

Abstract
Compelling genetic evidence links the amyloid precursor protein (APP) to Alzheimer’s disease (AD). A leading hypothesis proposes that a small amphipathic fragment of APP, the amyloid β-protein (Aβ), self-associates to form soluble assemblies loosely referred to as “oligomers” and that these are primary mediators of synaptic dysfunction. As such, Aβ, and specifically Aβ oligomers, are targets for disease modifying therapies. Currently, the most advanced experimental treatment for AD relies on the use of anti-Aβ antibodies. In this study, we tested the ability of the monomer-preferring antibody, m266 and a novel aggregate-preferring antibody, 1C22, to attenuate spatial reference memory impairments in J20 mice. Chronic treatment with m266 resulted in a ~70-fold increase in Aβ detected in the bloodstream, and a ~50% increase in water-soluble brain Aβ – and in both cases Aβ was bound to m266. In contrast, 1C22 increased the levels of free Aβ in the bloodstream, and bound to amyloid deposits in J20 brain. However, neither 1C22 nor m266 attenuated the cognitive deficits evident in 12 month old J20 mice. Moreover, both antibodies failed to alter the levels of soluble Aβ oligomers in J20 brain. These results suggest that Aβ oligomers may mediate the behavioral deficits seen in J20 mice and highlight the need for the development of aggregate-preferring antibodies that can reach the brain in sufficient levels to neutralize bioactive Aβ oligomers.
Aside from the lack of positive effect of m266 and 1C22 on cognition a substantial number of deaths occurred in m266- and 1C22-immunized J20 mice. These fatalities were specific to anti-Aβ antibodies and to the J20 mouse line since treatment of wild type or PDAPP mice with these antibodies did not cause any deaths. These and other recent results indicate that J20 mice are particularly susceptible to targeting of the APP/Aβ/tau axis. Notwithstanding the specificity of fatalities for J20 mice, it is worrying that the murine precursor (m266) of a lead experimental therapeutic, Solanezumab, did not engage with putatively pathogenic Aβ oligomers.
Keywords: Alzheimer’s disease, amyloid β-protein, immunotherapy, oligomers, APP transgenic mice
INTRODUCTION
Alzheimer’s disease (AD) is characterized by amyloid deposition, neurofibrillary tangles, synaptic loss, neuronal loss, reactive gliosis and memory impairment. Several transgenic human amyloid precursor protein (hAPP) mouse models reproduce certain features of AD and have been used to assess the efficacy of therapeutic interventions, including use of anti-Aβ antibodies. Early studies demonstrated that the anti-Aβ monoclonal antibody (mAb), 3D6 (raised to Aβ1–5), reduced cortical Aβ burden by ~86% in PDAPP mice (Bard et al., 2000), whereas when PDAPP mice were treated with the mid-region specific mAb, m266, it had little effect on Aβ deposition but dramatically increased circulating levels of antibody-bound Aβ (DeMattos et al., 2001). Subsequent studies using other anti-Aβ mAbs and mouse models also demonstrated significant reductions in amyloid burden (Levites et al., 2006; Schroeter et al., 2008). In addition, several studies found that passive administration of certain anti-Aβ antibodies protected or restored cognition in hAPP mice (Basi et al., 2010; Dodart et al., 2002; Karlnoski et al., 2008; Kotilinek et al., 2002; Oddo et al., 2006; Wilcock et al., 2006; Wilcock et al., 2004a; Wilcock et al., 2004b; Zago et al., 2012). Specifically, m266, was shown to improve object recognition memory in 11 mo PDAPP mice 24 hr after a single antibody administration (Dodart et al., 2002). The mechanism of this striking short-term improvement is uncertain, but has been suggested to result due to either: (1) direct neutralization of soluble toxic forms of cerebral Aβ, or (2) m266 sequestering Aβ in the bloodstream and causing an efflux of soluble toxic Aβ species into the blood.
Although mAbs can engage Aβ when administered to man, the success of mAb immunization in preclinical AD models has not translated well to humans (Blennow et al., 2012; Farlow et al., 2012; Salloway et al., 2014; Salloway, 2012). Solanezumab, the humanized version of m266, is the only antibody with published results reporting benefit in humans. Even with Solanezumab the cognitive benefit was marginal, with extracted analysis of a phase III trial data revealing only a modest attenuation of cognitive decline in mild AD patients and no effect in individuals with moderate AD (Doody et al., 2014).
Despite millions of dollars being invested in clinical trials, there is limited published data on the preclinical testing of m266 on cognition, and to-date all such studies have used a single mouse model–PDAPP mice. Moreover, no prior published study directly tested the effects of m266 versus another anti-Aβ antibody, nor assessed the effect of treatment on cerebral Aβ oligomers. Here we tested m266 alongside a recently described aggregate-preferring antibody, 1C22 (O’Nuallain et al., 2014; Yang et al., 2015) employing the well-characterized J20 mouse model (Cheng et al., 2007; Chin et al., 2005; Karl et al., 2012; Mably et al., 2015; Palop et al., 2003; Roberson et al., 2011; Roberson et al., 2007; Wright et al., 2013). The J20 model was chosen because it has important similarities to the PDAPP mouse model which is the only model in which m266 has ever been tested. Specifically, both the PDAPP and J20 mice express similar APP copy numbers, the transgene has a similar minigene structure and both are under the control of the PDGF promoter (Hsia et al., 1999). In prior studies we tested the performance of J20 mice and littermate Wt controls from the same colony as used here at 3 different ages (4, 8, and 12 months) in 5 different tasks: (1) open field, (2) spontaneous alternation Y-maze, (3) radial arm maze (RAM), (4) novel object recognition, and (5) contextual fear conditioning (Mably et al., 2015). In agreement with several other reports (Karl et al., 2012; Kim et al., 2013b; Wright et al., 2013) we found that J20 mice exhibited hyperactivity which waned with increasing age and decreased performance on the RAM which became more prominent with increasing age (Mably et al., 2015). However, J20 and Wt mice performed at comparable levels on Y-maze, contextual fear conditioning and novel objection recognition. In an attempt to mimic current human trials in individuals with mild AD, mice began immunotherapy at an age (9.5 months) when they had some amyloid deposition and mild impairment of spatial reference memory. We found m266 bound to certain water-soluble forms of Aβ in the brain and dramatically elevated circulating levels of Aβ in blood, most of which was bound to antibody. In contrast, 1C22 bound to plaque Aβ and although it promoted an increase in the levels of circulating Aβ none of this was bound to 1C22. Despite clear evidence of engagement of m266 with Aβ monomer, and 1C22 with plaques, neither antibody affected cerebral Aβ oligomer levels or attenuated deficits in spatial reference memory. These results suggest that removal of Aβ oligomers may be necessary to overcome the spatial reference memory deficits evident in J20 mice. Furthermore, these findings indicate that in the complex milieu of brain removal of oligomers can only be achieved by antibodies with minimal reactivity to monomers or plaques.
