

Abstract
Increased dietary consumption of docosahexaenoic acid (DHA) is associated with decreased risk for Alzheimer’s disease (AD). These effects have been postulated to arise from DHA’s pleiotropic effects on AD pathophysiology, including its effects on β-amyloid (Aβ) production, aggregation, and toxicity. While in vitro studies suggest DHA may inhibit and reverse the formation of toxic Aβ oligomers, it remains uncertain whether these mechanisms operate in vivo at the physiological concentrations of DHA attainable through dietary supplementation. We sought to clarify the effects of dietary DHA supplementation on Aβ indices in a transgenic APP/PS1 rat model of AD. Animals maintained on a DHA-supplemented diet exhibited reductions in hippocampal Aβ plaque density and modest improvements on behavioral testing relative to those maintained on a DHA-depleted diet. However, DHA supplementation also increased overall soluble Aβ oligomer levels in the hippocampus. Further quantification of specific conformational populations of Aβ oligomers indicated that DHA supplementation increased fibrillar (i.e. putatively less toxic) Aβ oligomers and decreased prefibrillar (i.e. putatively more toxic) Aβ oligomers. These results provide in vivo evidence suggesting that DHA can modulate Aβ aggregation by stabilizing soluble fibrillar Aβ oligomers and thus reduce the formation of both Aβ plaques and prefibrillar Aβ oligomers. However, since fibrillar Aβ oligomers still retain inherent neurotoxicity, DHA may need to be combined with other interventions that can additionally reduce fibrillar Aβ oligomer levels for more effective prevention of AD in clinical settings.
Keywords: docosahexaenoic acid, β-amyloid, aggregation, oligomers, fibrillar, prefibrillar, hippocampus, transgenic, Alzheimer’s disease
INTRODUCTION
Docosahexaenoic acid (DHA) is an ω-3 fatty acid that is an important structural component of cell membranes in the brain. Humans synthesize only a small fraction of their DHA requirements, with the remainder obtained from dietary sources, primarily fish oils. Dietary DHA consumption appears to be associated with Alzheimer’s disease (AD) risk. Epidemiological studies have demonstrated that increased fish consumption correlates with decreased prevalence of AD and dementia, and that increased blood DHA levels correlate with slower cognitive decline (Cole et al., 2009). While clinical trials examining the effects of DHA supplementation in AD dementia have not demonstrated clear benefits in mild-to-moderate AD dementia (Freund-Levi et al., 2006; Quinn et al., 2010), other smaller studies have shown that fish oil or DHA supplementation improves cognition in mild cognitive impairment (Chiu et al., 2008; Kotani et al., 2006; Lee et al., 2013; Rondanelli et al., 2012; Sinn et al., 2012) and age-associated memory impairment (Vakhapova et al., 2010; Yurko-Mauro et al., 2010). These findings suggest that DHA supplementation may be most beneficial at earlier stages of AD pathogenesis and more effective for the prevention of AD than its treatment.
The exact mechanisms that underlie the beneficial effects of DHA supplementation in AD remain somewhat uncertain (see Bazinet and Laye, 2014 for review). Studies in animal models suggest that DHA is likely to exert pleotropic effects on AD pathology that help ameliorate associated behavioral deficits (Cole et al., 2009). Of particular interest, given prior work indicating that β-amyloid (Aβ) pathology is often the earliest neuropathological manifestation of AD (Querfurth and LaFerla, 2010) and is necessary, though not sufficient, for the development of AD-associated cognitive impairment (Knopman, 2014), are the interactions between DHA and Aβ production, aggregation, and toxicity.
Prior studies of DHA supplementation in transgenic mouse models of AD that overexpress autosomal dominant human AD mutations in amyloid precursor protein (APP) and/or presenilin 1 (PS1) have produced mixed results. Although some investigators report that DHA supplementation decreases Aβ accumulation in these models (Lim et al., 2005; Oksman et al., 2006), particularly at earlier stages of disease progression (Broersen et al., 2013; Green et al., 2007; Perez et al., 2010), others have failed to show such effects (Arendash et al., 2007; Koivisto et al., 2014), possibly due to differences in specific dietary regimens between studies. The anti-Aβ effects of DHA supplementation have primarily been attributed to its ability to decrease Aβ production through various mechanisms, including modulating APP localization and decreasing α- and β-secretase activity (Lim et al., 2005), decreasing PS1 levels (Green et al., 2007), or decreasing β-and γ-secretase activity and increasing α-secretase activity (Grimm et al., 2011).
Several in vitro studies suggest that DHA may also decrease Aβ-associated neurotoxicity (Florent et al., 2006; Hashimoto et al., 2011; Hossain et al., 2009; Veszelka et al., 2013; Wang et al., 2010), although others have reported conflicting results (Bate et al., 2008). One hypothesis regarding the Aβ-specific neuroprotective effects of DHA proposes that it may directly modulate Aβ aggregation (Hossain et al., 2009). Soluble oligomeric Aβ species are more neurotoxic than monomeric Aβ species (Larson and Lesne, 2012), and conformational studies indicate that soluble Aβ oligomers exist in multiple conformations, including prefibrillar and fibrillar forms (Kayed et al., 2007), with the former demonstrating greater neurotoxicity (Ahmed et al., 2010). Multiple in vitro studies suggest that DHA inhibits and reverses Aβ aggregation (Hashimoto et al., 2009; Hashimoto et al., 2008; Hossain et al., 2009) and reduces the concentration of prefibrillar Aβ oligomers such as those recognized by the A11 antibody (Kayed et al., 2003). It remains less certain whether the anti-Aβ aggregation properties of DHA contribute to its beneficial effects in vivo. Although a trend towards less A11 immunoreactivity was seen in 3xTgAD mice chronically maintained on DHA-enriched diets, this effect failed to reach statistical significance (Green et al., 2007).
In the current study, we sought to clarify the effects of dietary DHA supplementation on brain and cerebrospinal fluid (CSF) Aβ indices in a transgenic rat model of AD that exhibits age-associated Aβ deposition and cognitive impairment (Flood et al., 2009; Liu et al., 2008; Teng et al., 2011). The use of a rat (as opposed to a mouse) model of AD offers a number of advantages, including longitudinal CSF sampling, more robust behavioral indices, and closer homology to humans (Do Carmo and Cuello, 2013). In order to more closely mimic human clinical trials of DHA in mild cognitive impairment, we initiated chronic DHA supplementation after the onset of Aβ-associated pathology. Based on the prior work described above, our initial hypothesis was that DHA supplementation in this model would decrease Aβ production, oligomerization, and deposition.
METHODS
Experimental Animals
The APP/PS1 transgenic rat model used in these experiments is homozygous for three gene constructs: 1) human APP 695 with the K670N/M671L mutation (rat synapsin-1 promoter); 2) human APP minigene with the K670N/M671L and V717F mutations (platelet derived growth factor β promoter); and 3) human PS-1 with the M146V mutation (rat synapsin-1 promoter) that are expressed on a Sprague-Dawley background (Flood et al., 2009). Sparse parenchymal Aβ plaques begin to appear between 7 to 9 months of age, with progressive age-associated increases in plaque density seen in cortical and hippocampal regions (but not the cerebellum) that plateau by 18 to 20 months of age (Flood et al., 2009; Liu et al., 2008; Teng et al., 2011). Breeding pairs originally obtained from Cephalon (West Chester, PA) were bred and aged at the UCLA School of Medicine vivarium facility.
