregates evolved from small, diffuse cytoplasmic inclusions at 30 days to large, dense perinuclear LB-like α-syn inclusions at the 60-day time-point (Fig. 4a). Although α-syn aggregates were not observed in the SNpr at any time point, neuritic pathology became apparent at the 180d time point (Fig. 4a) when a majority of the aggregates within the SN had diminished, and pathology was more pronounced in the striatum. Accumulation of pSyn positive aggregates in the SNpc was always observed ipsilateral to intrastriatal injection and were most often observed within the more vulnerable ventral tier of the SNpc [43, 44] (Fig. 5b) where they continued to accumulate until the 180d time point, at which time they diminished (Fig. 5c). For the 1-site groups, there was a significant effect of time for aggregate counts within the SNpc (F (1, 14) = 24.80, p = 0.0002). Post hoc analysis indicates the number of aggregates significantly increased at the early time-points and then decreased at the 180-day time-point (Fig. 4b) when nigral dopamine neuron loss was apparent. There was a more pronounced decrease in aggregates at 180 days in rats that received the 2-site intrastriatal injection of PFF compared to the 1-site group (t(9) = 2.326, p < 0.05) (Fig. 4c).
Figure 5. Seeded α-synuclein pathology leads to progressive bilateral nigral dopamine neuron degeneration.
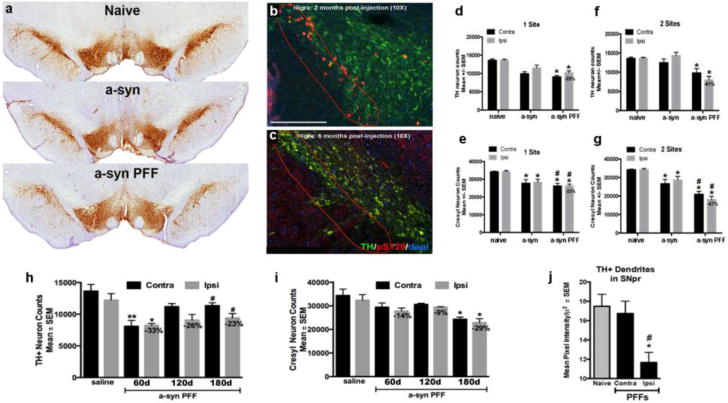
Rats were injected intrastriatally with equal amounts (8μg) of recombinant α-syn or PFF at 1-site or 2-sites. (a). Panel illustrates substantia nigra dopamine neurons visualized using tyrosine hydroxylase (TH) immunohistochemistry in both the recombinant α-syn or α-syn PFF treatment groups compared to naïve age-match control 6 months post injection. Rats in both α-syn groups developed substantial α-syn pathology over time in the substantia nigra; however, only rats in the α-syn PFF injected groups exhibited significant nigral dopamine neuron degeneration at the 6 month time-point. (b). Representative images of pSYN pathology in the more vulnerable ventral tier (red dashed line) of the substantia nigra pars compacta (SNpc) illustrating the accumulation of α-syn aggregates at two months and their subsequent reduction at 6 months (c) pi. (d). Stereology results show significant bilateral dopamine (TH+) neuron loss in both the 1- (d) and 2-site (f) PFF injected animals. * p<0.002 compared to either naïve or recombinant α-syn. (e) and (g). Both recombinant α-syn and PFF injections resulted in neuronal loss (cresyl counts) however, PFF-induced neuronal loss was significantly greater than recombinant α-syn injected rats. * p<0.001 compared to naïve. #p < 0.05 compared to recombinant α-syn. (h). Stereological assessment of TH+ neuron loss at 60d, 120d and 180d following unilateral 1-site (8μg) α-syn PFF or saline injection. Significant bilateral reductions in TH+ neurons were observed as early as 60d pi. * p<0.01, ** p<0.001, # p<0.05 compared to saline. (i) Stereological assessment of neuronal loss at 60, 120, and 180 days following unilateral 1-site (8μg) α-syn PFF or saline injection. Significant bilateral neuron loss was not observed until 180d following α-syn PFF injection. *p≤0.003 compared to saline. (j). Optical density analysis of TH+ immunoreactivity within the substantia nigra pars reticulata 180d after 1- or 2-site α-syn PFF injection. Within the SNpr, TH+ dendrites were significantly decreased on the side of the injection, but were unaffected on the contralateral side. Data represent mean +/−SEM. Abbreviations: pS129 = phosphorylated α-syn at the serine 129 site; PFF = α-syn pre-formed fibril; contra=contralateral hemisphere (relative to injection site); ipsi=ipsilateral hemisphere (relative to injection site); TH=tyrosine hydroxylase; α-syn=recombinant α-syn; SEM=standard error of the mean; pi=post injection.
Unilateral PFF injection results in progressive bilateral degeneration of nigral dopamine neurons
At the 180-day time-point, there was an appreciable difference in the appearance of the SNpc in the α-syn PFF treated animals compared to an aged-matched naïve control (Fig. 5a). To assess the extent of nigral dopamine neuron loss, we conducted unbiased stereology for TH+ (dopamine neurons) and cresyl+ (neurons) neurons 180d post-PFF injection. One-way ANOVA revealed no effect of side for any treatment group in either TH+ neurons (F (8, 37) = 10.58, p>0.05) or cresyl+ neurons (F (8, 35) = 13.69, p>0.05). Further, ipsilateral and contralateral 1- and 2- site recombinant α-syn treatment groups also were not significantly different from each other and were combined into a single treatment group for subsequent repeated measures analysis. For TH+ neurons, there was a significant effect of treatment (F (3, 19) = 14.547, p<0.05), both 1-site and 2-site PFF rats possessed significantly fewer TH+ neurons compared to naïve rats (p<0.001) and recombinant α-syn treated rats (p<0.002) but were statistically similar to each other (p>0.05; Fig. 5 d, f). No significant differences in TH+ neurons were observed between naïve and recombinant α-syn injected rats (p>0.05). For cresyl+ neurons a significant effect of treatment was also observed (F (3,18) = 19.25, p <0.0001). Recombinant α-syn solution, 1-site PFF and 2-site PFF injections all resulted in significantly fewer cresyl+ neurons compared to naïve rats (p<0.001; Fig. 5e, g). Further, both PFF-injected treatment groups possessed fewer cresyl+ neurons than recombinant α-syn-injected rats (p<0.05) but PFF treatment groups were not significantly different from each other (p=0.07).
