

Abstract
Hyperphosphatemic familial tumoral calcinosis (HFTC, OMIM #211900) is an autosomal recessive metabolic disorder characterized by hyperphosphatemia, tooth root defects, and the progressive deposition of calcium phosphate crystals in periarticular spaces, soft tissues, and sometimes bone.1 In this HFTC case report, we document the dental phenotype associated with a homozygous missense mutation (g.29077 C>T; c.484 C>T; p.Arg162*) in GALNT3 (OMIM 6017563), a gene encoding UDP-GalNAc transferase 3 that catalyzes the first step of O-linked oligosaccharide biosynthesis in the Golgi. The medical and dental pathology is believed to be caused primarily by high serum phosphate levels (hyperphosphatemia), which, in turn, is caused by failure of GALNT3 to glycosylate the phosphate regulator protein FGF23, impairing its ability inhibit reabsorption of filtered phosphate in the kidneys.
Dentists are increasingly called upon to diagnose rare inherited conditions affecting tooth development. Until recently, the genetic etiologies of most dental anomalies were unknown. Although it was appreciated that a tooth phenotype might be one feature of a broader syndrome, no diagnostic tests were available. The revolution in genetics has changed this and offers dentists both the opportunity and the responsibility to contribute to the diagnosis of inherited conditions in their patients, especially when the first manifestation of the disease is in the dentition and when early treatment might palliate subsequent pathology. In this case, we encountered a patient with generalized short roots, pulp stones, and partial pulp obliteration. Although the proband was the only family member affected, the general (rather than localized) distribution of the dental malformations strongly suggested a genetic etiology.
The most common inherited defects of tooth dentin are autosomal dominant conditions classified as three forms of dentinogenesis imperfecta (DGI) and 2 forms of dentin dysplasia (DD).2 DGI-I is caused by defects in the genes encoding type-I collagen.3 DGI-II, DGI-III, and DD-II are caused by mutations in the dentin-specific gene DSPP.4 These nonsyndromic inherited conditions are relatively common in the population, having an incidence of 1 in every 6000 to 8000 persons.5,6 The dental condition in our patient resembled DD-I.7 The genetic etiologies of DD-I are unknown. Despite its rarity (~1/100,000),8 DD-I appears to be a collection of several inherited conditions. Variability in the clinical presentation suggests that there are (at least) four different DD-I subtypes.9-11 In addition, some cases of DD-I have been reported to show a recessive pattern of inheritance.12
Generalized short roots are observed in multiple syndromes,13 but most of these are rare. In hyperphosphatemic familial tumoral calcinosis (HFTC), the dental root malformations, which include pulp stones and pulp obliteration and a local thickening of the permanent tooth roots, are somewhat specific for this disorder and can serve as a phenotypic marker;14 these malformations seem to fit somewhere between the classic descriptions for DD-I and DD-II.15 The hallmark of hyperphosphatemic diseases is tumoral calcinosis. Ectopic calcifications are often observed in the thyroid cartilage and the large muscles of the lower extremities but can occur at many sites, such as the placenta, iliac vessels, and bone (resembling osteoarthritis).16 HFTC is a group of autosomal recessive soft tissue calcification conditions caused by a breakdown of the FGF23 system for lowering elevated serum phosphate, which is accomplished primarily by reducing the reabsorption of filtered phosphate in the kidneys. Loss of function mutations in both alleles of GALNT3,17 FGF23,1,18-20 and KL (KLOTHO)21 cause tumoral calcinosis.22 It is important that dentists recognize selected tooth root malformations as a potential sign of hyperphosphatemia because dental radiographs can be the first sign of the disease.23,24 Patients are currently placed on a low phosphate diet and medicated with phosphate binders. Additional treatment strategies are showing promise when the normal regimen is ineffective.23,25
CASE REPORT