All animals were housed under a 12-hour light/dark cycle and had access to rat chow ad libitum. Rats were maintained on a standard breeder diet (S-2335; Harlan Teklad, Madison WI) until 13–14 months of age. After baseline CSF sampling, animals were assigned to either a safflower oil-based diet depleted of ω-3 polyunsaturated fatty acids (DHA-group; TD.00522; Harlan Teklad, Madison WI) or supplemented with 0.6% (w/w) DHA in triglyceride form from algal sources (KSF58 powder, Martek Bioscience, Columbia MD) that substituted for a portion of the safflower oil input (DHA+ group; TD.09584; Harlan Teklad, Madison WI). Detailed dietary information is included in Supplementary Table 1. Animals remained on these diets for 4 months and underwent behavioral testing and additional CSF sampling prior to euthanasia at 17–18 months of age. The choice of a DHA-depleted diet as the control condition reflects the relative deficiency of ω-3 fatty acids in the typical Western diet (Cole et al., 2009). The UCLA Chancellor’s Animal Research Committee approved this study and all animal experiments were conducted in compliance with its guidelines.
Behavioral testing
At 17–18 months of age, all animals were tested on the Morris water maze (MWM), which has previously been shown to be sensitive to hippocampal dysfunction (Morris et al., 1982). Animals were trained for 4 trials per day over 10 days to learn to locate and swim to a hidden 6-inch square escape platform submerged below the surface of the water in a 6-foot diameter circular tank. A maximum of 90 seconds was permitted for each trial, after which the animal was placed onto the escape platform. Twenty-four hours after the final training trial, animals were returned to the water tank and allowed to freely swim for 60 seconds during a probe trial in which the escape platform was removed. Video tracking software (SMART 2.5; Panlab, Barcelona, Spain) was used to determine the latency for each training trial and the number of crossings over the former platform position during the probe trial.
CSF sampling
Animals underwent CSF sampling at 13–14 months of age and 17–18 months of age. Under inhaled isoflurane anesthesia, the posterior aspect of each animal’s skull and neck was shaved, and the animal was positioned on its abdomen with its head gently flexed. A 27-gauge butterfly needle affixed to a 1 ml syringe was inserted through the skin at the location of the atlanto-occipital membrane and slowly advanced with constant backpressure until CSF was detected in the needle. A total of 75 to 100 μl of CSF was slowly aspirated from each animal. CSF samples without visible blood contamination were flash frozen in liquid nitrogen and stored at −80° C.
Biochemical and immunohistochemical analyses
After CSF sampling at 17–18 months of age, animals were terminally sedated with pentobarbital and perfused with a HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer containing protease and phosphatase inhibitors (Calon et al., 2004). Brains were removed immediately and hemisected, with one hemisphere prepared for biochemical analyses and the other for immunohistochemical analyses.
For hemispheres prepared for biochemical analyses, the hippocampus and cortical regions were dissected out and flash frozen in liquid nitrogen. The hippocampus was serially extracted into tris-buffered saline (TBS), lysis, and guanidine fractions (pH for all buffers=8.0) as previously described (Calon et al., 2004; Yang et al., 2005). Total protein concentrations for each fraction were determined with BCA (TBS and lysis; Thermo Scientific Pierce; Rockford IL) or Bradford (guanidine; Bio-Rad Laboratories, Hercules CA) protein assay kits.
Total Aβ42 levels from CSF samples and hippocampal fractions were measured with a microsphere-based multiplex flow-cytometry method that incorporates a sandwich-type immunoassay (xMAP; Luminex) using a commercially available kit (Aβ42 Human Singleplex Bead Kit; Invitrogen, Carlsbad CA). Total concentrations of Aβ oligomers in soluble hippocampal fractions were measured with a sandwich ELISA that utilizes the same monoclonal anti-Aβ antibody (10G4; Yang et al., 1994) as both the capture and reporter antibody. A 7-point standard curve (ranging from 1.95 to 125 ng/ml) was derived from photochemically cross-linked synthetic Aβ40 oligomers (Rosensweig et al., 2012).
Western blotting was used for additional analyses of Aβ oligomers and to investigate APP processing. Equal amounts of protein (20 μg) from each sample were mixed with Novex Tris-glycine SDS sample buffer (Invitrogen; Carlsbad CA) and 100mM dithiothreitol (DTT; Expedeon, Harston UK), then boiled, resolved by SDS-PAGE on a 10–20% Tris-glycine gradient gel (Invitrogen; Carlsbad CA) and transferred to a PVDF membrane (Millipore; Billerica MA). To insure equal protein loading and transfer quality, membranes were stained with Ponceau S; only membranes with equal loading were quantified. After blocking with 3% BSA/PBS (1 hr at RT), membranes were probed with 10G4 (for Aβ) or O443 (for APP, recognizes amino acids 751–770; Millipore; Billerica MA). Relative concentrations of different conformational species of Aβ oligomers were determined using dot blot assays that incorporated either anti-prefibrillar (A11; Millipore; Billerica MA) or anti-fibrillar (M87; provided by C. Glabe) Aβ oligomer amyloid antibodies (Kayed et al., 2003; Nussbaum et al., 2012). Soluble hippocampal fractions [1 μg (A11) or 2.5 μg (M87) total protein per well] were applied to rehydrated 0.2 μm nitrocellulose membranes (Bio-Rad Laboratories, Hercules CA) using a Bio-Dot microfiltration apparatus (Bio-Rad Laboratories, Hercules CA). After blocking with 10% fat-free dry milk in TBS with 0.01% Tween 20 (1 hour at RT), membranes were probed with A11 or M87. Chemiluminescent signals from Western and dot blots were detected with Super Signal West Femto substrate (Thermo Scientific Pierce; Rockford, IL) using a BioSpectrum 600 imaging system (UVP; Upland, CA) and quantified using VisionWorks software (UVP; Upland, CA).
Separate Western blots were used to quantify synaptic and microglial markers. Eighty μg of protein from each sample were mixed with a Laemmli’s sample loading buffer with 2.5% 2-mercaptoethanol (2-ME; Fisher Scientific), then boiled, resolved by SDS-PAGE on a 6–12% Tris-glycine gradient gel and transferred to a PVDF membrane (Millipore; Billerica MA). Only membranes with equal loading as determined by Coomassie stain were quantified. After blocking with 10% milk/0.1% gelatin/PBS, membranes were probed with antibodies for drebrin (M2F6; MBL International, Woburn MA), NR2B (BWJHL; Millipore, Billerica MA), CD33 (EPR4423; Abcam, Cambridge MA), or Iba-1 (016-20001; Wako Chemicals, Richmond VA).
For brain fatty acid analyses, lipids were extracted from flash frozen mixed (parietal, occipital, and temporal) cortex (Bligh and Dyer, 1954) with all solvents containing butylated hydroxytoluene and samples routinely purged with nitrogen to minimize oxidation. Methyl esters of fatty acids were prepared using boron trifluoride/methanol and 23:0 fatty acid serving as an internal standard. Quantification of 29 fatty acids was conducted by capillary column gas chromatography with flame ionization as previously described (Hoffman et al., 2000).
Hemispheres prepared for immunohistochemical analyses were immersion fixed in a 5% paraformaldehyde solution, serially cryoprotected in 10% and 20% sucrose solutions, flash-frozen with liquid nitrogen, and cryostat-sectioned into 10 μm coronal sections. Selected sections from each animal were immunolabeled with DAE, a rabbit polyclonal antibody against synthetic peptide Aβ1–13 as previously described (Lim et al., 2001). Hippocampal Aβ deposition was quantified via measurements of DAE-labeled plaque density with NIH Image (NIH; Bethesda MD).