Our studies also revealed an important negative effect of m266 and 1C22, that is, ~20% of immunized J20 mice died. These studies follow on from our recent report that a mid-region anti-tau mAb also caused death in J20 mice (Mably et al., 2015), and suggest that J20 mice are particularly susceptible to targeting of the APP/Aβ/tau axis. To the best of our knowledge this is the first report that anti-Aβ antibodies can have deleterious effects on animal viability, and that monomer-preferring and aggregating-preferring mAbs are unable to reduce the levels of water-soluble Aβ oligomers in brain. How generalizable these results are to humans, or indeed other mouse models, is as yet unclear. Nonetheless, the fact that a lead experimental therapeutic did not reduce the levels of a putatively pathogenic form of Aβ might explain its very modest efficacy in the clinic.
MATERIALS AND METHODS
Mice and antibody administration
Mice were housed under a 12 hr light:dark cycle (lights on 7 am, lights off 7 pm). Ad libitum food (standard chow; LabDiet, Richmond, IN) was provided unless otherwise indicated. Male hemizygous hAPPSwe/Ind mice (J20) were obtained from Jackson Laboratories (Bar Harbor, ME) and crossed with C57BL/6J female mice to produce hemizygous J20 mice or wild type (Wt) littermate controls. Only male hemizygous J20 mice and wild type (Wt) littermate controls were used for the study, and were the F1 progeny of 7 male J20 mice. J20 mice over-express hAPP carrying the Swedish (KM670/671NL) and Indiana (V717F) mutations (Mucke et al., 2000). Pups were weaned at 20 – 21 days old, male progeny were tail snipped and genotyped. Mice were group housed (2 – 4 animals per cage) until 5 days prior to behavioral testing, and after this time mice were housed individually. Female PDAPP mice and Wt littermate controls were a kind gift from Janssen Alzheimer Immunotherapy (South San Francisco, CA). PDAPP mice arrived at 9 mo and were housed (2 – 4 animals per cage) according to genotype. PDAPP mice over-express hAPP carrying the Indiana mutation (Games et al., 1995), PDAPP mice and littermates were on a hybrid background representing a combination of 3 strains: (1) Swiss-Webster, (2) C57BL/6J, and (3) DBA/2J. Five days prior to behavior testing, mice were individually housed. The 46-4-treated J20 and Wt mice reported here have previously been reported as a control for a separate immunization study that was conducted in parallel with the current study (Mably et al., 2015).
Exactly the same immunization paradigm was employed for all transgenic and Wt mice. Beginning at 9.5 mo animals received 11 weekly intraperitoneal (i.p.) infusions of endotoxin-free antibody (250 μl of 1 mg/ml antibody in sterile PBS) (Figure 1). To ensure levels of circulating antibody were kept at a maximum, throughout behavioral testing mice received an additional 3 250 μg antibody injections (Figure 1). Antibody infusions took place in the afternoon (2 – 5 pm). On days where behavioral testing had taken place, antibody infusions were carried out at least 2 hr after training. Injections were administered by a person other than the investigator carrying out the behavioral testing. The investigator carrying out the behavioral testing was blind to the treatment groups. All animal procedures were approved by the Harvard Medical School Institutional Animal Care and Use Committee (Protocol number 04869).
Figure 1. Immunization paradigm and behavioral testing.

Mice began receiving antibody injections (250 μg, i.p) at 9.5 months of age. Animals received one weekly antibody administration for 10 wk, prior to behavioral assessment commencing at 12 mo. Following the 11th antibody infusion testing in the open field arena took place. Five days later, mice received their 12th i.p infusion of antibody; the following day habituation to the RAM began. The 13th and 14th i.p infusions of antibody took place on the final day of RAM habituation and on the 2nd day of RAM training, respectively. All antibody injections were carried out in the afternoon (2 – 5 pm). Injections on days during behavioral testing were done at least 2 hr after testing. Blood, CSF and brain samples were collected immediately following completion of behavioral testing.
Antibodies used for passive immunization studies
Three IgG1 antibodies were used in this study: (i) monoclonal antibody (mAb) 1C22 which was raised in-house and preferentially recognizes aggregated forms of Aβ (Supplementary Figure 1) (O’Nuallain et al., 2014; Yang et al., 2015), (ii) mAb m266 which recognizes soluble, predominantly monomeric Aβ (Supplementary Figure 1) (Yamada et al., 2009), and (iii) mAb 46-4 which was raised to HIV coat protein 1 (Reeves et al., 1995). m266 was a generous gift from Dr. F. Bard Janssen Alzheimer Immunotherapy. All antibodies were purified using Protein G (PrG) and their purity determined using SDS-PAGE and Coomassie staining. Antibodies were ≥95% pure and contained ≤1 U/mg of endotoxin (LAL Chromogenic Endotoxin quantitation kit; Thermo Scientific).
Preparation of soluble Aβ assemblies and solid-phase ELISA used to determine mAb binding
AβS26C DAEFRHDSGY-EVHHQKLVFF-AEDVGCNKGA-IIGLMVGGVV in which serine 26 was substituted with cysteine were synthesized and purified by Dr. James I Elliott at Yale University (New Haven, CT). Mass spectrometric (MS) analysis and reverse-phase HPLC confirmed that the peptide had the correct mass and was >90% pure. Cross-linking of Aβ1-40S26C was achieved by atmospheric oxidation of ~20 μM peptide in 10 mM ammonium bicarbonate, pH 8.2, and (Aβ1-40S26C)2 dimers were isolated from unreacted monomer and higher molecular weight aggregates by SEC using a Superdex™ 75 10/300 HR. Meta-stable (Aβ1-40S26C)2 protofibrils were generated by incubating (Aβ1-40S26C)2 in 20 mM sodium phosphate, pH 7.4, at 37°C for 3 days and characterized as described previously (Colgin and Moser, 2010; O’Nuallain et al., 2010). Binding of 1C22 and m266 to PFs was determined using a solid phase ELISA. Briefly, mAbs were serially diluted in triplicate with assay buffer (1% BSA in phosphate buffer saline (PBS) containing 0.05% Tween 20, pH 7.4 (PBS-Tw)) into microtiter plate wells (#3369, COSTAR, Corning, NY) that were coated with 200 ng of (Aβ1-40S26C)2 PFS and blocked with 1% BSA (Sigma-Aldrich, Saint Louis, MO) in PBSA. Biotinylated goat anti-mouse IgG (γ-chain specific, Sigma-Aldrich, Saint Louis, MO) served as the secondary antibody, and the detection system consisted of streptavidin-horse radish peroxidase (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) and 3,3′,5,5′ –tetramethylbenzidine (TMB) substrate (SureBlue Reserve™; KPL, Gaithersburg, MD, USA). Antibody binding curves were fitted using a standard 3-parameter sigmoid (logistic) function (SigmaPlot 2000, version 6; Systat Software, Chicago, IL) with the lower limit of detection set as the signal 3 times higher than the buffer blank.