In a follow-up study, we examined the time course of nigral dopamine neuron degeneration by conducting unbiased stereology for TH+ (dopamine) and cresyl+ (neurons) neurons in an additional cohort of rats at 60d, 120d or 180d post 1-site injection of either α-syn PFF or saline. For TH+ neurons (Fig 5h), there was a significant effect of treatment (F (7, 26) = 5.278, p<0.001). There was no effect of side/hemisphere on number of TH+ neurons within a specific time point for any treatment group (p>0.05). Saline injected rats exhibited a non-significant reduction in TH+ neurons in the SN ipsilateral to injection (−13%, p>0.05) demonstrating non-specific damage due to intrastriatal surgical injection. Further, no effect of time pi was observed between any saline injected treatment group (p>0.05); therefore, all saline injected rats were combined into one treatment group for further posthoc comparisons. 1-site α-syn PFF injected rats possessed significantly fewer TH+ neurons compared to saline injected rats at 120d (−33%, p<0.001) and 180d (−23%, p<0.05) pi, with a non-significant similar magnitude of reduction in TH+ neurons observed at 120d (−26%, p<0.08). The magnitude of TH+ neuron loss in the SN ipsilateral to 1-site α-syn PFF injection at 180d was similar to the preceding 1-site α-syn PFF experiment (Experiment 1 = 25% loss, Fig. 5d, Experiment #2 = 23% loss). For cresyl+ neurons (Fig. 5i) a significant effect of treatment was observed (F (7,26) = 3.378, p <0.01). A significant loss of cresyl+ neurons was not observed from 1-site α-syn PFF injection until the 180d time point (p<0.003 compared to saline injection). Both nigral hemispheres demonstrated a similar magnitude of cresyl+ neuron loss (≈ 29%) 180d pi, which was similar to the loss observed 180d after the 1-site α-syn PFF injection in the previous study (≈ 23%; Fig. 5e). Smaller, non-significant reductions in cresyl+ neurons and were observed at 60d (≈ 14%) and 120d (≈ 9%), after 1-site α-syn PFF injection (p>0.05).
Together, these results indicate that PFF injections induce a loss of dopamine neurons that exceeded the degeneration produced by non-fibrillized recombinant α-syn or saline injections. Further, although α-syn accumulation was only observed in the SNpc ipsilateral to the injected striatum, we observed significant bilateral dopamine neuron loss occurs in both the 1-site and 2-site PFF groups. This finding suggests that factors beyond α-syn accumulation/aggregation in the SNpc may contribute to nigral degeneration in the rat PFF model. Lastly, in our follow-up time course study, we observed a significant loss of nigral TH phenotype as early as 60d after 1-site PFF injection whereas significant nigral neuronal loss was not evident until 180d.
Finally, we assessed the impact of α-syn PFF injection on TH+ dendritic density in the SNpr. Results from a one-way ANOVA show a significant effect between groups (F(2,27)=6.970, p<0.005). Posthoc analysis indicated a significant loss of dendrites in the ipsilateral SNpr when compared to either the contralateral SNpr (p=0.01) or naïve controls (p=0.02; Fig. 5j).
Unilateral PFF injection results in bilateral reductions in striatal innervation, but not striatal neuron loss
Rats that received intrastriatal injection of PFF developed substantial α-syn pathology within the ipsilateral and contralateral striatum by the 180d time point compared to naïve control animals (Fig. 6a, b). Using near infrared integrated intensity measurements, we evaluated the extent of TH+ denervation in the striatum resulting from 1-site PFF injections at 30d, 60d and 180d compared to naïve control. One-way ANOVA results indicate there was a significant effect of treatment on TH intensity between groups (F(6,21)=56.939, p<0.0001). Post hoc analysis revealed a significant bilateral increase in TH intensity 30d after PFF injection (+54% ipsi, p<0.0001, +51% ipsi, p<0.0001) compared to TH intensity levels in naïve controls (Fig. 6c). At 60d after PFF injection a significant bilateral reduction in striatal TH intensity was observed compared to naïve (−80% ipsi, p<0.0001, −71% contra, p<0.0001). At 180d post PFF injection, TH intensity in both the ipsilateral and contralateral striatum remained significantly decreased compared to naïve controls (−49% ipsi, p<0.0001; −46% contra, p<0.004). At 180d, contralateral striatal TH intensity was significantly greater than both ipsilateral and contralateral TH intensity at 60d (p<0.03) however striatal TH intensity in the ipsilateral striatum at 180d was not significantly different from either striatal hemisphere at 60d (p>0.05) (Fig. 6c). No significant differences were observed between hemispheres of PFF-injected within the same time point (p>0.05).
Figure 6. Bilateral α-syn pathology in the striatum ultimately results in reduced striatal innervation, but not neuronal cell death.
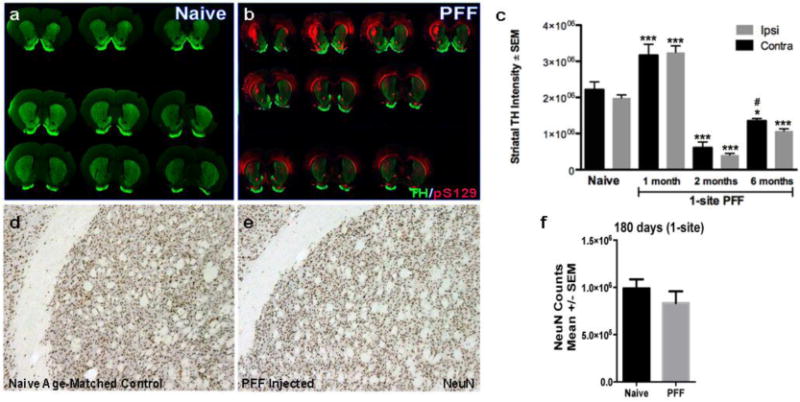
(a, b). Representative images of serial coronal sections proceeding from rostral to caudal, left to right, top to bottom, illustrating the pSyn immunoreactivity (red) and TH intensity (green) 6-months post injection (pi) in a naïve control rat (a) and a α-syn PFF 1-site rat. (c). Using near infrared integrated intensity measurements, we evaluated the extent of dopamine denervation in the striatum resulting from 1-site PFF injected animals at 30d, 60d and 180d and compared to naïve control. In PFF injected rats there was a significant bilateral increase in TH intensity at 30d pi followed by a significant bilateral reduction in the TH intensity at 60d and 180d compared to naïve animals. *** p < 0.0001 compared to naïve, * p<0.01 compared to naive, # p<0.03 compared to either ipsi or contra striatum at 2 months. (d, e). To determine whether pSYN accumulation in the striatum results in neuronal cell death, we utilized stereology to determine the total neuronal population (NeuN) throughout the rostro-caudal striatum. NeuN staining showed no significant differences between the naïve-age-matched striatum (d) and the PFF-injected striatum (e). (f). Stereological quantification of NeuN counts in the striatum (ipsilateral to injection site) 6 months post-PFF injection (1-site) compared to naïve control. Data represent mean +/−SEM. Abbreviations: PFF= pre-formed α-syn fibrils, TH=tyrosine hydroxylase, pS129= α-syn phosphorylation at serine 129, contra=contralateral hemisphere (relative to injection site), ipsi=ipsilateral hemisphere (relative to injection site, NeuN=neuronal nuclei.