Data analyses
Statistical analyses comparing behavioral, biochemical, and immunohistochemical indices between the DHA− and DHA+ groups were performed using SPSS 22 for Macintosh (IBM, Armonk NY). Levene’s test was used to assess for homogeneity of variance. Indices fulfilling the assumptions of homogeneity of variance were analyzed with parametric statistics [unpaired t-tests, paired t-tests, or Pearson’s correlation coefficient]. Indices that violated these assumptions were analyzed with nonparametric statistics (Mann-Whitney U tests).
RESULTS
Weights and dietary consumption
The average weights and daily food consumption for the animals maintained on the DHA+ and DHA− diets are shown in Table 1. Animals in both groups were similar in weight at both 13–14 months of age and 17–18 months of age (p’s>0.05). Likewise, average daily food consumption from 13–14 months of age through 17–18 months of age was similar between groups (p’s>0.05).
Table 1.
Average body weights and daily dietary consumption for animals maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets. *df=8 for males; df=10 for females. Parentheses denote standard deviation.
| DHA− | DHA+ | t* | |
|---|---|---|---|
| N (M/F) | 9 (4 M/5 F) | 13 (6 M/7 F) | |
| Weight at 13–14 months (g) | |||
| Males | 602.0 (25.6) | 607.5 (5.2) | −0.43 |
| Females | 368.4 (16.5) | 384.6 (50.4) | −0.68 |
| Weight at 17–18 months (g) | |||
| Males | 595.5 (32.92) | 621.7 (15.1) | −1.73 |
| Females | 394.8 (44.21) | 394.1 (63.83) | 0.02 |
| Daily feed consumption (g) | |||
| Males | 20.9 (1.4) | 20.4 (1.1) | 0.58 |
| Females | 19.1 (4.3) | 16.0 (0.8) | 1.91 |
Brain polyunsaturated fatty acid levels
Polyunsaturated fatty acid (PUFA) levels measured from mixed cortex are shown in Table 2. Animals maintained on the DHA+ diet had significantly higher levels of DHA (p<0.001) and significantly lower levels of arachidonic acid, adrenic acid, and ω-6 docosapentaenoic acid (all p’s<0.001) than those maintained on the DHA− diet. Overall, the DHA+ diet resulted in higher total levels of ω-3 PUFAs, lower total levels of ω-6 PUFAs, and a lower ω-6 PUFA/ω-3 PUFA ratio (all p’s<0.001) than the DHA− diet. These results confirm prior work with Tg2576 mice that indicate these dietary regimens can modulate brain fatty acid composition even when initiated at older ages (Calon et al., 2004).
Table 2.
Polyunsaturated fatty acid (PUFA) content in mixed cortex expressed as mean percentage of total fatty acid. Parentheses denote standard deviation; *p<0.05.
| DHA− | DHA+ | t(17) | |
|---|---|---|---|
|
| |||
| N | 9 | 10 | |
| ω-3 PUFA total | 13.24% (0.60) | 16.50% (0.66) | −11.21* |
|
| |||
| 18:3 ω-3 (ALA) | 0.01% (0.01) | 0.02 (0.01) | −1.01 |
| 18:4 ω-3 (SDA) | 0.02% (0.02) | 0.03% (0.02) | −1.64 |
| 20:3 ω-3 (ETE) | 0.007% (0.006) | 0.015% (0.007) | −2.88* |
| 20:5 ω-3 (EPA) | 0.004% (0.003) | 0.04% (0.02) | −4.85* |
| 22:3 ω-3 (DTA) | 0.05% (0.02) | 0.07% (0.03) | −2.38* |
| 22:5 ω-3 (DPA) | 0.10% (0.02) | 0.24% (0.05) | −8.18* |
| 22:6 ω-3 (DHA) | 13.05% (0.61) | 16.08% (0.62) | −10.69* |
|
| |||
| ω-6 PUFA total | 19.59% (0.88) | 13.99 (0.83) | 14.26* |
|
| |||
| 18:2 ω-6 (LA) | 0.82% (0.08) | 0.94% (0.10) | −2.79* |
| 18:3 ω-6 (GLA) | 0.002% (0.001) | 0.003% (0.003) | −1.54 |
| 20:2 ω-6 (EDA) | 0.07% (0.02) | 0.06% (0.01) | 1.17 |
| 20:3 ω-6 (DGLA) | 0.22% (0.03) | 0.35% (0.05) | −7.80* |
| 20:4 ω-6 (ARA) | 11.86% (0.54) | 9.59% (0.58) | 8.79* |
| 22:2 ω-6 (DDA) | 0.04% (0.02) | 0.04% (0.03) | −0.08 |
| 22:4 ω-6 (ADA) | 3.78% (0.23) | 2.46 (0.19) | 13.79* |
| 22:5 ω-6 (DPA) | 2.81% (0.21) | 0.56% (0.09) | 30.69* |
|
| |||
| ω-6/ω-3 ratio | 1.48 (0.10) | 0.85 (0.06) | 16.99* |
ALA: α-linolenic acid; SDA: stearidonic acid; ETE: eicosatrienoic acid; EPA: eicosapentaenoic acid; DTA: docosatrienoic acid; DPA: docosapentaenoic acid; DHA docosahexaenoic acid; LA: linoleic acid; GLA: γ-linolenic acid; EDA: eicosadienoic acid; DGLA: dihomo-gamma-linolenic acid; ARA: arachidonic acid; DDA: docosadienoic acid; ADA: adrenic acid
MWM performance
Previous behavioral studies of this transgenic APP/PS1 rat model of AD indicate that deficits on the MWM emerge by 7 months of age and progressively worsen with increasing age (Liu et al., 2008). In particular, marked deficits are seen by 18–20 months of age (Bilousova et al., 2015). Average swim latencies for the two treatment groups at 17–18 months of age across 10 days of training trials are shown in Figure 1A. The DHA+ group reached asymptotic levels of performance more rapidly than the DHA− group. Across training days 4–7, the DHA− group had significantly more variable performance than the DHA+ group [F(20)=5.33, p=0.032], prompting the use of non-parametric statistics, which revealed significantly better performance by the DHA+ group over this interval [Z(22)=−2.30, p=0.021]. While both groups performed similarly across training days 8–10 [Z(22)=−0.03, p=0.97], the DHA+ group swam across the former platform location during the probe trial more frequently than the DHA− group [Figure 1B; t(20)=−2.76, p=0.012], suggesting better retention of Morris water maze training.
Figure 1.
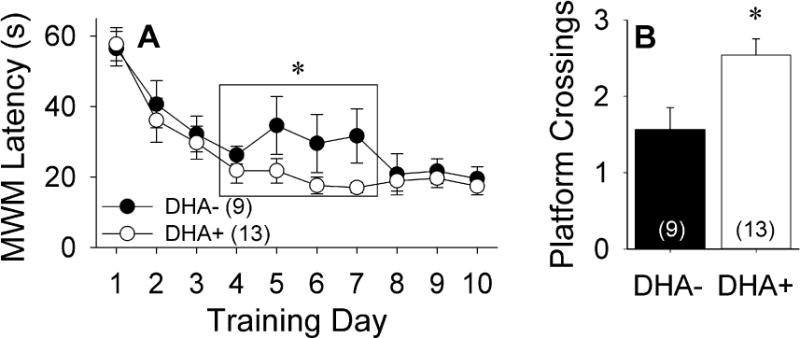
Behavioral performance of APP/PS1 rats maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets. *p<0.05 vs. DHA− group. A) Latency to finding the hidden platform on the Morris water maze (MWM) task at 17–18 months of age. B) Number of crossings over the prior platform location during the MWM probe trial.