A 16-year-old South American boy of African descent presented with short roots and generalized tooth mobility and was referred to the Medical Genetic Clinic of Hospital de Clinicas de Porto Alegre in Brazil. No acute health problems were evident clinically. The patient (proband) was reported to be the only person in his family who was affected. At the initial visit, the patient presented with the age-appropriate height of 163 cm and weight of 56 kg and was normotensive. He complained about loose teeth; occasional fatigue; and mild, episodic soreness of his leg muscles and joints, especially his hip, which prevented him from engaging in strenuous physical activity. The patient had a negative history of bone fractures and denied bone fragility in family members. A limited pedigree was drawn up on the basis of the information provided by the patient’s mother (Figure 1A). The simplex pedigree (only one individual in the family being affected) did not allow deduction of a pattern of inheritance or even help determine if the condition was genetic. A review of systems revealed an ectomorphic body type without dysmorphic features or dysfunction. Intraorally, his teeth appeared to be normal in color, size, and shape but had increased surface roughness (Figure 1B). Radiographically, the patient had a full complement of permanent teeth and an overretained primary maxillary cuspid (tooth C). On radiographs, the enamel layer contrasted well with dentin in most teeth (Figures 2A and 2B) and appeared to be slightly thin but within the normal range for thickness. Pulp chambers were obliterated or filled with pulp stones, whereas the tooth roots were shortened and misshaped and appeared to show signs of external root resorption (Figure 2C). Single-rooted teeth exhibited midroot bulges with apical thinning (Figure 2D). By comparing the panoramic radiographs of the same patient taken 2 years apart, it was evident that the root anomalies were progressive. Over time, the midroot bulging, apical thinning, and decreasing crown-to-root ratio become more apparent. None of the teeth had abscessed. There was mild but apparent incisal attrition, particularly on the maxillary lateral incisors. Together with the enamel surface roughness, tooth wear contributed to the impression of modestly altered enamel integrity. A tentative clinical diagnosis of dentin dysplasia was made, and the patient was referred for genetic analyses.
Fig. 1.

Family pedigree and oral photograph of proband. A, Simplex pedigree showing the proband is the only affected individual in the nuclear family. B, Oral photo taken at age 16 years shows that the color and size of teeth were within normal limits. The height of contour of the incisors, which is normally near the incisal edge, appears to be nearer to the cervical margin.
Fig. 2.

Panoramic radiographs. A, Panoramic radiograph at age 14 years. B, Panoramic radiograph at age 16 years. All 32 permanent teeth were visible, although the left maxillary cuspid (tooth #11) was impacted, with retention of tooth C. The tooth roots were shortened and misshaped. Pulp stones and obliterated pulp chambers are observed throughout. The enamel layer contrasted well with dentin in most teeth. Subtle radiolucencies were associated with some mandibular roots, although these teeth were vital and asymptomatic. C, Details of mandibular molars. Some roots (particularly #30 and 31) showed signs of external root resorption. D, Details of mandibular bicuspids. Single-rooted teeth exhibited midroot bulges with apical thinning.
The human study protocol and subject consent were reviewed and approved by the Ethics Committee at the Medical Genetic Clinic of Hospital de Clínicas de Porto Alegre in Brazil, by the IRB of University of Pittsburgh School of Dental Medicine, and the University of Michigan School of Dentistry. The study participant signed an appropriate written consent after its contents had been explained and questions about the study answered. Genomic DNA was extracted from a blood sample and characterized by whole exome sequencing (WES) and informatics analyses following established protocols.26 The WES produced 78.8 M reads at a total of 80× coverage. We identified a truncation mutation in both alleles of GALNT3 (polypeptide N-acetylgalactosaminyl transferase 3; NCBI gene reference NC_000002.12: G.29077 C>T; NCBI cDNA reference NM_004482.3: C.484 C>T; p.Arg162*). The presence of this sequence variation was confirmed by Sanger sequencing of polymerase chain reaction products amplified from genomic DNA (Figure 3A). This rare sequence variation (rs137853086 with allele frequency of 0.0002 validated by 1000 Genome) was previously reported to cause HFTC (OMIM 211900).17 Serum and urine analyses of our patient supported a diagnosis of hyperphosphatemia (Figure 3B).