Due to the increased α-syn accumulation within the striatum, we examined whether striatal neurons degenerated in response to aggregation. No difference in NeuN immunoreactive neurons was observed within the striatum of PFF treated and naïve rats (Fig. 6d, e). Unbiased stereological analysis was conducted to evaluate the total number of neurons (NeuN) in the striatum (ipsilateral to injection site) of PFF-treated and naïve animals at the 180-day time point (Fig. 6f). Results indicate no significant differences between the PFF-injected animals (1-site paradigm) and the naïve controls, suggesting α-syn accumulation does not elicit striatal neuron death at this time point and with the doses of α-syn PFF injected here.
Unilateral PFF injection results in ipsilateral reductions in striatal dopamine and DOPAC, and bilateral reductions in HVA
HPLC was utilized to assess striatal dopamine and metabolite levels 180 days following 2-site recombinant α-syn or PFF injection and directly compared to levels from the striatum of naïve age-matched controls. There was a significant effect for treatment (F (2, 26)=6.01, p=0.007) and hemisphere (F (1, 26)=14.67, p=0.0007) for DA levels (Fig. 7a), indicating a significant decrease in striatal DA levels in the injected hemisphere of α-syn PFF treated rats. There was also a significant effect of treatment for DA metabolites: 3,4-Dihydroxyphenylacetic acid [DOPAC; (F (2, 26) = 15.61, p < 0.0001); Fig. 7b] and homovanillic acid [HVA; (F (2, 26) = 27.61, p < 0.0001) Fig. 7c]. HPLC posthoc results indicated significant reductions in striatal dopamine (−53%; p < 0.01) and its metabolite DOPAC (−58%; p < 0.01) in the injected striatum of rats that received 2-site α-syn PFF injections compared to naïve age-matched controls. Also, HVA levels were significantly decreased in both the ipsilateral hemisphere (−60%; p < 0.0001), and to a lesser extent in the contralateral hemisphere (−28%; p < 0.05). Furthermore, dopamine turnover (HVA/DA ratio) was significantly reduced in the ipsilateral striatum of recombinant α-syn-injected rats (−29%, p < 0.01) as well as the contralateral striatum of PFF treated rats (−43%; p < 0.001) when compared to naïve control (Fig. 7d). There were no significant differences between groups for striatal levels of norepinephrine (NE) and serotonin (5HT) or their respective metabolites, 3-Methoxy-4-hydroxyphenylglycol (MHPG) and 5-Hydroxyindoleacetic acid (5HIAA) (data not shown).
Figure 7. Striatal dopamine levels and motor performance in recombinant α-syn and PFF injected rats over time.
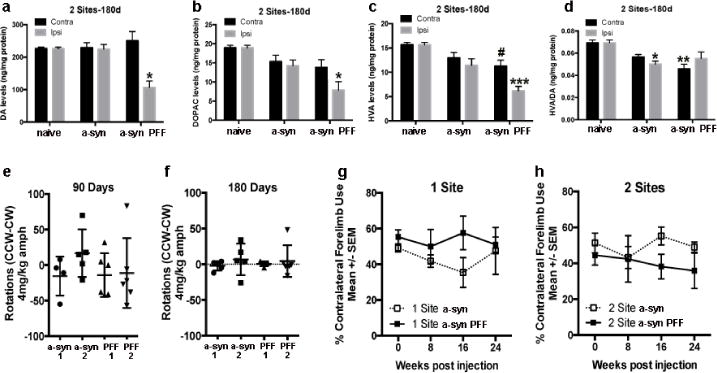
Increased α-syn pathology was associated with a loss of dopamine input to the striatum. HPLC results indicate levels of dopamine (a) and its metabolites 3,4-Dihydroxyphenylacetic acid (DOPAC; b) and homovanillic acid (HVA; c) were significantly decreased in the ipsilateral dorsal striatum 180 days following 2-site α-syn PFF injection. *p < 0.01, *** p < 0.0001, compared to intact naïve controls. (c). HVA levels were also significantly decreased in the contralateral striatum. #p<0.05 compared to intact naïve controls. (d). Dopamine turnover was significantly reduced in the contralateral striatum in α-syn PFF-injected rats as well as in the ipsilateral side of recombinant α-syn-injected rats. **p < 0.001, *p < 0.01, compared to intact naïve controls. (e, f). The disparity in dopamine levels between the 2-site α-syn PFF injected and contralateral striatum was not sufficient to induce amphetamine-induced rotational asymmetry at either 90 (e) or 180 days (f). (g, h). Contralateral forelimb use was not significantly impaired in the cylinder task for either the 1-site (g) or 2-site (h) groups at any time-point. Data represent mean +/-SEM. Abbreviations: α-syn 1= recombinant α-syn 1 site, α-syn 2= recombinant α-syn 2 site, d = days, DA = dopamine, ng = nanogram, mg = milligram, α-syn = recombinant α-syn, contra=contralateral hemisphere (relative to injection site), kg=kilogram, CCW=counter-clockwise, CW=clockwise, amph=amphetamine, PFF=pre-formed fibrilliar alpha-synuclein, PFF1=PFF 1 site, PFF2=PFF 2 site.
Motor function was assessed over the course of the study by assessment of amphetamine-induced rotational asymmetry and contralateral forelimb use in the cylinder task in both 1- and 2-site recombinant α-syn and PFF-injected rats. Dopamine asymmetry was not sufficient to elicit rotational behavior after amphetamine challenge at 90 (Fig. 7e) and 180 days (Fig. 7f). Dopamine asymmetry at these time points also was not sufficient to elicit significant contralateral forelimb deficits in the cylinder task for either the 1-site (Fig. 7g) or 2-site (Fig. 7h) groups. However in the 2-site PFF group, 2 of the 5 rats exhibited profound contralateral forelimb akinesia (<16% use) at the 180d time point.