CSF Aβ42 levels
At 13–14 months of age, prior to initiation of the dietary interventions, the DHA+ and DHA− groups had similar CSF Aβ42 levels [Figure 2; t(17)=−0.63, p=0.54]. CSF Aβ42 levels were numerically lower in the DHA+ group at 17–18 months of age, but this difference failed to reach statistical significance [t(17)=1.63, p=0.12]. However, longitudinal analyses indicated that CSF Aβ42 levels significantly declined from 13–14 months of age to 17–18 months of age in the DHA+ group [t(9)=2.28, p=0.049], but not in the DHA− group [t(8)=0.15, p=0.79].
Figure 2.
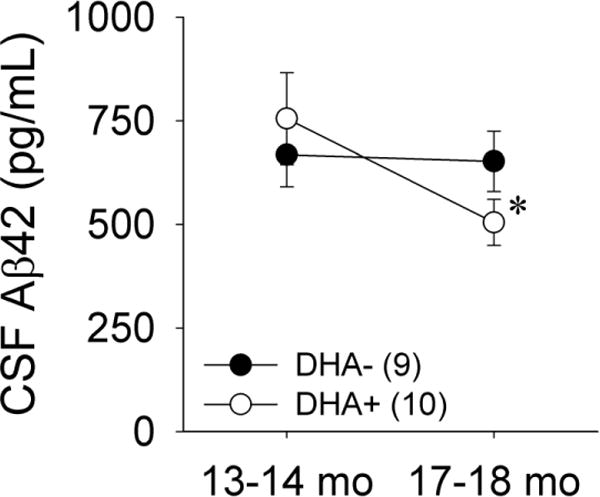
Cerebrospinal fluid (CSF) Aβ42 levels in APP/PS1 rats maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets. *p<0.05 vs. 13–14 months of age.
Hippocampal Aβ plaque density
Immunohistochemical analyses were performed to determine whether DHA supplementation altered Aβ plaque deposition. Figures 3A and 3B are representative coronal hippocampal sections from animals in DHA− and DHA+ groups immunolabeled for Aβ with DAE. The variance in Aβ plaque density was significantly higher in the DHA− group relative to the DHA+ group [F(18)=4.73, p=0.043], prompting the use of non-parametric statistics. Significantly higher Aβ plaque density was seen in the DHA− group relative to the DHA+ group [Figure 3C; Z(20)=−2.70, p=0.006].
Figure 3.
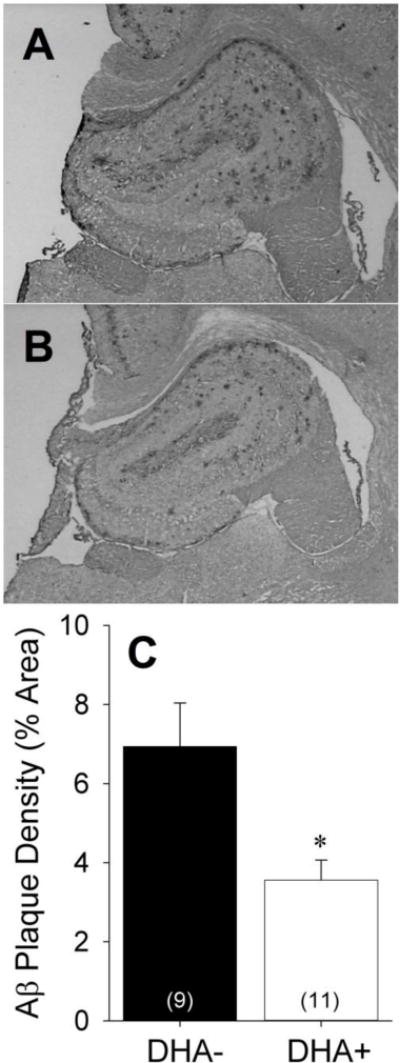
Representative hippocampal sections from APP/PS1 rats maintained on A) DHA-depleted (DHA−) or B) DHA-supplemented (DHA+) diets labeled with the anti-Aβ antibody DAE. C) Aβ plaque density in the DHA− and DHA+ groups expressed as percentage of hippocampal area. *p<0.05 vs. DHA− group.
APP processing
Western blots were performed on the lysis (membrane-associated) hippocampal fractions to examine the effects of DHA supplementation on APP processing (Figure 4). Expression of full-length APP [~110–130 kDa; t(11)=−0.42, p=0.68] and the α-secretase carboxyl-terminal fragment [~9 kDa; t(9)=−0.51, p=0.62] did not differ between groups. Although expression of the β-secretase carboxyl-terminal fragment [~11 kDa] was numerically higher in the DHA+ group than the DHA− group, this difference fell short of statistical significance [t(9)=−2.18, p=0.06].
Figure 4.
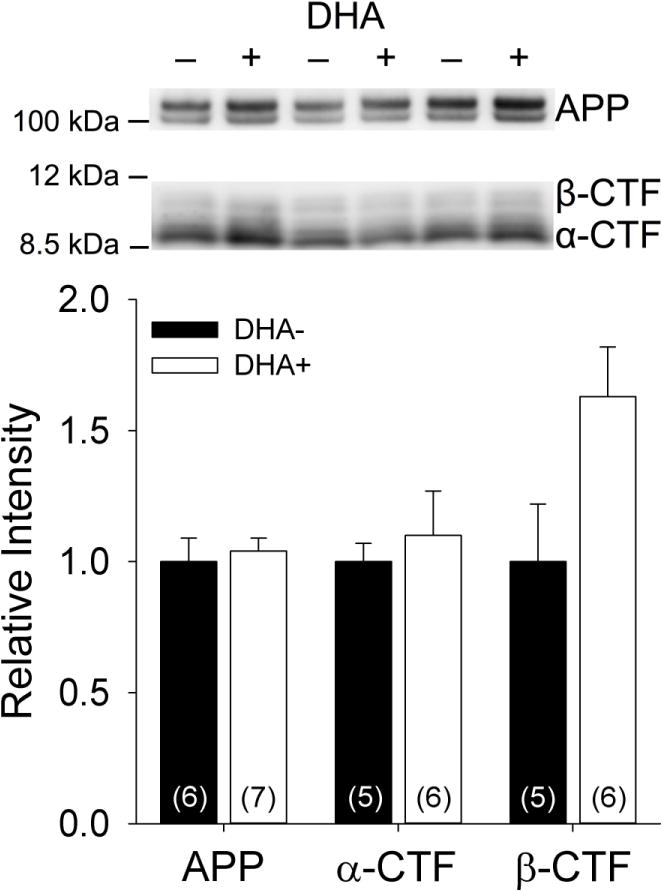
Representative and quantified Western blots of membrane-associated hippocampal fractions from APP/PS1 rats maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets using the anti-APP antibody O443 to detect full-length APP, the β-secretase carboxyl-terminal fragment (β-CTF), and the α-secretase carboxyl-terminal fragment (α-CTF).
Total hippocampal Aβ42 levels
Biochemical analyses were performed to determine the effects of DHA supplementation on total Aβ42 levels in the TBS (soluble), lysis (membrane-associated), and guanidine (insoluble) hippocampal fractions (Figure 5). Animals maintained on the DHA+ and DHA− diets had similar Aβ42 levels in the TBS [t(17)=−0.99, p=0.34] and lysis [t(18)=−0.43, p=0.67] fractions. Aβ42 measurements from the guanidine fraction were notable for marginally higher variance in the DHA+ group [F(17)=4.21, p=0.056], prompting further analyses with non-parametric statistics. However, similar Aβ42 levels were also seen between groups in this fraction [Z(19)=−1.23, p=0.22].
Figure 5.