Fig. 3.

Verification of diagnosis. A, Sanger sequencing chromatograms. Left is the forward (sense) sequence; right is the reverse sequence. An arrowhead marks the mutated nucleotide that introduced a TGA translation termination codon (asterisk). B, Blood and urine chemistry results at age 16 years. *Urine analyses. †Reference values are from the Mayo Clinic, Mayo Medical Laboratories, http://www.mayomedicallaboratories.com/. ALPL, alkaline phosphatase; iPTH, intact parathyroid hormone; WNL, within normal limits.
DISCUSSION
The identical mutation in both alleles of GALNT3 introduced a premature translation termination codon (TGA) in exon 2. GALNT3 has 11 exons; the first exon is not coding. Upstream termination codons generally activate nonsense-mediated decay and degrade mRNA expressed from mutant alleles.27 Even if the GALNT3 mRNA was not degraded, there would definitely be a loss of function because only 162 out of 633 amino acids would be translated, deleting both the glycosyl transferase (aa 188–374) and ricin-B-lectin domains (aa 497–630).16 In any case, there did not appear to be any significant dominant negative effects caused by the truncated protein, as the condition is recessive (no disease phenotype is observed in heterozygotes (the parents). Therefore it is apparent that pathogenesis resulted from a loss of GALNT3 function. GALNT3 catalyzes the first step of O-linked oligosaccharide biosynthesis in the Golgi. The full range of proteins that are glycosylated by GALNT3 in vivo is not known. It appears certain that FGF23 is normally glycosylated by GALNT3 and that failure to glycosylate FGF23 impairs its function, possibly by making it susceptible to proteolytic cleavage (Figure 4). In patients with HFTC, intact serum FGF23 is typically decreased or undetectable, whereas its C-terminal cleavage product is highly elevated.28
Fig. 4.
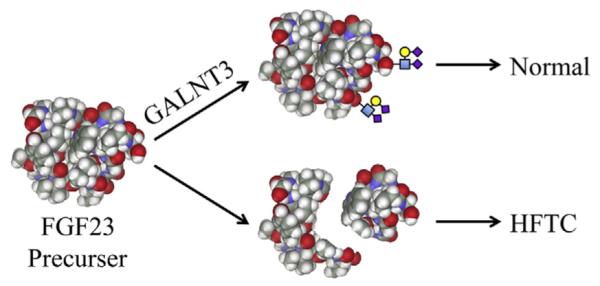
Mechanism for GALNT3 mutations causing hyperphosphatemic familial tumoral calcinosis (HFTC). HFTC can be caused by mutations in GALNT3, FGF23, or KL (FGF23 receptor klotho). GALNT3 is a Golgi enzyme that is required for the O-glycosylation of FGF23. Glycosylation of FGF23 protects it from proteolytic inactivation that results in elevated serum phosphate (causing HFTC).
Loss of function mutations in FGF23, GALNT3, or KLOTHO dysregulates the reabsorption of filtered phosphate in the kidneys, which increases renal phosphate reabsorption and causes hyperphosphatemia. Chronic hyperphosphatemia results in ectopic calcifications within the interstitial spaces, and this is often the cardinal sign that leads to the initial diagnosis of the disorder, often in adulthood. However, the abnormal dental pulp and root structure are typically evident during the late mixed dentition stage (~age 12 years). In the absence of systemic signs, a patient presenting with apparently normal dental crowns with short, blunted roots and pulpal obliteration may easily be misdiagnosed as having dentin dysplasia. Except for the midroot bulge, the dental clinical and radiographic presentation resembles DD-I. In this case, the patient was initially thought to have DD-I until the WES analysis identified the homozygous mutation in GALNT3. Radiographic evaluation revealed hip dysplasia at age 13 years. Blood and urine test results validated the hyperphosphatemic condition, with vitamin D3, parathyroid hormone and calcium in the normal range. The patient will be re-evaluated periodically and will soon start treatment with a phosphate-binding drug.