Pathological α-syn accumulation produces deficits in ultrasonic vocalizations (USVs)
USVs elicited from naïve control rats and rats injected with recombinant α-syn solution or PFF (180 days) were analyzed for the following acoustic parameters: duration, bandwidth, and peak frequency for simple calls (Table 2). Schematic illustration of a variety of simple and complex calls recorded from a male rat after the female is removed from the cage is presented in Fig. 8a. Initially, the 1 and 2 site groups were analyzed separately, but no significant differences were found; therefore, data from both groups were combined to increase statistical power. Additionally, data were analyzed for both simple and compound calls as previously described [27]; however, there were not enough complex calls made for quantification; therefore, data are presented for simple calls only. Two-way ANOVA results show a significant interaction on the number of calls [F(2, 44) = 4.656, p < 0.01] between treatment [F2, 44) = 7.522, p < 0.01] and call type [F(1,44) = 43.16, p < 0.0001]. Post hoc analysis indicated that α-syn PFF rats make fewer simple calls than naïve control rats (Fig. 8b). There was a significant effect of treatment on the call rate [F(2, 23) = 6.09, p < 0.001], where PFF treated animals made significantly fewer calls per second than naïve animals (Fig. 8c). There was also a significant effect of treatment on the duration of calls [maxF(2, 23) = 3.788, p < 0.05, top10F(2, 23) = 12.00, p = 0.0003], suggesting α-syn PFF significantly impacted call duration (Fig. 8d). Post hoc analysis indicates both recombinant α-syn and PFF treated animals had a significant reduction in max call duration compared to naïve rats. Additionally, there was an overall effect of treatment on call frequency [F(2, 23) = 3.50, p < 0.05], indicating a significant reduction in the max peak frequency in PFF treated rats compared to naïve animals (Fig. 8e). There were no significant differences between groups for call intensity (Fig. 8f) or latency (Fig. 8g).
Table 2.
Acoustic parameters of frequency modulated calls in naïve control, recombinant α-syn and PFF α-syn rats.
| Parameter | Naïve Control | Monomer | PFF |
|---|---|---|---|
| Simple calls (%) | 82.21±2.58 | 73.02±3.92 | 73.65±3.93 |
| Duration (ms) | |||
| Max | 0.08±0.007 | 0.06±0.003 | 0.06±0.006* |
| Mean | 0.03±0.001 | 0.03±0.001 | 0.03±0.002 |
| Top 10 | 0.06±0.003 | 0.04±0.002** | 0.04±0.004*** |
| Bandwidth (Hz) | |||
| Max | 33200±2983 | 32029±3940 | 25150±2350 |
| Mean | 8157±516 | 9988±1215 | 8361±597 |
| Top 10 | 21824±2321 | 19897±3306 | 14533±1375 |
| Peak Frequency (Hz) | |||
| Max | 75367±3806 | 67886±2722 | 65630±1540* |
| Mean | 57976±847 | 55236±1479 | 56476±807 |
| Top 10 | 66109±1456 | 61366±2266 | 60604±1178* |
p<0.05,
p<0.01,
p<0.001 compared to naïve control values.
Figure 8. Ultrasonic Vocalizations (USVs) are reduced after recombinant α-syn and α-syn PFF injection.
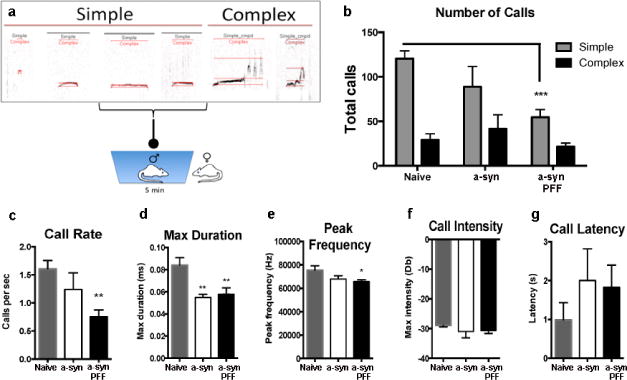
(a). Schematic illustration of a variety of simple and complex calls recorded from male rats after female is removed from cage Results show a reduction in the number of simple calls (b), call rate (c), call duration (d) and frequency (e) of calls from rats that received α-syn PFF injections compared to naïve age-matched rats 6-months post-PFF injection. No significant differences were seen between groups for call intensity (f) or latency (g). Rats that received recombinant α-syn injections also exhibited significant reductions in call duration (d). Data represent mean +/−SEM from both the 1- and 2-site groups combined. *p < 0.05, **p < 0.01, ***p < 0.001 compared to naïve age-matched rats. Abbreviations: α-syn = recombinant α-syn, PFF = preformed α-syn fibrils, s/sec = second, ms = millisecond, Hz = hertz, Db = decibels
Discussion
We show here the successful establishment and characterization of a rat α-synucleinopathy model that recapitulates many features of sporadic PD including progressive spread and bilateral death of nigral neurons across a protracted time course. This model is based on direct injection of the PFF form of α-syn that triggers the pathological conversion of endogenous α-syn, which is associated with progressive PD-like neurodegeneration. Our time-course study demonstrates that at early time-points (30 and 60 days post-injection), PFF-injected rats exhibited hyper-phosphorylated α-syn intraneuronal accumulations (i.e., diffuse LN- and LB-like inclusions) in several brain regions that innervate the striatum, most prominently in the frontal and insular cortices, the amygdala, and the SNpc, suggesting α-syn pathology initially extends to multiple brain regions distal from, but that project to, the injection site. At early time-points the overwhelming majority of hyper-phosphorylated α-syn in the striatum appears as accumulations in neuritic processes, similar to what had observed at early time-points in the Luk et al. (2012) mouse model. Although α-syn accumulation spreads bilaterally in cortical areas, Lewy-like inclusions in the SNpc were found only on the side of injection, illustrating that the spread of α-syn pathology is mediated by synaptic innervation of brain structures [45].
Specifically, based on the pattern of α-syn accumulation observed, it suggests that PFF were initially retrogradely, but not anterogradely, transported from the striatum since hyper-phosphorylated α-syn intraneuronal accumulations did not develop within the target structures of the striatum: SNpr, globus palladus (GP), and the entopeduncular nucleus (EP). At 180d, hyper-phosphorylated α-syn intraneuronal accumulations persisted in structures in which they were originally observed, albeit less abundant in the SNpc, and at this time-point the profile of α-syn pathology in the striatum shifted from neuritic to intraneuronal. We speculate that the delayed intraneuronal striatal α-syn pathology results from “secondary seeding” of striatonigral terminals in the SNpr. It is possible that the hyper-phosphorylated α-syn intraneuronal accumulations were released from degenerating SNpc ventral tier neurons and resulted in α-syn intraneuronal accumulations in the striatonigral pathway. Further studies will examine this issue.