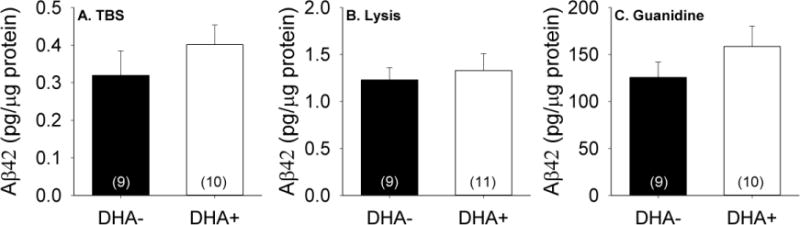
Aβ42 levels measured in A) soluble (TBS), B) membrane-associated (lysis), and C) insoluble hippocampal fractions from APP/PS1 rats maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets.
Hippocampal oligomeric Aβ levels
Since prior studies have reported that DHA modulates in vitro Aβ oligomerization (Hashimoto et al., 2009; Hashimoto et al., 2008; Hossain et al., 2009; Johansson et al., 2007), we sought to compare soluble Aβ oligomer levels between the DHA+ and DHA− groups. Aβ oligomer levels measured from the hippocampal TBS fraction by ELISA are shown in Figure 6. Somewhat surprisingly, given the better behavioral performances and lower hippocampal Aβ plaque densities seen with DHA supplementation, significantly higher Aβ oligomer levels were seen in the DHA+ group relative to the DHA− group [t(16)=−2.65, p=0.018]. Aβ oligomer levels correlated significantly with Aβ plaque density in the DHA+ group [r(8)=0.88, p=0.004], but not in the DHA− group [r(9)=0.27, p=0.49]. Our results from the aggregated Aβ ELISA were confirmed by Western blots probed with the anti-Aβ antibody 10G4 (Figure 7), which demonstrated significantly higher levels of presumed dimeric Aβ (~8 kDa band) in the DHA+ group relative to the DHA− group [t(12)=−2.69, p=0.020].
Figure 6.
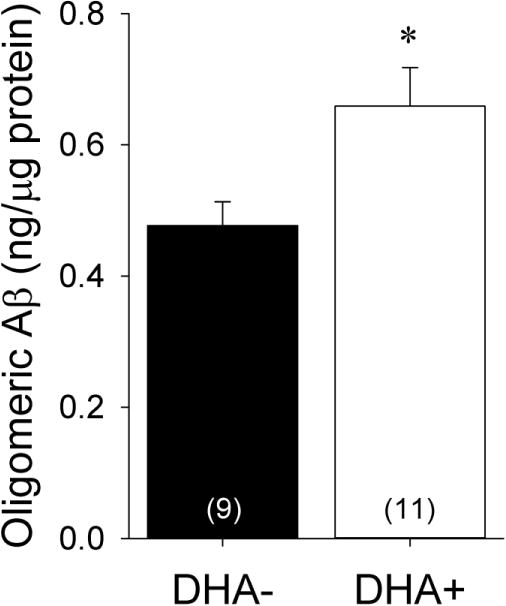
Hippocampal oligomeric Aβ levels measured by ELISA in soluble fractions from APP/PS1 rats maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets. *p<0.05 vs. DHA− group.
Figure 7.

Representative and quantified Western blots of soluble hippocampal fractions from APP/PS1 rats maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets using the anti-Aβ antibody 10G4. sAPP: soluble amyloid precursor protein; Aβ*56: Aβ dodecamer; *p<0.05 vs. DHA− group.
These unexpected findings prompted us to determine whether DHA supplementation might have shifted the conformational distribution of pre-fibrillar versus fibrillar Aβ oligomers. Relative to fibrillar Aβ oligomers, which are recognized by M87 antibody (Nussbaum et al., 2012), prefibrillar Aβ oligomers, which are recognized by A11 antibody (Kayed et al., 2003), appear to be significantly more cytotoxic (Ahmed et al., 2010). Representative dot blots for M87 and A11 using hippocampal TBS fractions from the DHA+ and DHA− groups are shown in Figure 8. Quantification of the M87 dot blots revealed significantly higher concentrations of fibrillar Aβ oligomers in the DHA+ group relative to the DHA− group [t(16)=−3.09, p=0.007]. In contrast, quantification of the A11 dot blots indicated significantly lower concentrations of prefibrillar Aβ oligomers in the DHA+ group relative to the DHA− group [t(17)=2.15, p=0.046]. As expected, M87 and A11 dot blot intensities exhibited an inverse correlation [r(18)=−0.65, p=0.004], confirming that these two conformationally-specific antibodies identify distinct populations of Aβ oligomers. Furthermore, total Aβ oligomer levels measured by ELISA were positively correlated with M87 dot blot intensity [r(18)=0.83, p<0.001] and negatively correlated with A11 dot blot intensity [r(18)=−0.48, p=0.044], indicating that the higher Aβ oligomer levels seen after DHA supplementation were primarily driven by increasing concentrations of fibrillar Aβ oligomers. However, none of the hippocampal Aβ oligomer indices were significantly correlated with any of the measured behavioral outcomes (p’s>0.10).
Figure 8.
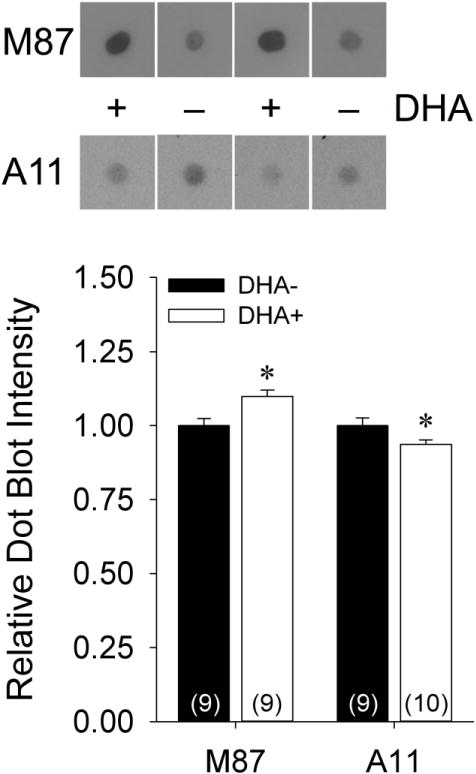
Representative and quantified dot blots of soluble hippocampal fractions from APP/PS1 rats maintained on DHA-depleted (DHA−) and DHA-supplemented (DHA+) diets using conformationally-specific antibodies for fibrillar (M87) and pre-fibrillar (A11) Aβ. *p<0.05 vs. DHA− group.
Synaptic and microglial indices
Prior work in transgenic mouse models of AD has suggested that DHA supplementation may selectively prevent the loss of some synaptic markers (i.e. drebrin) but not others [i.e. the NR2B subunit of the N-methyl-D-aspartate (NMDA) receptor] and that these effects may modulate the beneficial effects of DHA (Calon et al., 2005; Calon et al., 2004; Perez et al., 2010). However, Western blots of the hippocampal lysis fraction showed similar expression of both drebrin [t(16)=−0.92, p=0.37] and NR2B [t(16)=0.77, p=0.45] in the DHA+ and DHA− groups (Supplemental Figure 1). Alternatively, it has also been suggested that DHA supplementation may facilitate microglial Aβ phagocytosis (Hjorth et al., 2013). Higher levels of CD33 expression have been shown to inhibit microglial Aβ phagocytosis and correlate with increased insoluble Aβ levels (Griciuc et al., 2013). However, Western blots of the hippocampal lysis fraction did not reveal any differences in CD33 levels [t(16)=−0.32, p=0.75], Iba-1 levels [t(16)=−0.33, p=0.74], or CD33/Iba-1 expression ratio [t(16)=−0.07, p=0.95], between the DHA+ and DHA− groups (Supplemental Figure 2).