CONCLUSIONS
Advances in our understanding of genetic diseases affecting the dentition create new opportunities for dentists to recognize inherited conditions and order genetic consultations in order to make a specific diagnosis, which can lead to early and appropriate intervention. In this case, root anomalies detected on dental radiographs led to a genetic consultation that resulted in the diagnosis and treatment of HFTC. In other cases such as generalized enamel malformations29 or tooth agenesis and taurodontism,30 a dental phenotype inspired the genetic consultation that resulted in a specific diagnosis.
Acknowledgments
We thank Dr. Shrikant Mane at the Yale Center for Mendelian Genomics for WES technical support. We thank the participant in this study and the Oral Pathology team and the Medical Genetics Service, Hospital de Clinicas de Porto Alegre, RS, Brazil, for management of the reported case.
This study was supported by NIDCR/NIH research grant DE015846 and by grants from the Bio & Medical Technology Development Program (2011-0027790), the Science Research Center grant to Bone Metabolism Research Center (2012-0000487) by the Korea Research Foundation Grant funded by the Korean Government (MEST), and the Yale Center for Mendelian Genomics (NIH U54 HG006504). The funding sources had no involvement in the study or its publication.
REFERENCES
- 1.Chefetz I, Heller R, Galli-Tsinopoulou A, et al. A novel homozygous missense mutation in FGF23 causes familial tumoral calcinosis associated with disseminated visceral calcification. Hum Genet. 2005;118:261–266. doi: 10.1007/s00439-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 2.Shields ED, Bixler D, el-Kafrawy AM. A proposed classification for heritable human dentine defects with a description of a new entity. Arch Oral Biol. 1973;18:543–553. doi: 10.1016/0003-9969(73)90075-7. [DOI] [PubMed] [Google Scholar]
- 3.Basel D, Steiner RD. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet Med. 2009;11:375–385. doi: 10.1097/GIM.0b013e3181a1ff7b. [DOI] [PubMed] [Google Scholar]
- 4.Kim JW, Simmer JP. Hereditary dentin defects. J Dent Res. 2007;86:392–399. doi: 10.1177/154405910708600502. [DOI] [PubMed] [Google Scholar]
- 5.Witkop CJ. Hereditary defects in enamel and dentin. Acta Genet Stat Med. 1957;7:236–239. doi: 10.1159/000150974. [DOI] [PubMed] [Google Scholar]
- 6.Aplin HM, Hirst KL, Crosby AH, Dixon MJ. Mapping of the human dentin matrix acidic phosphoprotein gene (DMP1) to the dentinogenesis imperfecta type II critical region at chromosome 4 q21. Genomics. 1995;30:347–349. doi: 10.1006/geno.1995.9867. [DOI] [PubMed] [Google Scholar]
- 7.Kalk WW, Batenburg RH, Vissink A. Dentin dysplasia type I: five cases within one family. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;86:175–178. doi: 10.1016/s1079-2104(98)90121-4. [DOI] [PubMed] [Google Scholar]
- 8.Witkop CJ., Jr Hereditary defects of dentin. Dent Clin North Am. 1975;19:25–45. [PubMed] [Google Scholar]
- 9.Scola SM, Watts PG. Dentinal dysplasia type I. A subclassification. Br J Orthod. 1987;14:175–179. doi: 10.1179/bjo.14.3.175. [DOI] [PubMed] [Google Scholar]
- 10.Carroll M, Duncan WK, Perkins TM. Dentin dysplasia: review of the literature and a proposed subclassification based on radiographic findings. Oral Surg Oral Med Oral Pathol. 1991;72:119–125. doi: 10.1016/0030-4220(91)90202-n. [DOI] [PubMed] [Google Scholar]
- 11.Ozer L, Karasu H, Aras K, Tokman B, Ersoy E. Dentin dysplasia type I: report of atypical cases in the permanent and mixed dentitions. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;98:85–90. doi: 10.1016/j.tripleo.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Suman SV, Jayam R, Kumar BV, Dirasantchu S, Kumar KV, Sk S. Typical radiographic findings of dentin dysplasia type 1 b with dental fluorosis. Case Rep Dent. 2013;2013:902861. doi: 10.1155/2013/902861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valladares Neto J, Rino Neto J, de Paiva JB. Orthodontic movement of teeth with short root anomaly: should it be avoided, faced or ignored? Dental Press J Orthod. 2013;18:72–85. doi: 10.1590/s2176-94512013000600012. [DOI] [PubMed] [Google Scholar]