The pSyn antibody utilized in our study (81a) identified aggregates that co-localized with endogenous α-syn and also expressed markers of insoluble (ThioS+/proteinase-K resistant), aggregated Lewy-like pathology (ubiquitin and p62), indicating they share common properties with human LBs/LNs. It is notable that the striatum displayed significant α-syn accumulation at the 180d time-point in the absence of neuronal cell loss. This finding suggests that α-syn aggregates are not directly toxic (at least within the same timeframe), and/or that some cell types may be more or less vulnerable to α-syn accumulation and toxicity under the specific α-syn dose and survival conditions used here.
The time-course, pattern of α-syn accumulation, and magnitude of neurodegeneration we observed is similar to that seen in the Luk et al. (2012) mouse model, suggesting that intrastriatal α-syn PFF seeding results in similar pathology in the rat. Similar to Luk et al. we also observed α-syn PFF injection induces a loss of TH phenotype that precedes overt loss of nigrostriatal dopamine neurons. We scaled up the total amount of PFF protein injected into the rat by 40% (5 μg to 8 μg) based on the relative size of the rat brain compared to the mouse. Additionally, we examined two different injection paradigms to assess whether spatial distribution (1×4 μl vs. 2×2 μl; 8 μg protein total) would change the extent of α-syn pathology and accompanying degeneration. Both paradigms exhibited similar bilateral accumulation of α-syn in specific cortical regions and ipsilateral accumulation in the SNpc, suggesting there were not substantial differences observed in the extent of α-syn accumulation. Unbiased stereology revealed bilateral decreases in TH+/cresyl+ counts in the SNpc for both paradigms, suggesting both injection paradigms induced bilateral dopamine neuron loss, not merely a down-regulation of TH phenotype. However, the magnitude of dopamine neuron degeneration appeared greater in the 2-site injection paradigm. As PFF induced α-syn accumulations have been associated with microglia [46], it is possible that the 2-site PFF injection paradigm resulted in enhanced neuroinflammation that contributed to nigrostriatal degeneration. Similar to PFF injections in the mouse, α-syn inclusions gradually increased within the ventral tier of the SNpc during the first 60d however at the 180d time-point, although α-syn inclusions persisted, a drastic reduction in pSyn immunoreactivity was observed. Indeed, the peak of SNpc α-syn inclusions (60d) appeared to correlate with loss of SNpc TH immunoreactivity that was temporally dissociated from ultimate neurodegeneration. The pattern of ventral tier degeneration may be indicative of placement within the striatum or a selective pattern of α-syn uptake and/or degradation [47]. It is also known that ventral tier SNpc neurons are more vulnerable in PD compared to dorsal tier neurons [44]. This decrease in aggregate number was more pronounced in the 2-site PFF-injected rats corresponding to the difference in nigral dopamine neuron loss between groups, and consistent with the notion that inclusion-containing cells represent the population of cells that ultimately succumbed to degeneration. Overall, our present results suggest that future studies in which spatial distribution of PFF, perhaps by increasing the number of nigral terminals accessible to PFF, may result in more robust bilateral dopamine neuron degeneration.
In line with previous findings in the mouse [21], distant monoaminergic cell groups that innervate the striatum (e.g., locus coerulus (LC) and dorsal raphe) did not develop α-syn pathology, suggesting susceptibility to α-syn accumulation may be region-specific or require a longer incubation period (beyond 180d). Alternatively, the pattern of pathology may be directly related to the degree of innervation, as both the LC and dorsal raphe have relatively sparse input to the dorsal striatum [48]. The observation that SNpc α-syn pathology continues to accumulate during the early time points (30–60d) but then is decreased by the 180d time point as neurons die, suggests that the α-syn does not propagate (i.e., may not be spreading in a “prion-like” manner) and that α-syn pathology results from the direct uptake of the injected protein. A longer time-course may be required to observe second or third order spread from affected sites since the amount of α-syn being released from dying cells would be substantially less than the initial bolus injection. Additional studies will be necessary to determine the extent to which exogenous PFF travel from the site of injection and whether the spread of α-syn stops at the point when affected cells die. Another important factor to consider is that this process occurs in naïve, young, healthy animals that do not exhibit the comorbid factors implicated in PD such as age, inflammation, mitochondrial insufficiencies, etc., which could conceivably exacerbate the spread of α-syn pathology in the human condition.
Although the rat PFF model faithfully recapitulated many of the features seen previously in the mouse, there are several notable differences between the two models. First, in the previous study mice received mouse α-syn PFF, therefore the species of the α-syn PFF was identical to the host. In contrast, the present study utilized mouse α-syn PFF injected into rats. We chose to use mouse α-syn PFF as they have been extensively characterized [18, 21, 31] and so that we could make direct comparisons to the Luk et al. study. Rat α-syn differs from mouse α-syn by a single amino acid change (rat = S121, UniProt P37377; mouse = G121, UniProt O55042). Clearly, if we had observed that mouse α-syn PFF did not seed accumulation of phosphorylated endogenous rat α-syn, this protein difference between species could be implicated. Second, the rat intraneuronal striatal pathology appeared to develop at a slower rate than the mouse and was not observed beyond the injection site until the 180d time-point, despite the 40% increase in total PFF protein injected. Since PFF pathology is dependent on sufficient levels of endogenous α-syn [49] and does not develop in mice lacking α-syn [21], differences in endogenous expression of striatal α-syn between the mouse and rat could affect the rate of α-syn accumulation. Alternatively, mouse α-syn fibrils were injected into a different host species, which could make them less efficient at recruiting endogenous α-syn in the rat, resulting in a slower rate of spread. It is also possible that the larger scale of the rat brain compared to the mouse influenced the rate of pathological accumulation. Third, in the striatum we observed an early upregulation of TH immunoreactivity at the earliest time point examined, 30d, prior to marked loss of TH signal. This early event in the degenerative process induced by α-syn PFF injection may reflect a failed attempt of the nigrostriatal system to maintain dopamine homeostasis. Fourth, we observed that rats injected with non-fibrillized recombinant α-syn solution also developed neuropathology, albeit at a much slower rate and to a lesser extent. To characterize the composition of the recombinant α-syn solution and PFF solution used in our study, we directly compared the two. Although our analysis did not indicate the presence of high molecular weight α-syn in the non-fibrillized recombinant α-syn solution, we have no knowledge as to the aggregation state of the non-fibrillized recombinant α-syn injectate prior to injection. It is possible that aggregates were present in this injectate or may have converted into either oligomers or fibrils following the injection of such a large quantity of α-syn (8ug) into an environment conducive to misfolding (37°C)[50]. In fact, injection of a large quantity of α-syn solution may be akin to α-syn overexpression albeit temporally limited, in which case some toxicity would be expected due to the presumed increase in intracellular α-syn (i.e. concentration dependent kinetics). Future studies in the rat should take into account that the non-fibrillized recombinant α-syn injection is not equivalent to a naive control condition for PFF injections in the rat. Fourth, and most significant, we observed significant bilateral nigral degeneration in the rat PFF model, whereas this did not occur following PFF injection into mice.