DISCUSSION
These results confirm prior reports that dietary DHA supplementation in Aβ-overexpressing transgenic rodent mouse models of AD can reduce Aβ plaque deposition (Lim et al., 2005) and modestly improve behavioral deficits (Calon et al., 2004), even when initiated after the onset of neuropathological and behavioral changes. However, DHA supplementation did not result in reductions in total Aβ42 levels measured from soluble, membrane-associated, or insoluble hippocampal fractions, which have been seen in some (Green et al., 2007; Lim et al., 2005; Oksman et al., 2006) but not all (Arendash et al., 2007; Arsenault et al., 2011; Koivisto et al., 2014) prior DHA studies.
Furthermore, in contrast to our initial predictions, DHA supplementation increased rather than decreased the concentration of soluble Aβ oligomers in the hippocampus, a paradoxical finding given that oligomers are postulated to be more neurotoxic than other forms of Aβ (Larson and Lesne, 2012). Western blotting using SDS-PAGE gels also suggested that the most prominent oligomeric Aβ species in the DHA+ group were dimers, which have previously been shown to be intrinsically neurotoxic (Shankar et al., 2008). However, it remains uncertain whether the low molecular weight oligomers that are most commonly identified on SDS-PAGE gels represent actual in vivo Aβ oligomers or artifactual constructs that arise during Western blotting (Benilova et al., 2012) through the dissociation of larger Aβ oligomers (Johansson et al., 2007).
However, our further investigations into the structural conformation of the soluble Aβ oligomers suggested that the higher Aβ oligomer levels in the DHA+ group primarily reflected higher levels of fibrillar Aβ oligomers, which may be relatively less toxic than prefibrillar Aβ oligomers (Ahmed et al., 2010). Indeed, DHA supplementation resulted in relative reductions in prefibrillar Aβ oligomers identified by the A11 antibody, a finding that is concordant with a previous study of DHA supplementation in 3xTgAD mice in which treated animals had numerically lower (though not statistically significant) levels of A11-reactive Aβ oligomers (Green et al., 2007). Passive immunization with the A11 antibody reduces cognitive deficits and Aβ deposition in the 3xTgAD model (Rasool et al., 2013), emphasizing the contribution of prefibrillar Aβ oligomers to the pathological features of transgenic rodent models of AD. These results suggest that the specific conformations of Aβ oligomers rather than their overall concentration may be more important for determining their relative neurotoxicity, and in turn, their impact on behavioral outcomes.
Our findings parallel prior analyses of Aβ indices from a human neuropathological cohort, which showed that while AD patients and non-demented high pathology controls exhibited similar overall levels of Aβ monomers and oligomers, AD patients demonstrated significantly higher levels of Aβ oligomers labeled by the conformation-specific NAB61 antibody (Perez-Nievas et al., 2013). Nevertheless, while fibrillar Aβ oligomers may be less neurotoxic than prefibrillar Aβ oligomers, they are not entirely benign. Fibrillar Aβ oligomer levels are elevated in brains from AD patients relative to cognitively normal controls and inversely correlated with cognitive performance (Tomic et al., 2009). Indeed, the elevated levels of fibrillar Aβ oligomers in DHA+ group relative to DHA− group may explain why the behavioral benefits of this intervention remain quite modest, despite the significant reductions in both Aβ plaque load and the more neurotoxic prefibrillar Aβ oligomers.
These findings provide in vivo evidence suggesting that DHA can modulate Aβ aggregation and shift the distribution of different Aβ oligomer conformations. Prior in vitro studies have indicated that DHA inhibits Aβ fibril formation and destabilizes existing Aβ fibrils (Hashimoto et al., 2009; Hashimoto et al., 2008; Hossain et al., 2009) while stabilizing soluble fibrillar oligomers (Johansson et al., 2007). Therefore, one possible explanation for our results is that diet-related increases in DHA levels may favor Aβ aggregation into less neurotoxic fibrillar oligomers relative to more neurotoxic prefibrillar oligomers. Johansson and colleagues postulated that DHA micelles interact with Aβ to facilitate the nucleation of oligomers, accelerating the formation of smaller fibrillar aggregates while simultaneously inhibiting the formation of larger fibrils (Johansson et al., 2007). Alternatively, Hashimoto and colleagues suggested that DHA’s effects on Aβ oligomerization might be independent of micelle formation (since they occur below critical micelle concentration) and revolve around the disruption of hydrophobic intra- and inter-chain amino acid interactions (Hashimoto et al., 2009; Hashimoto et al., 2008; Hossain et al., 2009). However, directly extrapolating these in vitro findings to in vivo settings remains challenging given that the free DHA concentrations necessary to modulate Aβ aggregation in vitro (10 μM to 50 μM) (Hashimoto et al., 2009; Hashimoto et al., 2008; Hossain et al., 2009; Johansson et al., 2007) are far higher than those measured from human CSF (185 nM; Pilitsis et al., 2001).
DHA supplementation did not significantly modulate APP processing, nor did it affect total Aβ42 levels measured from hippocampal fractions, suggesting that the reductions in Aβ plaque density and modest behavioral improvements seen in the DHA+ group were not directly attributable to DHA-mediated inhibition of Aβ production (Green et al., 2007; Grimm et al., 2011; Lim et al., 2005). While prior work in an APP/PS1 transgenic mouse model raised the possibility that DHA effects were sex-specific and more prominent in female animals (Perez et al., 2010), we did not detect interactions between sex and diet on MWM performance, cortical DHA levels, or hippocampal Aβ indices. It has previously been hypothesized that while the effects of DHA on Aβ synthesis may be more important at younger ages (i.e. earlier stages of disease) in transgenic rodent models of AD, other mechanisms may become more prominent at older ages (i.e. more advanced stages of disease), particularly in animals that overexpress both APP and PS1 mutations (Koivisto et al., 2014). Indeed, while our experiments provide compelling evidence that DHA supplementation modulates Aβ oligomer levels, it remains likely, given the pleotropic effects of DHA on AD pathophysiology (Cole et al., 2009), that this is but one of several mechanisms by which DHA ameliorates the cognitive and synaptic deficits associated with Aβ overexpression. The absence of direct correlations between hippocampal Aβ oligomer indices and behavioral outcomes in this study suggests that the behavioral benefits of DHA supplementation may arise at least in part from other mechanisms downstream of Aβ oligomerization (Arsenault et al., 2011; Calon et al., 2005; Calon et al., 2004), and/or the impact of modulating Aβ oligomer conformations in other brain regions.
Cell culture studies have suggested that DHA may also stimulate microglial phagocytosis of Aβ species (Hjorth et al., 2013). This represents an alternative mechanism by which dietary DHA supplementation may contribute to reductions in hippocampal Aβ plaque density in the APP/PS1 rat model. In particular, Aβ fibrils (but not soluble Aβ oligomers) have been shown to increase microglia-mediated Aβ phagocytosis (Pan et al., 2011), which is reduced in APP and APP/PS1 mice relative to wild-type controls in an age- and region-dependent fashion (Krabbe et al., 2013). A potential link between DHA-related increases in microglial phagocytosis of Aβ fibrils and increased levels of soluble fibrillar Aβ oligomers in the DHA-treated animals in the current study is implied by recent work that suggests that microglia may release Aβ oligomers after Aβ phagocytosis (Joshi et al., 2014). This possibility is supported by our finding of significant correlations between total soluble Aβ oligomer levels and Aβ plaque density in the DHA+ group but not in the DHA− group. While our experiments did not directly demonstrate DHA-associated differences in CD33 expression, which is one factor that modulates microglial Aβ phagocytosis (Griciuc et al., 2013), further investigations of the effects of DHA supplementation on CD36 expression (Vallve et al., 2002), which appears to mediate microglial Aβ phagocytosis induced by PPAR-γ and prostaglandin E2 receptor subtype 2 signaling (Li et al., 2015; Yamanaka et al., 2012) appear to be warranted.