Behavioral tests
Mice were single housed and handled daily for 5 days prior to beginning behavioral testing. Testing always took place during the light photoperiod and mice were acclimatized to testing rooms for at least 20 min before testing began.
Open Field
Mice were tested in an open field chamber (27.3 cm × 27.3 cm × 20.3 cm; Med Associates, St. Albans, VT) for a total of 60 min, as described previously (Dillon et al., 2008; Kornecook et al., 2010; Mably et al., 2015). Infrared beams across the chamber track the animal’s placement and movement within the chamber. Data were analyzed using Activity Monitor software (Med Associates, St. Albans, VT) to calculate the distance travelled. Zone analysis gathered using the same system allowed calculation of percentage time spent in the center of the open field chamber.
Radial Arm Maze
Testing in the radial arm maze (RAM) was carried out exactly as previously described (Mably et al., 2015). Briefly, mice were placed on a food restricted diet (consisting of rationed amounts of standard chow (LabDiet, Richmond, IN)) 5 days prior to testing in the RAM to lower their body weight to ~85% of their free-feeding weight and maintained at this weight level during habituation and testing in the RAM. Mice were habituated to the apparatus for 2 days, followed by 8 consecutive days of spatial reference memory testing, 6 trials a day with a ~1 min inter-trial interval. Once an arm had been traversed it was blocked by a motorized door, which remained in place for the remainder of that trial, eliminating the working memory component of the task (Hodges, 1996; Wenk, 2004). Three arms of the RAM were baited with 20 mg casein pellets (BioServ, Frenchtown, NJ), each trial lasted until the mouse collected all 3 food pellets or for a maximum of 5 min. The maze was cleaned with 70% ethanol between each trial and between mice. Any mice that consistently failed to explore the maze and eat the food pellets during habituation were removed from the study. Two measures of behavior were recorded over the training days: (1) the percentage of 45° turns made; and (2) the number of incorrect arm entries made (SRM errors). The radial arm maze requires that animals both learn and recall the task, such that the first trials require that animals learn the task, and after learning the task the animals can recall that experience and thus exhibit improved performance on subsequent trials. In this regard, the most important read out is the number of reference errors (Wenk, 2004), whereas 45° turns are used to assess the navigation strategy that the mice are using to search the maze, and subsequently whether they are using knowledge of the spatial environment (Hodges, 1996; Wenk, 2004). We and others have demonstrated that J20 mice exhibit an age-dependent decrease in RAM performance that predominantly effects the number of reference errors. The time required for testing necessitated the use of 8 interleaved genotype- and treatment-balanced groups. As a precaution we compared the performance of Wt mice in each of the groups to each other (data not shown) and to a cohort of previously tested unimmunized 12 mo Wt mice (Supplementary Figure 2). The purpose of this exercise was two-fold: (i) to confirm that Wt mice across all groups adequately acquired the RAM task, and (ii) to ensure that chronic infusion of mAb did not perturb or delay the acquisition of spatial reference memory in Wt mice. It was clear from the high level of reproducibility that all animals acquired the task and that the infusion of 1C22 and 46-4 did not alter task performance.
Euthanasia and tissue collection
Immediately following completion of behavioral testing mice were anesthetized with ketamine/xylazine/acepromazine (100/10/2 mg/kg). Terminal blood was collected by cardiac puncture into ethylenediaminetetraacetic acid (EDTA) charged polypropylene tubes, centrifuged at 3500 g for 15 min and the plasma fractionated, transferred to clean tubes and stored at −80°C until required for analysis. Mice were intracardially perfused with ~20 ml ice-cold PBS using a 30 ml syringe. The brain was removed and cut down the mid-line. The left hemisphere was immediately frozen on dry ice for biochemical analysis, and the right hemisphere was drop-fixed in 10% formalin for 2 hr. Tail snips were collected and genotype re-tested.
Tissue preparation
The olfactory bulbs and cerebellum were dissected from frozen hemi-brains and the remaining tissue weighed on a top pan balance. Thereafter the tissue was cut into small pieces using a razor blade and homogenized in 5 volumes (w/v) Tris buffered saline (TBS), pH 7.4, plus protease inhibitors (5 mM EDTA, 1 mM ethylene glycol tetraacetic acid (EGTA), 10 mg/ml leupeptin, 1 mg/ml aprotinin, 1 mg/ml pepstatin A, 1 mM pefabloc and 2 mM 1,10 phenanthroline) using a dounce homogenizer fitted to an overhead stirrer (Wheaton, Millville, NJ). Samples were centrifuged at 90,000 g and 4°C for 1 hr. The entire supernatant (TBS extract) was removed to clean polypropylene tubes and aliquots stored at −80°C pending analysis. The resulting pellet was re-homogenized in the presence of 5 volumes (w/v) TBS containing 1% Triton X-100 and protease inhibitors, centrifuged at 90,000 g and 4°C for 1 hr, and the supernatant (TBS-TX extract) removed to clean tubes and stored at −80°C. The final pellet was resuspended in 0.5 volume (w/v) 88% formic acid and agitated overnight. Samples were centrifuged at 14,000 g for 15 min and the entire supernatant (formic acid extract) removed to clean tubes and stored at −80 °C pending analysis. Immediately prior to analysis formic acid samples were neutralized by diluting 1:27 (v/v) with unbuffered 1M Tris.
Separation of antibody-bound Aβ from free Aβ
Aβ bound to antibody was recovered using Protein G-agarose beads (PrG; Roche Diagnostics, Indianapolis, IN). Plasma (150 μl) and TBS or TBS-TX brain extracts (200 μl), but not formic acid extracts were incubated with PrG (20 μl) on a nutator at 4°C for 14 hr. The beads were recovered by centrifugation at 5000 g for 10 min and the supernatant which contained free Aβ was transferred to clean tubes pending analysis. Beads were washed 2 × 15 min with 500 μl TBS to remove adventitiously-associated Aβ. Antibody-Aβ complexes were released from the PrG by boiling the beads in TBS (150 μl for plasma samples and 200 μl for brain extracts) for 15 min. Immediately thereafter beads were pelleted by centrifugation (5000 g for 10 min) and the supernatant containing Aβ and denatured antibody quickly transferred to a clean tube and stored at 4°C pending analysis.