Despite differences in the overall profile of nigrostriatal pathology between hemispheres (Table 1), unilateral intrastriatal injection of α-syn PFF resulted in a progressive loss of ≈45% dopamine neurons within both hemispheres of the SNpc. Specifically, ipsilateral to PFF injection we observed phosphorylated α-syn accumulations in the SNpc, loss of dopamine neurons and dendrites in the SN, and reductions in striatal dopaminergic terminal density and striatal dopamine levels. Contralateral to PFF injection, no nigral α-syn accumulations were observed, yet pathology developed in the cortices and striatum, and significant loss of nigral dopamine neurons occurred, accompanied by significant deficits in striatal dopamine turnover. The fact that α-syn pathology in the SN was restricted to the ipsilateral hemisphere suggests that dopamine neuron degeneration in the contralateral SN was mediated by a different mechanism, one not directly attributable to phosphorylated α-syn accumulation. Indeed, the dissociation of dopamine neuron degeneration from the presence of a-syn aggregates raises interesting questions. While the cause of the observed contralateral nigral degeneration is unknown, it may be the result of increased striatal pathology at the 180d time point. Alternatively, we speculate that the intrahemispheric imbalance in dopamine levels in the PFF injected striatum may result in an overactivity of glutamatergic projections to both the ipsilateral and contralateral SN. In support of this it has been shown that unilateral lesioning of the SNpc with 6-OHDA results in increased firing rates of the SNpr, subthalamic nucleus (STN) and pedunculopontine nucleus (PPN) neurons within both hemispheres [49]. Dopamine depletion in the ipsilateral striatum may set off a chronic slow wave of excitotoxicity that ultimately leads to degeneration in the contralateral SNpc. Similar to our present findings, unilateral nigral injections of an excitotoxic/oxidative stress agent to rats also results in an early wave of ipsilateral nigral dopamine neuron degeneration of similar magnitude, followed several weeks later by contralateral nigral neuron degeneration [50]. Whereas secondary neurodegeneration mediated by glutamatergic activity has been previously suggested to play a role in basal ganglia structures distant to the SN [51], we hypothesize that both the ipsilateral and contralateral SNpc may be impacted by this excitotoxicity as well. Neuroinflammation induced by ipsilateral α-syn accumulations [46] may also have contributed to nigral degeneration within the contralateral hemisphere.
Table 1.
Bilateral nigrostriatal pathology observed after unilateral intrastriatal PFF injection.
| Regional Effect | Timepoint | Ipsilateral | Contralateral |
|---|---|---|---|
| Substantia Nigra | |||
| Hyper-phosphorylated α-syn accumulations | 1, 2 & 6 Months | Y | N |
| TH+ Neuron Loss | 6 Months | Y | Y |
| TH+ Dendrite Loss | 6 Months | Y | N |
| Striatum | |||
| TH+ Terminal Density Reduction | 6 Months | Y | Y |
| Diminished Dopamine Levels | 6 Months | Y | N |
| Dopamine Metabolite Deficits | 6 Months | Y (HVA) Y (DOPAC) |
Y (HVA) N (DOPAC) |
| Dopamine Turnover Deficits | 6 Months | N | Y |
Abbreviations: Y=yes/present; N=no/not present; HVA=homovanillic acid; DOPAC= 3,4-Dihydroxyphenylacetic acid; d=days; α-syn=alpha-synuclein
The mechanisms underlying PD pathogenesis and progression may be different, insults responsible for disease initiation may be specific to α-syn mediated toxicity, whereas continued degeneration may result from non-specific oxidative stress, neuroinflammation or excitotoxicity. While the exact mechanisms of the bilateral nigral degeneration observed following unilateral α-syn PFF remain to be elucidated, the end result is clear-showing that PFF initiate a process that ultimately results in nigral cell death and loss of striatal tone. Further studies will examine the time course of unilateral vs. bilateral nigral degeneration induced by unilateral α-syn PFF injection. Should a pattern of initial asymmetry leading to eventual symmetry of nigrostriatal dysfunction emerge, as is observed in PD [51, 52], this would strengthen the face validity of the unilateral α-syn PFF rat model. Taken together, the differences observed between mouse and rat highlight important factors to consider when developing an experimental paradigm for future studies.
We did not observe significant motor impairment following PFF injection using behavioral assessments sensitive to pronounced asymmetry in striatal DA release. It is likely that the bilateral dopaminergic dysfunction observed minimizes asymmetry contributing to our behavioral results. Previous studies suggest that functional impairments on the behavioral measures used here are associated with higher levels of unilateral nigrostriatal denervation (~60–80%)[53, 54]. Although we previously observed significant effects in forelimb asymmetry in our AAV- α-syn overexpression model [27], the rate, extent and asymmetry of dopaminergic cell loss was greater. In light of the bilateral nigrostriatal degeneration we observed, future studies should incorporate motor assessments to detect deficits not dependent on asymmetry, i.e. general locomotor activity. Ongoing studies will determine whether increasing the concentration or injection sites of PFF or extending the length of time pi can increase the magnitude of degeneration, thereby allowing functional deficits to emerge.
USV deficits have been suggested to model the speech difficulties reported by early PD patients [55, 56]. Rats in both recombinant α-syn solution and PFF groups exhibited significant reductions in their ability to vocalize. Communication deficits are common in PD, with dysfunction evident early in the disease process, often prior to the motor deficits that result from striatal dopamine depletion [57]. USVs are postulated to be controlled primarily by the nucleus ambiguous, the hypoglossal nucleus and the periaqueductal grey [58], regions in which we do not observe α-syn pathology. However, targeted nigrostriatal dopamine depletion has also been shown to produce USV deficits, implicating this circuitry as well [55, 56, 59]. We found that the ability to vocalize was impacted following injection of either recombinant α-syn or α-syn PFF, but vocalization deficits were more pronounced in the PFF treated animals where nigral pathology was most pronounced. Previous findings have demonstrated that overexpression of WT α-syn can similarly impair USVs [27, 58]. In our present study, the total number of simple calls made by the rats was significantly reduced following PFF injection. Further, the rate, duration and peak frequency of simple calls were significantly decreased, but bandwidth and intensity were not. Previous studies demonstrate that the quality of calls (bandwidth and intensity) can be impacted by large magnitude striatal dopamine depletion [55, 56, 59]. It is possible that the level of dopamine depletion induced in our 2-site PFF model did not achieve a threshold of deficiency required to observe an impact on the quality of the acoustic signal. Taken together, these results suggest α-syn accumulation and resulting pathology impacts USVs in rats and suggest that α-syn accumulations in the SNpc may underlie the speech difficulties experienced by PD patients.