Our use of a transgenic APP/PS1 rat model also allowed us to analyze CSF Aβ42 levels, which showed significant longitudinal decreases in animals maintained on the DHA+ diet but not those maintained on the DHA− diet. While age-associated reductions in CSF Aβ42 levels have been reported in a number of transgenic mouse models of AD and attributed to increased Aβ sequestration in the brain parenchyma as plaques or other insoluble forms (Kawarabayashi et al., 2001; Liu et al., 2004; Maia et al., 2013), dietary DHA supplementation in this model decreased rather than increased hippocampal plaque density. These findings also do not appear to reflect DHA-associated reductions in Aβ production, since, as noted above, the DHA+ and DHA− diets resulted in similar hippocampal Aβ levels across compartments. Instead, longitudinal reductions in CSF Aβ42 in the DHA+ group may be a direct consequence of the relative increase in fibrillar Aβ oligomers in these animals. One possibility is that oligomeric Aβ may be cleared into the CSF less efficiently than monomeric Aβ due to its sequestration in the brain parenchyma (Hong et al., 2014). Alternatively, DHA-associated reductions in measured CSF Aβ42 levels may be caused by Aβ oligomerization-related masking of the C-terminal epitopes recognized by the antibodies used in commercially available Aβ42 immunoassays (Englund et al., 2009).
Taken together, our data suggest that dietary DHA supplementation modulates Aβ aggregates in a transgenic rat model of AD by shifting the equilibrium towards higher concentrations of less toxic fibrillar oligomers and lower concentrations of more toxic prefibrillar oligomers. This shift in conformational oligomer distributions represents yet another potential mechanism by which DHA may disrupt AD pathogenesis, but the associated increases in total Aβ oligomer levels raise the possibility that DHA supplementation alone may be insufficient for the prevention and/or treatment of AD in clinical populations. Further studies of DHA supplementation in conjunction with other interventions that also reduce fibrillar Aβ oligomers may yield more robust improvements in behavioral and neuropathological endpoints (Cole and Frautschy, 2010; Yang et al., 2005).
Supplementary Material
Research Highlights.
We studied dietary DHA supplementation in a transgenic APP/PS1 rat model of AD.
DHA reduced hippocampal Aβ plaque density and improved behavioral outcomes.
DHA increased soluble Aβ oligomer levels but did not affect total Aβ levels.
DHA increased fibrillar Aβ oligomers and decreased prefibrillar oligomers.
DHA-associated modulation of Aβ aggregation may reduce AD pathology.
Acknowledgments
This work was supported by the National Institute on Aging (K08 AG34628 [to ET; jointly sponsored by NIA, AFAR, the John A. Hartford Foundation, the Atlantic Philanthropies, the Starr Foundation and an anonymous donor] and RC1 AG035878 [to GMC]). We would like to thank Eric Y. Haden for cross-linked Aβ oligomers, Christopher C. Giza for behavioral testing space, and Ta-Lee Teng for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed M, et al. Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat Struct Mol Biol. 2010;17:561–7. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendash GW, et al. A diet high in omega-3 fatty acids does not improve or protect cognitive performance in Alzheimer’s transgenic mice. Neuroscience. 2007;149:286–302. doi: 10.1016/j.neuroscience.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Arsenault D, et al. DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PLoS One. 2011;6:e17397. doi: 10.1371/journal.pone.0017397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate C, et al. Docosahexaenoic and eicosapentaenoic acids increase neuronal death in response to HuPrP82-146 and Abeta 1–42. Neuropharmacology. 2008;54:934–43. doi: 10.1016/j.neuropharm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Bazinet RP, Laye S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci. 2014;15:771–85. doi: 10.1038/nrn3820. [DOI] [PubMed] [Google Scholar]
- Benilova I, et al. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–57. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Bilousova T, et al. Parallel age-associated changes in brain and plasma neuronal pentraxin receptor levels in a transgenic APP/PS1 rat model of Alzheimer’s disease. Neurobiol Dis. 2015;74:32–40. doi: 10.1016/j.nbd.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1954;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Broersen LM, et al. A specific multi-nutrient diet reduces Alzheimer-like pathology in young adult AbetaPPswe/PS1dE9 mice. J Alzheimers Dis. 2013;33:177–90. doi: 10.3233/JAD-2012-112039. [DOI] [PubMed] [Google Scholar]
- Calon F, et al. Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer’s disease. Eur J Neurosci. 2005;22:617–26. doi: 10.1111/j.1460-9568.2005.04253.x. [DOI] [PubMed] [Google Scholar]
- Calon F, et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron. 2004;43:633–45. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CC, et al. The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: a preliminary randomized double-blind placebo-controlled study. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1538–44. doi: 10.1016/j.pnpbp.2008.05.015. [DOI] [PubMed] [Google Scholar]
- Cole GM, Frautschy SA. DHA may prevent age-related dementia. J Nutr. 2010;140:869–74. doi: 10.3945/jn.109.113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole GM, et al. Omega-3 fatty acids and dementia. Prostaglandins Leukot Essent Fatty Acids. 2009;81:213–21. doi: 10.1016/j.plefa.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Carmo S, Cuello AC. Modeling Alzheimer’s disease in transgenic rats. Mol Neurodegener. 2013;8:37. doi: 10.1186/1750-1326-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund H, et al. Oligomerization partially explains the lowering of Abeta42 in Alzheimer’s disease cerebrospinal fluid. Neurodegener Dis. 2009;6:139–47. doi: 10.1159/000225376. [DOI] [PubMed] [Google Scholar]
- Flood DG, et al. A transgenic rat model of Alzheimer’s disease with extracellular Abeta deposition. Neurobiol Aging. 2009;30:1078–90. doi: 10.1016/j.neurobiolaging.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Florent S, et al. Docosahexaenoic acid prevents neuronal apoptosis induced by soluble amyloid-beta oligomers. J Neurochem. 2006;96:385–95. doi: 10.1111/j.1471-4159.2005.03541.x. [DOI] [PubMed] [Google Scholar]
- Freund-Levi Y, et al. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch Neurol. 2006;63:1402–8. doi: 10.1001/archneur.63.10.1402. [DOI] [PubMed] [Google Scholar]
- Green KN, et al. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci. 2007;27:4385–95. doi: 10.1523/JNEUROSCI.0055-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–43. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm MO, et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J Biol Chem. 2011;286:14028–39. doi: 10.1074/jbc.M110.182329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, et al. Docosahexaenoic acid withstands the Abeta25–35-induced neurotoxicity in SH-SY5Y cells. J Nutr Biochem. 2011;22:22–9. doi: 10.1016/j.jnutbio.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, et al. Effects of docosahexaenoic acid on in vitro amyloid beta peptide 25–35 fibrillation. Biochim Biophys Acta. 2009;1791:289–96. doi: 10.1016/j.bbalip.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, et al. Docosahexaenoic acid disrupts in vitro amyloid beta(1–40) fibrillation and concomitantly inhibits amyloid levels in cerebral cortex of Alzheimer’s disease model rats. J Neurochem. 2008;107:1634–46. doi: 10.1111/j.1471-4159.2008.05731.x. [DOI] [PubMed] [Google Scholar]
- Hjorth E, et al. Omega-3 fatty acids enhance phagocytosis of Alzheimer’s disease-related amyloid-beta42 by human microglia and decrease inflammatory markers. J Alzheimers Dis. 2013;35:697–713. doi: 10.3233/JAD-130131. [DOI] [PubMed] [Google Scholar]