Enzyme-link immunosorbant assays (ELISA)
MULTI-ARRAY® 96 well small-spot black microplates (Meso Scale Discovery, Rockville, MD) were coated with mAb m266 (Table 1, 3 μg/ml) in PBS and incubated at room temperature for 18 hr. Antibody m266 recognizes an epitope within Aβ13–26, thus enabling detection of both N- and C-terminally heterogeneous Aβ species. Wells were washed with PBS-Tw for 15 min × 4 and blocked with 150 μl of 5% Blocker A (Meso Scale Discovery, Rockville, MD) in PBS-Tw for 1 hr at room temperature with shaking. Plates were then washed 3 times with PBS-Tw. Samples were diluted to give a final concentration of 1X ELISA diluent (1% Blocker A/PBS-Tw) and added to the appropriate wells. Twenty-five micro liters of samples or synthetic Aβ1–40 or Aβ1–42 standards (Meso Scale Discovery, Rockville, MD) were added and incubated for 2 hr at room temperature with shaking. All synthetic peptides were aliquoted and stored at −80 °C prior to use, and diluted in plasma/ELISA diluent or TBS/ELISA diluent, so as to correspond with the buffer composition of the samples tested. After capture, wells were washed 3 times with PBS-Tw and incubated with biotinylated anti-Aβ antibody. To allow the detection of Aβ species beginning at Asp1 and terminating at Ala42 mAb 3D6 and 21F12 (Table 1, 1 μg/ml) were used, respectively. Simultaneously, 1 μg/ml of the reporter reagent (SULFO-TAG Labeled Streptavadin) (Meso Scale Discovery, Rockville, MD) was added in ELISA diluent and incubated for 2 hr at room temperature with gentle agitation. Finally, wells were washed 3 times with PBS-Tw and 150 μl of 2X MSD read buffer was added to allow for electrochemiluminecence detection (Meso Scale Discovery, Rockville, MD). A SECTOR imager (Meso Scale Discovery, Rockville, MD) was used to measure the intensity of emitted light, thus allowing quantitative measurement of analytes present in the samples. To control for detection of endogenous murine Aβ and reactivity with non-Aβ material we were careful to include 2 important controls – (1) at least one or more extracts from an age-matched Wt mouse brain, and (2) extracts from 12 mo J20 mouse brain immunodepleted of Aβ. Extracts were immunodepleted of Aβ using the polyclonal anti-Aβ antibody, AW7, as previously described (Shankar et al., 2011). All samples were diluted so the concentration of Aβ present fell within the linear range of the standard curve and the dilutions were optimized so that values fell within the LLoQ and upper limit of quantitaion (i.e. the value of the highest standard with a CV ≤ 20% and a recovery of 100 ± 20%) of the respective assay. The fact that several dilutions of the same sample yielded closely similar Aβ concentrations indicates that these ELISAs were not influenced by matrix effects under the conditions used. For a given assay all samples were analyzed on the same day. The lower limit of reliable quantitation (LLoQ) of the x-42 and 1-x assays was 17 pg/ml and 15 pg/ml, respectively.
Table 1.
Primary antibodies used for Western blot, ELISA, IHC and immunization
| Antibody | Immunogen/epitope | Host | Application (dilution) | IHC pretreatment | IHC secondary antibody | Source/Reference |
|---|---|---|---|---|---|---|
| 22C11 | APP residues 66–81 | Mouse monoclonal | Western blot (2 μg/ml) | N/A | N/A | Millipore (Hilbich et al., 1993; Weidemann et al., 1989) |
| 8E5 | APP residues 444–592 | Mouse monoclonal | IHC (0.1 μg/ml) | Microwaveb | Biotinylated anti-mouse IgG | Elan Pharmaecuticals (Games et al., 1995) |
| 3D6 | Aβ 1–5, requires free Asp1 of Aβ | Mouse monoclonal | ELISA (1 μg/ml) | N/A | N/A | Elan Pharmaecuticals (Johnson-Wood et al., 1997) |
| 6E10 | Amino acids 3–8 within Aβ sequence | Mouse monoclonal | Western blot (1 μg/ml) | N/A | N/A | Covance |
| m266 | Aβ 16–23 | Mouse monoclonal | ELISA (3 μg/ml) Immunization (1 mg/ml, 250 μl injection) | N/A | N/A | Elan Pharmaecuticals (Seubert et al., 1992) |
| 2G3 | Aβ33–40 | Mouse monoclonal | IHC (1 μg/ml) | 88% Formic Acida | Biotinylated anti-mse IgG | Elan Pharmaecuticals (Johnson-Wood et al., 1997) |
| 21F12 | Aβ 33–42 | Mouse monoclonal | IHC (1 μg/ml) ELISA (1 μg/ml) | 88% Formic Acida | Biotinylated anti-mse IgG | Elan Pharmaecuticals (Johnson-Wood et al., 1997) |
| AW7 | Aβ 1–40 | Rabbit polyclonal PrA (purified) | IHC (1 μg/ml) | 88% Formic Acida | Biotinylated goat, anti-rabbit | Walsh lab (McDonald et al., 2012) |
| 1C22 | Aβ [S26C]2 protofibrils | Mouse monoclonal | ELISA (3 μg/ml) Immunization (1 mg/ml, 250 μl injection) | N/A | N/A | Walsh lab (Yang et al., 2015) |
| NAB61 | Aß1–40 treated with peroxynitrate | Mouse monoclonal | ELISA (3 μg/ml) | N/A | N/A | Lee lab (Lee et al., 2006) |
| C1/6.1 | APP residues 676-695 | Rabbit polyclonal | Western blot (3.4 μg/ml) | N/A | N/A | Mathews lab (Mathews et al., 2002) |
| AT8 | Phospho-tau Ser202/Thr205 | Mouse monoclonal | IHC (5 μg/ml) | Microwaveb | Biotinylated goat, anti-mouse IgG1 | Pierce (Mercken et al., 1992) |
| CD45 | Mouse B-cells | Rat monoclonal | IHC (0.2 μg/ml) | 88% Formic Acida | Biotinylated goat, anti-rat, mouse adsorbed | AbD Serotec |
| 46-4 | HIV glycoprotein 120 | Mouse monoclonal (IgG1) | Immunization (1 mg/ml, 250 μl injection) | N/A | N/A | ATCC (Reeves et al., 1995) |
N/A, not applicable; IHC, immunohistochemistry; ELISA, enzyme-linked immunosorbant assay; PrA, protein A sepharose.
IHC pretreatments: incubated in 88% Formic acid (8 min room temperature)
Boiled in Sodium citrate buffer (pH 6.0; BioGenex, San Ramon, CA)
Oligomer-specific ELISA (oELISA)
The presence and amount of oligomers in the aqueous phase of mouse brain was measured using 2 different MSD-based oELISAs. The first assay employed the aggregate-preferring anti-Aβ mAb, NAB61, for capture and 3D6 for detection (Yang et al., 2013) and has an LLoQ of ~ 156 pg/ml. In the second assay the aggregate-preferring mAb, 1C22, was used for capture and 3D6 for detection. The 1C22-3D6 assay is >37,000-fold more sensitive for Aβ oligomers than Aβ monomer and has an LLoQ of ~78 pg/ml (Mc Donald et al., 2015). Both assays reliably detects Aβ oligomers of a variety of different sizes (Yang et al., 2013; Yang et al., 2015) and were performed essentially as described for the x-42 and 1-x assays, but ADDLs were used as the calibrant.