In summary, we demonstrate that an intrastriatal injection of recombinant mouse α-syn triggers a neurodegenerative cascade in the rat, characterized by the “seed-initiated” retrograde accumulation of intracellular α-syn and bilateral nigrostriatal dopamine neuron degeneration. This model elicits widespread α-syn pathology and diminished unilateral striatal dopamine and results in significant disruption of vocalizations. Accordingly, we have established a reproducible, progressive, neurodegenerative model of synucleinopathy in rats. We contend that establishment of this model offers distinct advantages for in vivo modeling, as rats exhibit greater similarities to humans than mice in their fine motor coordination, complex synaptic organization and pharmacokinetics. The rat PFF model reproduces some important features of the human sporadic PD condition and can provide an important platform for the evaluation of novel therapeutic strategies and investigations into the specific mechanisms involved in the pathogenesis of PD.
Supplementary Material
Highlights.
Striatal injection of fibrillized mouse α-syn triggers a neurodegenerative cascade in the rat
Ipsilateral α-syn accumulations induce bilateral degeneration in the nigra
α-syn accumulations result in significant disruption of ultrasonic vocalizations
Pattern of α-syn accumulation suggests only retrograde transmission
Rat PFF model reproduces important features of the human sporadic PD condition
Acknowledgments
This work was supported by the Saint Mary’s Foundation, and the Morris K. Udall Centers of Excellence for Parkinson’s Disease Research at Michigan State University (NS 058830) and The University of Pennsylvania (NS 053488). The authors also acknowledge Dr. Michelle Ciucci for her expert advice and statistical help with the USV analyses.
Funding: This work was supported by the Saint Mary’s Foundation, and the Morris K. Udall Centers of Excellence for Parkinson’s Disease Research at Michigan State University (NS 058830) and The University of Pennsylvania (NS 053488).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Relevant conflicts of interest/financial disclosures: All funding sources provided unrestricted support and had no role in the oversight or review of the research data or reporting.
References
- 1.Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 2.Stefanis L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harbor perspectives in medicine. 2012;2:a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, et al. alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013;125:753–69. doi: 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 5.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 6.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 7.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–20. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 8.Yonetani M, Nonaka T, Masuda M, Inukai Y, Oikawa T, Hisanaga S, et al. Conversion of wild-type alpha-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem. 2009;284:7940–50. doi: 10.1074/jbc.M807482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braak E, Sandmann-Keil D, Rub U, Gai WP, de Vos RA, Steur EN, et al. alpha-synuclein immunopositive Parkinson’s disease-related inclusion bodies in lower brain stem nuclei. Acta Neuropathol. 2001;101:195–201. doi: 10.1007/s004010000247. [DOI] [PubMed] [Google Scholar]
- 10.Muller CM, de Vos RA, Maurage CA, Thal DR, Tolnay M, Braak H. Staging of sporadic Parkinson disease-related alpha-synuclein pathology: inter- and intra-rater reliability. J Neuropathol Exp Neurol. 2005;64:623–8. doi: 10.1097/01.jnen.0000171652.40083.15. [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 12.Ulusoy A, Rusconi R, Perez-Revuelta BI, Musgrove RE, Helwig M, Winzen-Reichert B, et al. Caudorostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol Med. 2013;5:1051–9. doi: 10.1002/emmm.201302475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bancher C, Braak H, Fischer P, Jellinger KA. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci Lett. 1993;162:179–82. doi: 10.1016/0304-3940(93)90590-h. [DOI] [PubMed] [Google Scholar]
- 14.Chu Y, Kordower JH. Lewy body pathology in fetal grafts. Ann N Y Acad Sci. 2010;1184:55–67. doi: 10.1111/j.1749-6632.2009.05229.x. [DOI] [PubMed] [Google Scholar]
- 15.Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 16.Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, et al. Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis. 2011;43:552–7. doi: 10.1016/j.nbd.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–25. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freundt EC, Maynard N, Clancy EK, Roy S, Bousset L, Sourigues Y, et al. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol. 2012;72:517–24. doi: 10.1002/ana.23747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–47. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–62. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 23.Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–38. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan GA, Pal A, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Scientific reports. 2012;2:898. doi: 10.1038/srep00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antony PM, Diederich NJ, Balling R. Parkinson’s disease mouse models in translational research. Mamm Genome. 2011;22:401–19. doi: 10.1007/s00335-011-9330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koprich JB, Johnston TH, Reyes MG, Sun X, Brotchie JM. Expression of human A53T alpha-synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson’s disease. Mol Neurodegener. 2010;5:43. doi: 10.1186/1750-1326-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gombash SE, Manfredsson FP, Kemp CJ, Kuhn NC, Fleming SM, Egan AE, et al. Morphological and behavioral impact of AAV2/5-mediated overexpression of human wildtype alpha-synuclein in the rat nigrostriatal system. PloS one. 2013;8:e81426. doi: 10.1371/journal.pone.0081426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whishaw IQ, Metz GA, Kolb B, Pellis SM. Accelerated nervous system development contributes to behavioral efficiency in the laboratory mouse: a behavioral review and theoretical proposal. Dev Psychobiol. 2001;39:151–70. doi: 10.1002/dev.1041. [DOI] [PubMed] [Google Scholar]
- 29.Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- 30.Lin JH. Species similarities and differences in pharmacokinetics. Drug metabolism and disposition: the biological fate of chemicals. 1995;23:1008–21. [PubMed] [Google Scholar]