- Hoffman DR, et al. Impact of early dietary intake and blood lipid composition of long-chain polyunsaturated fatty acids on later visual development. J Pediatr Gastroenterol Nutr. 2000;31:540–553. doi: 10.1097/00005176-200011000-00016. [DOI] [PubMed] [Google Scholar]
- Hong S, et al. Soluble Aβ oligomers are rapidly sequestered from brain ISF and bind GM1 ganglioside on cellular membranes. Neuron. 2014;82:309–319. doi: 10.1016/j.neuron.2014.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain S, et al. Mechanism of docosahexaenoic acid-induced inhibition of in vitro Abeta1-42 fibrillation and Abeta1-42-induced toxicity in SH-S5Y5 cells. J Neurochem. 2009;111:568–79. doi: 10.1111/j.1471-4159.2009.06336.x. [DOI] [PubMed] [Google Scholar]
- Johansson AS, et al. Docosahexaenoic acid stabilizes soluble amyloid-beta protofibrils and sustains amyloid-beta-induced neurotoxicity in vitro. FEBS J. 2007;274:990–1000. doi: 10.1111/j.1742-4658.2007.05647.x. [DOI] [PubMed] [Google Scholar]
- Joshi P, et al. Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014;21:582–93. doi: 10.1038/cdd.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, et al. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–81. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Knopman DS. beta-Amyloidosis and neurodegeneration in Alzheimer disease: who’s on first? Neurology. 2014;82:1756–7. doi: 10.1212/WNL.0000000000000438. [DOI] [PubMed] [Google Scholar]
- Koivisto H, et al. Special lipid-based diets alleviate cognitive deficits in the APPswe/PS1dE9 transgenic mouse model of Alzheimer’s disease independent of brain amyloid deposition. J Nutr Biochem. 2014;25:157–69. doi: 10.1016/j.jnutbio.2013.09.015. [DOI] [PubMed] [Google Scholar]
- Kotani S, et al. Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci Res. 2006;56:159–64. doi: 10.1016/j.neures.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Krabbe G, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8:e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson ME, Lesne SE. Soluble Abeta oligomer production and toxicity. J Neurochem. 2012;120(Suppl 1):125–39. doi: 10.1111/j.1471-4159.2011.07478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LK, et al. Docosahexaenoic acid-concentrated fish oil supplementation in subjects with mild cognitive impairment (MCI): a 12-month randomised, double-blind, placebo-controlled trial. Psychopharmacology (Berl) 2013;225:605–12. doi: 10.1007/s00213-012-2848-0. [DOI] [PubMed] [Google Scholar]
- Li X, et al. Prostaglandin E2 receptor subtype 2 regulation of scavenger receptor CD36 modulates microglial Abeta42 phagocytosis. Am J Pathol. 2015;185:230–9. doi: 10.1016/j.ajpath.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, et al. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–40. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, et al. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–7. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, et al. Longitudinal observation on CSF Abeta42 levels in young to middle-aged amyloid precursor protein/presenilin-1 doubly transgenic mice. Neurobiol Dis. 2004;17:516–23. doi: 10.1016/j.nbd.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Liu L, et al. A transgenic rat that develops Alzheimer’s disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia LF, et al. Changes in amyloid-beta and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci Transl Med. 2013;5:194re2. doi: 10.1126/scitranslmed.3006446. [DOI] [PubMed] [Google Scholar]
- Morris RG, et al. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–3. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Nussbaum JM, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature. 2012;485:651–5. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksman M, et al. Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis. 2006;23:563–72. doi: 10.1016/j.nbd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Pan XD, et al. Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer’s disease. Mol Neurodegener. 2011;6:45. doi: 10.1186/1750-1326-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez SE, et al. DHA diet reduces AD pathology in young APPswe/PS1 Delta E9 transgenic mice: possible gender effects. J Neurosci Res. 2010;88:1026–40. doi: 10.1002/jnr.22266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Nievas BG, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain. 2013;136:2510–26. doi: 10.1093/brain/awt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilitsis JG, et al. Quantification of free fatty acids in human cerebrospinal fluid. Neurochem Res. 2001;26:1265–70. doi: 10.1023/a:1014227231130. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Quinn JF, et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA. 2010;304:1903–11. doi: 10.1001/jama.2010.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S, et al. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Abeta deposition and tau pathology in 3xTg-AD mice. J Neurochem. 2013;126:473–82. doi: 10.1111/jnc.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondanelli M, et al. Effects of a diet integration with an oily emulsion of DHA-phospholipids containing melatonin and tryptophan in elderly patients suffering from mild cognitive impairment. Nutr Neurosci. 2012;15:46–54. doi: 10.1179/1476830511Y.0000000032. [DOI] [PubMed] [Google Scholar]
- Rosensweig C, et al. Preparation of stable amyloid beta-protein oligomers of defined assembly order. Methods Mol Biol. 2012;849:23–31. doi: 10.1007/978-1-61779-551-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinn N, et al. Effects of n-3 fatty acids, EPA v. DHA, on depressive symptoms, quality of life, memory and executive function in older adults with mild cognitive impairment: a 6-month randomised controlled trial. Br J Nutr. 2012;107:1682–93. doi: 10.1017/S0007114511004788. [DOI] [PubMed] [Google Scholar]
- Teng E, et al. [F-18]FDDNP microPET imaging correlates with brain Abeta burden in a transgenic rat model of Alzheimer disease: effects of aging, in vivo blockade, and anti-Abeta antibody treatment. Neurobiol Dis. 2011;43:565–75. doi: 10.1016/j.nbd.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomic JL, et al. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol Dis. 2009;35:352–8. doi: 10.1016/j.nbd.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakhapova V, et al. Phosphatidylserine containing omega-3 fatty acids may improve memory abilities in non-demented elderly with memory complaints: a double-blind placebo-controlled trial. Dement Geriatr Cogn Disord. 2010;29:467–74. doi: 10.1159/000310330. [DOI] [PubMed] [Google Scholar]
- Vallve JC, et al. Unsaturated fatty acids and their oxidation products stimulate CD36 gene expression in human macrophages. Atherosclerosis. 2002;164:45–56. doi: 10.1016/s0021-9150(02)00046-1. [DOI] [PubMed] [Google Scholar]
- Veszelka S, et al. Docosahexaenoic acid reduces amyloid-beta induced toxicity in cells of the neurovascular unit. J Alzheimers Dis. 2013;36:487–501. doi: 10.3233/JAD-120163. [DOI] [PubMed] [Google Scholar]
- Wang PY, et al. Docosahexaenoic acid supplementation of primary rat hippocampal neurons attenuates the neurotoxicity induced by aggregated amyloid beta protein(42) and up-regulates cytoskeletal protein expression. J Nutr Biochem. 2010;21:345–50. doi: 10.1016/j.jnutbio.2009.01.012. [DOI] [PubMed] [Google Scholar]
- Yamanaka M, et al. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–31. doi: 10.1523/JNEUROSCI.1569-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Yang F, et al. Monoclonal antibody to the C-terminus of beta-amyloid. Neuroreport. 1994;5:2117–20. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]
- Yurko-Mauro K, et al. Beneficial effects of docosahexaenoic acid on cognition in age-related cognitive decline. Alzheimers Dement. 2010;6:456–64. doi: 10.1016/j.jalz.2010.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.