Immunohistochemistry and staining
All immunohistochemistry (IHC) and staining was carried out as previously described (Mably et al., 2015). Briefly, hemibrains were fixed for 2 hr with formalin and stored overnight in PBS prior to paraffin embedding, 10 μm sagittal sections were cut on a microtome (Leica Microsystems Inc., IL), and mounted onto microscope slides. Sections were de-paraffinized using Histo-clear (2 × 3 min; National Diagnostics, Somerville, NJ) and rehydrated. Incubation in 0.3% hydrogen peroxide in methanol (10 min, room temperature) served to quench endogenous peroxidise activity, and where necessary sections underwent the appropriate pre-treatment prior to IHC (Table 1).
Immunhistochemistry (IHC)
To minimize background staining the Vector Mouse-On-Mouse kit (Vector Laboratories Inc., Burlingame, CA) was used for mouse mAbs (8E5, 21F12 and 2G3), in accordance with manufacturer’s guidelines. For anti-tau mAb, AT8, sections were blocked with 5% non-fat milk in TBS for 1 hr at room temperature and incubated overnight in primary antibody (4°C). For polyclonal antibodies (AW7 and CD45) sections were blocked with 10% goat serum in TBS for 20 min at room temperature and incubated overnight at 4°C with primary antibodies. Sections (AT8, AW7 and CD45 staining) were washed with TBS and incubated with biotinylated secondary antibodies in the appropriate blocking solution (Table 1). Thereafter, all sections were incubated with Sterpavidin horseradish-peroxidase (Vector Elite ABC kit, Vector Laboratories Inc., Burlingame, CA) for 30 min at room temperature. Staining was visualized with diaminobenzidine (DAB; Sigma Chemical Co., St. Louis, MO) and development stopped by incubation in water. Sections were dehydrated, cleared by incubation in Histo-clear and coverslipped under Permount (Fischer Scientific).
Thioflavin S (ThS)
Tissue sections were stained by incubation in a 1% (w/v) ThS (Sigma) solution for 8 min at room temperature, followed by 3 min incubations in 80% ethanol (x2) and 95% ethanol (x1). Sections were rinsed in distilled water before coverslipping under Hydromount (National Diagnostics).
Hemosiderin
Microhemorrhages were identified by staining with Prussian blue to detect hemosiderin. Sections were de-paraffinized and rehydrated as above, followed by incubation in filtered 2% potassium hexacyano-ferrate (II) trihydrate in 2% hydrochloric acid solution for 20 min at room temperature. Sections were counterstained with hemotoxylin (Fischer Scientific), dehydrated through graded ethanol/water solutions (50%, 70%, 95% and 100%), cleared by incubation in Histo-clear and coverslipped under Permount (Fischer Scientific). Mouse spleen sections, which contain abundant hemosiderin deposits, were stained alongside as a positive control for hemosiderin staining.
Image analysis
Analysis was carried out on 6 sections taken from 3 equidistant planes (250 μm apart) by an investigator blind to the treatment condition. Staining was visualized and scored using an Olympus BX50 microscope (Olympus, Tokyo, Japan), and images collected using a QiCam digital camera (QImaging, Surrey, BC, Canada). Images from the entire hippocampus and the anterior region of the frontal cortex were analyzed using Image J software (NIH) as previously detailed (Mably et al., 2015). Briefly, following conversion of images to an 8-bit format and manual thresholding a region of interest was manually assigned. This region contained the entire hippocampus bordered dorsally by the corpus callosum. Within the region of interested the percent of positive pixels was analyzed using the calculation number of positive pixels/number of total pixels.
Statistical Analysis
Biostatisticians from the Center for Clinical Investigation, a joint Harvard Catalyst and Brigham and Women’s Hospital entity, were consulted on all statistical matters. Statistical analyses were carried out using GraphPad Prism 5 for Windows (GraphPad Software, Inc, La Jolla, CA). Differences between pairs of means were assessed by t-test. Differences between multiple means were assessed by 1-way ANOVA or 2-way ANOVA, as indicated. Values are presented as mean ± SEM. Significant differences between groups are indicated by ‘*’, (* p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001). P values are shown to 2 significant figures.
RESULTS
Chronic anti-Aβ immunization did not ameliorate behavioral disturbances or spatial reference memory deficits in J20 mice
The antibody immunization paradigm used is shown in Figure 1 and has been described previously (Mably et al., 2015). Beginning at 9.5 mo J20 mice received 11 weekly i.p. administrations (250 μg) of either 1C22, m266 or an isotype control IgG (46-4). To control for non-specific effects on learning and memory Wt littermate mice also received injections of 1C22 and 46-4. Approximately 20 hr following the 11th antibody injection (Figure 1) mice were placed in an open field arena for 60 min and the distance travelled along with the time spent in the center of the arena was recorded. J20 mice (irrespective of the antibody they received) were significantly more active than Wt mice (Figure 2A; t-test Wt + 46-4 vs. J20 + 46-4, p = 0.0031; t-test Wt + 1C22 vs. J20 + 1C22, p = 0.017). J20 mice treated with 1C22, m266, or 46-4 exhibited similar activity levels (Figure 2A) and spent a higher percentage of time in the center of the open field arena, compared to Wt controls (Figure 2B; t-test Wt + 46-4 vs. J20 + 46-4, p = 0.0006; t-test Wt + 1C22 vs. J20 + 1C22, p = 0.021). Thus anti-Aβ immunotherapy did not attenuate indices of increased activity and reduced anxiety in J20 mice.
Figure 2. Chronic anti-Aβ immunization does not improve behavioral deficits in J20 mice.