- 31.Volpicelli-Daley LA, Luk KC, Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nature protocols. 2014;9:2135–46. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schallert T. Behavioral tests for preclinical intervention assessment. NeuroRx. 2006;3:497–504. doi: 10.1016/j.nurx.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spieles-Engemann AL, Behbehani MM, Collier TJ, Wohlgenant SL, Steece-Collier K, Paumier K, et al. Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis. 2010;39:105–15. doi: 10.1016/j.nbd.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basken JN, Connor NP, Ciucci MR. Effect of aging on ultrasonic vocalizations and laryngeal sensorimotor neurons in rats. Experimental brain research. 2012;219:351–61. doi: 10.1007/s00221-012-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bialy M, Rydz M, Kaczmarek L. Precontact 50-kHz vocalizations in male rats during acquisition of sexual experience. Behavioral neuroscience. 2000;114:983–90. doi: 10.1037//0735-7044.114.5.983. [DOI] [PubMed] [Google Scholar]
- 36.Ringel LE, Basken JN, Grant LM, Ciucci MR. Dopamine D1 and D2 receptor antagonism effects on rat ultrasonic vocalizations. Behavioural brain research. 2013;252:252–9. doi: 10.1016/j.bbr.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gombash SE, Manfredsson FP, Mandel RJ, Collier TJ, Fischer DL, Kemp CJ, et al. Neuroprotective potential of pleiotrophin overexpression in the striatonigral pathway compared with overexpression in both the striatonigral and nigrostriatal pathways. Gene Ther. 2014;21:682–93. doi: 10.1038/gt.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelenyi G. Thioflavin S fluorescent and Congo red anisotropic stainings in the histologic demonstration of amyloid. Acta Neuropathol. 1967;7:336–48. doi: 10.1007/BF00688089. [DOI] [PubMed] [Google Scholar]
- 39.Gundersen HJ, Jensen EB, Kieu K, Nielsen J. The efficiency of systematic sampling in stereology–reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- 40.Koprich JB, Campbell NG, Lipton JW. Neonatal 3,4-methylenedioxymethamphetamine (ecstasy) alters dopamine and serotonin neurochemistry and increases brain-derived neurotrophic factor in the forebrain and brainstem of the rat. Brain Res Dev Brain Res. 2003;147:177–82. doi: 10.1016/s0165-3806(03)00219-0. [DOI] [PubMed] [Google Scholar]
- 41.Koprich JB, Chen EY, Kanaan NM, Campbell NG, Kordower JH, Lipton JW. Prenatal 3,4-methylenedioxymethamphetamine (ecstasy) alters exploratory behavior, reduces monoamine metabolism, and increases forebrain tyrosine hydroxylase fiber density of juvenile rats. Neurotoxicol Teratol. 2003;25:509–17. doi: 10.1016/s0892-0362(03)00091-6. [DOI] [PubMed] [Google Scholar]
- 42.Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, et al. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:2780–91. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Double KL, Reyes S, Werry EL, Halliday GM. Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Progress in neurobiology. 2010;92:316–29. doi: 10.1016/j.pneurobio.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Kanaan NM, Kordower JH, Collier TJ. Age-related changes in dopamine transporters and accumulation of 3-nitrotyrosine in rhesus monkey midbrain dopamine neurons: relevance in selective neuronal vulnerability to degeneration. The European journal of neuroscience. 2008;27:3205–15. doi: 10.1111/j.1460-9568.2008.06307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wall NR, De La Parra M, Callaway EM, Kreitzer AC. Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron. 2013;79:347–60. doi: 10.1016/j.neuron.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sacino AN, Brooks M, McKinney AB, Thomas MA, Shaw G, Golde TE, et al. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:12368–78. doi: 10.1523/JNEUROSCI.2102-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McNaught KS, Jnobaptiste R, Jackson T, Jengelley TA. The pattern of neuronal loss and survival may reflect differential expression of proteasome activators in Parkinson’s disease. Synapse. 2010;64:241–50. doi: 10.1002/syn.20719. [DOI] [PubMed] [Google Scholar]
- 48.Pasquier DA, Kemper TL, Forbes WB, Morgane PJ. Dorsal raphe, substantia nigra and locus coeruleus: interconnections with each other and the neostriatum. Brain Res Bull. 1977;2:323–39. doi: 10.1016/0361-9230(77)90066-1. [DOI] [PubMed] [Google Scholar]
- 49.Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, et al. Differential expression of alpha-synuclein in hippocampal neurons. PLoS One. 2014;9:e89327. doi: 10.1371/journal.pone.0089327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breydo L, Wu JW, Uversky VN. Alpha-synuclein misfolding and Parkinson’s disease. Biochim Biophys Acta. 2012;1822:261–85. doi: 10.1016/j.bbadis.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Marinus J, van Hilten JJ. The significance of motor (A)symmetry in Parkinson’s disease. Mov Disord. 2014 doi: 10.1002/mds.26107. [DOI] [PubMed] [Google Scholar]
- 52.Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, Lee CS, et al. Longitudinal progression of sporadic Parkinson’s disease: a multi-tracer positron emission tomography study. Brain. 2009;132:2970–9. doi: 10.1093/brain/awp209. [DOI] [PubMed] [Google Scholar]
- 53.Decressac M, Mattsson B, Lundblad M, Weikop P, Bjorklund A. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiology of disease. 2012;45:939–53. doi: 10.1016/j.nbd.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 54.Decressac M, Mattsson B, Bjorklund A. Comparison of the behavioural and histological characteristics of the 6-OHDA and alpha-synuclein rat models of Parkinson’s disease. Exp Neurol. 2012;235:306–15. doi: 10.1016/j.expneurol.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 55.Ciucci MR, Ahrens AM, Ma ST, Kane JR, Windham EB, Woodlee MT, et al. Reduction of dopamine synaptic activity: degradation of 50-kHz ultrasonic vocalization in rats. Behav Neurosci. 2009;123:328–36. doi: 10.1037/a0014593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ciucci MR, Ma ST, Fox C, Kane JR, Ramig LO, Schallert T. Qualitative changes in ultrasonic vocalization in rats after unilateral dopamine depletion or haloperidol: a preliminary study. Behav Brain Res. 2007;182:284–9. doi: 10.1016/j.bbr.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carneiro D, das Gracas Wanderley de Sales Coriolano M, Belo LR, de Marcos Rabelo AR, Asano AG, Lins OG. Quality of life related to swallowing in Parkinson’s disease. Dysphagia. 2014;29:578–82. doi: 10.1007/s00455-014-9548-3. [DOI] [PubMed] [Google Scholar]
- 58.Grant LM, Richter F, Miller JE, White SA, Fox CM, Zhu C, et al. Vocalization deficits in mice over-expressing alpha-synuclein, a model of pre-manifest Parkinson’s disease. Behav Neurosci. 2014;128:110–21. doi: 10.1037/a0035965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ciucci MR, Ma ST, Kane JR, Ahrens AM, Schallert T. Limb use and complex ultrasonic vocalization in a rat model of Parkinson’s disease: deficit-targeted training. Parkinsonism Relat Disord. 2008;14(Suppl 2):S172–5. doi: 10.1016/j.parkreldis.2008.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.