- 14.Lyles KW, Burkes EJ, Ellis GJ, Lucas KJ, Dolan EA, Drezner MK. Genetic transmission of tumoral calcinosis: autosomal dominant with variable clinical expressivity. J Clin Endocrinol Metab. 1985;60:1093–1096. doi: 10.1210/jcem-60-6-1093. [DOI] [PubMed] [Google Scholar]
- 15.Witcher SL, Jr, Drinkard DW, Shapiro RD, Schow CE., Jr Tumoral calcinosis with unusual dental radiographic findings. Oral Surg Oral Med Oral Pathol. 1989;68:104–107. doi: 10.1016/0030-4220(89)90123-0. [DOI] [PubMed] [Google Scholar]
- 16.Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation: case report and review of the literature. BMC Genet. 2014;15:98. doi: 10.1186/s12863-014-0098-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Topaz O, Shurman DL, Bergman R, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–581. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 18.Araya K, Fukumoto S, Backenroth R, et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:5523–5527. doi: 10.1210/jc.2005-0301. [DOI] [PubMed] [Google Scholar]
- 19.Larsson T, Yu X, Davis SI, et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:2424–2427. doi: 10.1210/jc.2004-2238. [DOI] [PubMed] [Google Scholar]
- 20.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 21.Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fathi I, Sakr M. Review of tumoral calcinosis: a rare clinicopathological entity. World J Clin Cases. 2014;2:409–414. doi: 10.12998/wjcc.v2.i9.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dumitrescu CE, Kelly MH, Khosravi A, et al. A case of familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome due to a compound heterozygous mutation in GALNT3 demonstrating new phenotypic features. Osteoporos Int. 2009;20:1273–1278. doi: 10.1007/s00198-008-0775-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burkes EJ, Jr, Lyles KW, Dolan EA, Giammara B, Hanker J. Dental lesions in tumoral calcinosis. J Oral Pathol Med. 1991;20:222–227. doi: 10.1111/j.1600-0714.1991.tb00423.x. [DOI] [PubMed] [Google Scholar]
- 25.Finer G, Price HE, Shore RM, White KE, Langman CB. Hyperphosphatemic familial tumoral calcinosis: response to acetazolamide and postulated mechanisms. Am J Med Genet A. 2014;164:1545–1549. doi: 10.1002/ajmg.a.36476. [DOI] [PubMed] [Google Scholar]
- 26.Wang S-K, Choi M, Richardson A, et al. STIM1 and SLC24 A4 are critical for enamel maturation. J Dent Res. 2014;93:94–100. doi: 10.1177/0022034514527971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller JN, Pearce DA. Nonsense-mediated decay in genetic disease: friend or foe? Mutat Res Rev Mutat Res. 2014;762:52–64. doi: 10.1016/j.mrrev.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichikawa S, Imel EA, Sorenson AH, et al. Tumoral calcinosis presenting with eyelid calcifications due to novel missense mutations in the glycosyl transferase domain of the GALNT3 gene. J Clin Endocrinol Metab. 2006;91:4472–4475. doi: 10.1210/jc.2006-1247. [DOI] [PubMed] [Google Scholar]
- 29.Herzog CR, Reid BM, Seymen F, et al. Hypomaturation amelogenesis imperfecta caused by a novel SLC24 A4 mutation. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;119:e77–e81. doi: 10.1016/j.oooo.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Wang S-H, Choi M, et al. Taurodontism, variations in tooth number, and misshapened crowns in Wnt10 a null mice and human kindreds. Mol Genet Genom Med. 2015;3:40–58. doi: 10.1002/mgg3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]