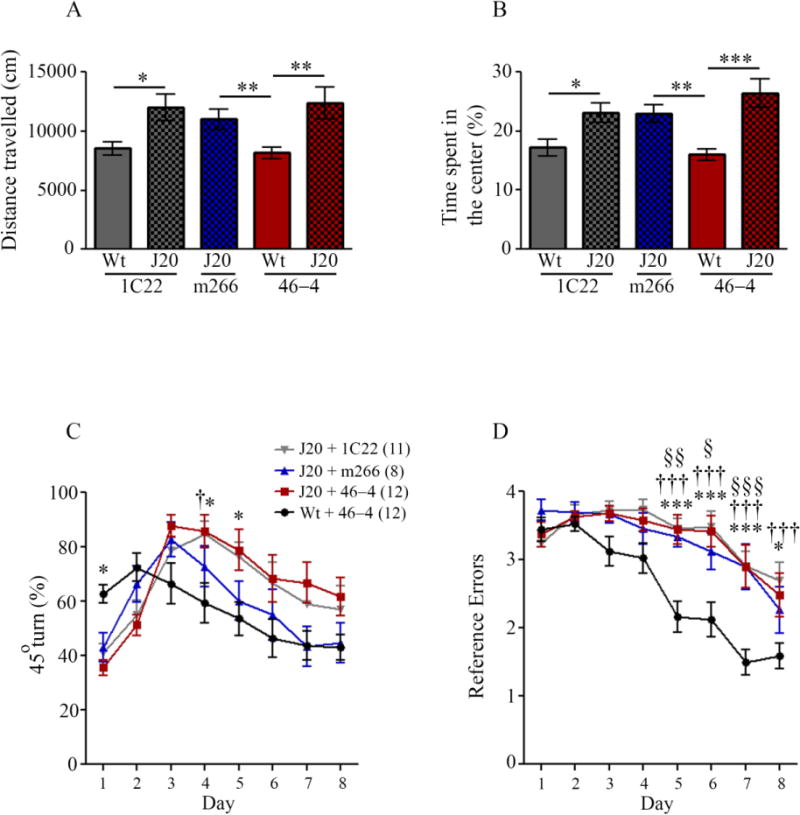
Following 10 weeks of antibody administration, 12 mo mice were tested in the open field arena and (A) distance travelled and (B) time spent in the center of the arena were measured. (A) J20 mice were consistently more active in the open field than Wt mice receiving 1C22 or 46-4 (t-test, Wt + 46-4 vs. J20 + 46-4, p = 0.0031; Wt + 1C22 vs. J20 + 1C22, p = 0.017; Wt + 46-4 vs. J20 + m266, p = 0.0098). (B) J20 mice spent more time in the center of the open field arena than treatment-matched Wt mice (t-test; Wt + 46-4 vs. J20 + 46-4, p = 0.0006; Wt + 1C22 vs. J20 + 1C22, p = 0.021; Wt + 46-4 vs. J20 + m266, p = 0.0049). Antibody treatment had no effect on J20 behavior as measured by activity and time spent in the center of the arena. Results are presented as mean values ± SEM for Wt and J20 mice treated with 1C22 (Wt n = 12, J20 n = 11), m266 (J20 n = 9), and 46-4 (Wt n = 11, J20 n = 11). Performance in the RAM was assessed by measuring (C) the percentage of 45° turns and (D) the number of incorrect arm entries. (C) Wt mice receiving 46-4 showed a different pattern of 45° turns made over the 8 days of training, compared to all J20 treatment groups (2-way ANOVA, Wt + 46-4 vs. J20 + 46-4, interaction p < 0.0001; Wt + 46-4 vs. J20 + 1C22, interaction p < 0.0001; Wt + 46-4 vs. J20 + m266, interaction p = 0.019). When comparing the J20 treatment groups, there was a significant interaction between day and treatment for m266 treated J20 mice compared to 46-4 treated J20 mice (2-way ANOVA, J20 + m266 vs. J20 + 46-4, interaction p = 0.003). There were no differences between J20 + 1C22 vs. J20 + 46-4 treatment groups. (D) J20 mice show impaired SRM compared to W mice (2-way ANOVA, Wt + 46-4 vs. J20 + 46-4, genotype p < 0.0001, interaction p < 0.0001; Wt + 46-4 vs. J20 + 1C22, genotype p < 0.0001, interaction p < 0.0001; Wt + 46-4 vs. J20 + m266, genotype p < 0.0001, interaction p = 0.025). 1C22- and m266-treated J20 mice were not significantly different to 46-4 treated J20 mice. (C) and (D) *, ** and *** denotes Wt + 46-4 vs. J20 + 46-4 statistics; †, †† and ††† denotes Wt + 46-4 vs. J20 + 1C22 statistics; §, §§ and §§§ denotes Wt + 46-4 vs. J20 + m266 statistics.
J20 mice that received 46-4 made more reference errors and 45° turns than Wt controls receiving 46-4 (Figure 2C, D; 45° turns, 2-way ANOVA, Wt + 46-4 vs. J20 + 46-4, interaction effect p < 0.0001; reference errors, 2-way ANOVA, Wt + 46-4 vs. J20 + 46-4, genotype effect p = 0.0004). The performance of Wt mice in the radial arm maze (RAM) task was similar irrespective of whether they received antibody, or the identity of the antibody they received. Since data for all groups completely overlapped only 46-4-treated Wt mice are shown (Supplementary Figure 2). With regard to 45° turns, there existed a significant interaction between treatment and day for m266-treated vs. 46-4-treated J20 mice (Figure 2C; 2-way ANOVA, J20 + m266 vs. J20 + 46-4, interaction effect p = 0.003). Although both m266- and 46-4-treated J20 mice show a similar percentage of 45° turns over days 1 – 3, over days 4 – 8 the percentage of 45° turns made by m266-treated J20 mice was lower than for 46-4-treated J20 mice, signifying adaptation to an allocentric navigational strategy more similar to that seen in Wt mice. Percentage 45° turns made by 1C22-treated J20 mice follow an almost identical pattern to those made by 46-4-treated J20 mice (Figure 2C), demonstrating that these mice used a predominately egocentric navigational strategy. J20 mice receiving m266, and the aggregate-preferring mAb, 1C22, made similar numbers of reference memory errors as J20 mice treated with control mAb 46-4 (Figure 2D). Thus, neither anti-Aβ antibody was capable of reversing the spatial memory impairment seen in J20 mice. These findings are in contrast to prior results obtained when PDAPP mice were immunized with m266. Specifically, Dodart et al. reported a recovery of novel object recognition memory in 11 month PDAPPs receiving a single mAb injection (360 μg of m266 3 hours before the familiarization session) and in 24 month male PDAPP mice following chronic treatment (360 μg once per week for 6 weeks) (Dodart et al., 2002) An improvement in spatial memory was seen in 11 month female PDAPP mice following a single antibody administration (360 μg of m266 24 hours before the first day of testing) (Dodart et al., 2002). Separately, Bales et al. found that a single injection of m266 reduced hyperactivity in 4 – 6 mo male PDAPPs (Bales et al., 2006).
Chronic anti-Aβ passive immunization increases plasma Aβ in J20 mice
To date there has been only one report indicating that m266 can rescue memory impairment in hAPP mice (Dodart et al., 2002). However there have been several reports which indicate that m266 dramatically increases peripheral levels of Aβ, and that m266 causes an efflux of Aβ from brain to plasma (DeMattos et al., 2001; DeMattos et al., 2002; Dodart et al., 2002). Thus, after we had confirmed that m266 and 1C22 were present and active in the plasma and brain (data not shown), it was critically important to ascertain if the lack of effect of either antibody on memory impairments was tied to a parallel lack of effect on cerebral and circulating Aβ.
To determine if either 1C22 or m266 altered circulating levels of Aβ, both free and antibody-bound Aβ were measured in plasma of J20 mice that received 1C22, m266 or 46-4. Plasma samples were incubated with Protein G (PrG) to separate free (supernatant) Aβ from antibody-bound (pellet) Aβ. To control for heterophilic antibody interactions which could compromise our ELISA measurement, and to allow us to discriminate between murine Aβ and transgene-derived human Aβ we also analyzed samples from Wt mice treated with the isotype control antibody 46-4 (Figure 3). Consistent with previous studies (DeMattos et al., 2001; DeMattos et al., 2002; Dodart et al., 2002), m266 did not alter the levels of free (supernatant) Aβ1-x and Aβx-42, but caused a massive increase in antibody-bound (pellet) Aβ (Figure 3B, D). For instance, antibody-bound Aβ was not detected in any of the other groups, whereas ~8 ng/ml Aβ1-x and ~0.3 ng/ml Aβx-42 were detected in the plasma of m266-treated mice. For Aβ1-x these values were, on average, 70-fold higher than total Aβ1-x (pellet plus supernatant) detected in 46-4-treated J20 mice (Figure 3B; 1-way ANOVA, p < 0.0001 post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.01). Similarly, the levels of m266-bound Aβx-42 were ~4 fold higher than total Aβx-42 detected in J20 mice that received 46-4 (Figure 3D; 1-way ANOVA, p < 0.0001; post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.001). Our method of isolating antibody-bound Aβ typically recover ~50% of bound Aβ (data not shown), so the overall increase in plasma Aβ induced by m266 is likely to be more than 100 times higher than that detected in J20 mice treated with 46-4. Thus m266 clearly caused a massive increase in the levels of circulating Aβ, the vast majority of which was bound to antibody. It seems likely that the increase in m266 bound Aβ is a consequence of antibody disrupting the equilibrium between brain and blood Aβ, such that the high concentration of m266 in the periphery causes an efflux of Aβ from brain.
Figure 3. Chronic administration of anti-Aβ mAbs increased plasma Aβ levels in J20 mice.

Plasma was incubated with PrG beads to separate free and antibody-bound Aβ and samples analyzed by ELISAs for Aβ1-x and Aβx-42. (A) Free Aβ1-x, i.e. material still present in the supernatant following PrG incubation, was significantly increased in J20 mice that received 1C22, compared to 46-4- and m266-treated J20 mice (1-way ANOVA, p < 0.0001; post tests, J20 + 1C22 vs. J20 + 46-4, p < 0.001; J20 + 1C22 vs. J20 + m266, p < 0.001). (B) Antibody-bound Aβ1-x was significantly increased in m266-treated J20 mice, compared to 46-4- and 1C22-treated J20 mice (1-way ANOVA, p < 0.0001; post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.01). Aβx-42 was present in plasma at lower levels than Aβ1-x, but was similarly affected by antibody treatment. (C) J20 mice treated with 1C22 had significantly increased levels of free Aβx-42 in their plasma compared to J20 mice treated with 46-4 (1-way ANOVA, p < 0.001; post tests, J20 + 1C22 vs. J20 + 46-4, p < 0.01; J20 + 1C22 vs. J20 + m266, p < 0.01). (D) Antibody-bound Aβx-42 was significantly increased in m266-treated mice compared to J20 mice that received either 46-4 or 1C22 antibodies (1-way ANOVA, p < 0.0001; post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.001). No Aβ was detected in the plasma of Wt mice. Error bars are SEM. 1C22 (J20 n = 11), m266 (J20 n = 9), and 46-4 (J20 n = 12, Wt n = 12).
1C22 also significantly altered circulating Aβ, but its effect was distinct from that of m266 (Figure 3). In animals treated with 1C22 the levels of free Aβ1-x and Aβx-42 were ~1.2 ng/ml and ~0.35 ng/ml, respectively. These levels correspond to an ~8 fold increase in Aβ1-x and a ~5 fold increase in Aβx-42 relative to total Aβ levels in 46-4-treated J20 mice. Levels of free Aβ1-x and Aβx-42 in 1C22-treated mice were also significantly increased compared to m266-treated J20 mice (Figure 3A, C; Aβ1-x; 1-way ANOVA, p < 0.0001; post tests, J20 + 1C22 vs. J20 + 46-4, p < 0.001; J20 + 1C22 vs. J20 + m266, p < 0.001; Aβx-42; 1-way ANOVA, p = 0.001; post tests, J20 + 1C22 vs. J20 + 46-4, p < 0.01; J20 + 1C22 vs. J20 + m266, p < 0.01). Although both 1C22 and m266 antibodies increased peripheral Aβ, their opposing effects on antibody-bound vs. free Aβ suggest that they mediate these outcomes by different mechanisms.
Chronic m266 administration increases antibody-bound Aβ in the water-soluble fraction of brain but neither m266 nor 1C22 altered the levels of Aβ oligomers
Given our findings that both m266 and 1C22 engaged with at least some form of Aβ, as evidenced by an increase in circulating Aβ, it was important to investigate how these antibodies affected the levels and forms of Aβ in J20 brain. For these experiments we used 4 distinct immunoassays: assays for Aβ1-x and Aβx-42 both of which preferentially detect Aβ monomer (Mc Donald et al., 2015), and two oligomer assays each of which recognize an array of both synthetic and human brain-derived soluble Aβ aggregates (Mc Donald et al., 2015; Yang et al., 2013; Yang et al., 2015). As with plasma, TBS and TBS-TX fractions were treated with PrG to separate the distinct pools of Aβ. There was no significant antibody effect on levels of free Aβ1-x and Aβx-42 (Figure 4A, C). Conversely, m266-treated J20 mice had ~8.5 ng/g antibody-bound Aβ1-x and ~6 ng/g antibody-bound Aβx-42, these levels represent a ~50% increase over total levels of Aβ (i.e. 1-x plus x-42) in 46-4-treated J20 brain, and were significantly higher than those of 1C22 or 46-4-treated J20 mice (Aβ1-x; Figure 4B; 1-way ANOVA, p = 0.0002 post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.01; Aβ1-x; Figure 4D; 1-way ANOVA, p = 0.0003; post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.05). Importantly, as the levels of m266-bound Aβ were very similar in the brain and periphery, and given that animals were perfused with PBS prior to brain collection it is clear that the Aβ found bound to m266 measured in brain extracts represent authentic cerebral antibody-Aβ complexes. No specific antibody-bound Aβ1-x or Aβx-42 was detected in 1C22-treated J20 mice; that is, the levels of Aβ precipitated with PrG beads were similar for 1C22- and 46-4-treated mice. The small signal detected in 46-4-treated mice is presumably due to non-specific binding of Aβ to PrG beads.
Figure 4. Chronic immunization with m266 increases antibody-bound Aβ in the water-soluble fraction.
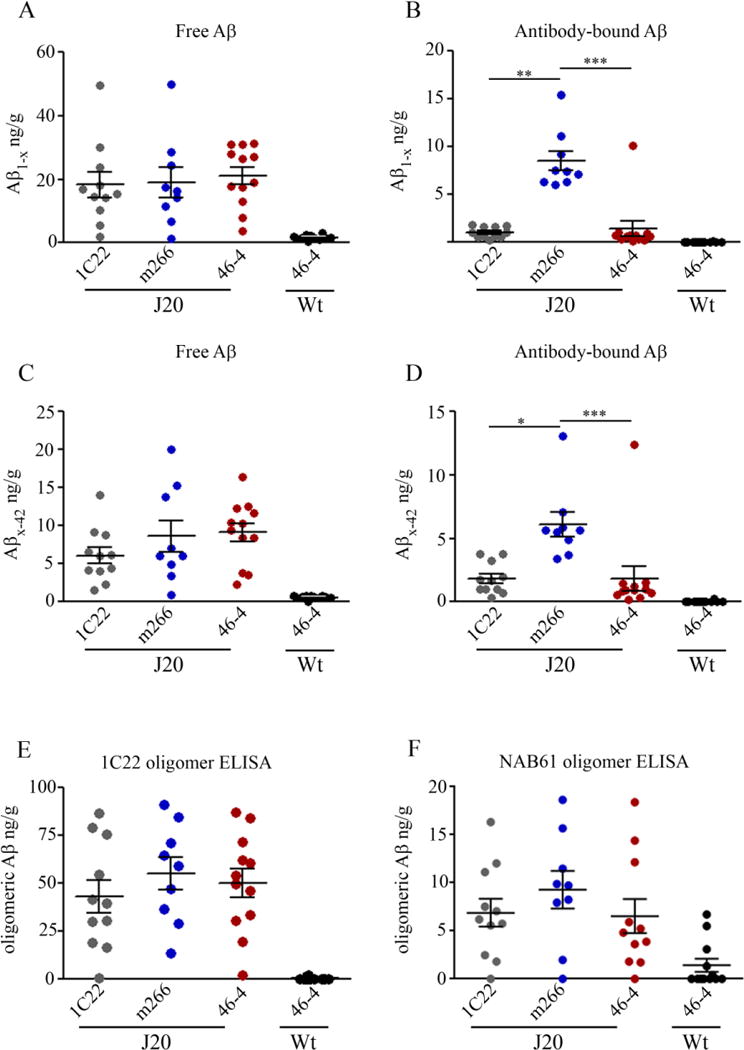
Hemi-brains were sequentially extracted to yield water-soluble, detergent-soluble and formic acid-soluble extracts. Water-soluble extracts were incubated with PrG to pull-down antibody-bound Aβ. Free and antibody-bound fractions of water-soluble Aβ were analyzed by ELISAs for Aβx-42 and Aβ1-x. (A) Free water-soluble Aβ1-x was unchanged in J20 mice that received either 1C22 or m266. (B) Antibody-bound Aβ1-x was significantly increased in m266-treated J20 mice compared to 1C22- or 46-4-treated J20 mice (1-way ANOVA, p = 0.0002; post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.01). (C) Free Aβx-42 was not significantly altered by anti-Aβ antibody treatment. (D) Antibody-bound water-soluble Aβx-42 was significantly increased in m266-treated J20 mice and tended to be increased in 1C22-treated J20 mice, compared to 46-4-treated J20 mice (1-way ANOVA, p = 0.0003; post tests, J20 + m266 vs. J20 + 46-4, p < 0.001; J20 + m266 vs. J20 + 1C22, p < 0.05). (E) Antibody treatment failed to alter the levels of soluble Aβ aggregates measured in unmanipulated TBS samples (1-way ANOVA, p = 0.59). (F) To ensure that 1C22 administered to the J20 mice did not interfer with the 1C22-based oligomer assay, we also measured oligomer levels using the NAB61-3D6 oELISA. Again, the levels of Aβ oligomers were unaffected by 1C22 or m266 treatment (1-way ANOVA, p = 0.502). Little or no Aβ was detected in Wt mouse brains. Error bars are SEM. 1C22 (J20 n = 11), m266 (J20 n ≥ 8), and 46-4 (J20 n = 12, Wt n = 12).
We also measured Aβ oligomers in aqueous extracts of mouse brain. Measurement of oligomeric Aβ using 2 distinct oligomer ELISAs in unmanipulated water-soluble extracts from immunized mice revealed no antibody effect on the levels of soluble Aβ aggregates (Figure 4E and F). Further analysis of free Aβ remaining after PrG pulldown indicated that neither m266 nor 1C22 were bound to significant levels of oligomeric brain Aβ (Supplementary Figure 3). Additionally, boiling the PrG pull-down in 1M GuHCl did not allow detection of additional Aβ (data not shown). These results provide further evidence that m266 and 1C22 do not bind appreciable amounts of Aβ oligomers in J20 brain.
Analysis of detergent-soluble Aβ1-x and Aβx-42 revealed no statistically significant changes in the levels of either antibody-bound or free Aβ regardless of antibody treatment (Supplementary Figure 4). However, the levels of Aβ in the PrG pellet tended to be higher in m266-treated mice, suggesting that in addition to binding water-soluble Aβ m266 may also engage with some membrane-associated Aβ. Because formic acid disrupts antibody-Aβ complexes it was only possible to measure total Aβ in this fraction (i.e. the sum of bound and free Aβ). Neither 1C22 nor m266 changed the amount of Aβ in the formic acid fraction (Supplementary Figure 4). Given that both 1% Trition X-100, and formic acid are likely to disrupt non-covalent assemblies oligomer level were not assessed in these fractions. Collectively these data indicate that m266 can directly engage with peripheral, water-soluble and membrane-bound Aβ but did not alter the levels of water-soluble Aβ oligomers. Given that 1C22 did not alter any of the cerebral forms of Aβ it is not clear how it mediated an increase in peripheral Aβ. To further examine the ability of anti-Aβ antibodies to engage with cerebral Aβ we used IHC to search for changes in amyloid burden and evidence of antibody binding to plaques.
Chronic infusion of 1C22 modestly reduced amyloid pathology in the hippocampus
Treatment with 1C22 caused a ~31% decrease in 21F12 reactive material in the hippocampus, compared with m266- and 46-4-treated J20 mice (Figure 5E – H; 1-way ANOVA, p = 0.0006; post tests, J20 + 1C22 vs. J20 + 46-4, p < 0.01; J20 + 1C22 vs. J20 + m266, p < 0.01). Additionally, there were ~23 % fewer AW7-positive Aβ deposits in the hippocampus of 1C22-treated J20 mice (Figure 5L; 1-way ANOVA, p = 0.033; post test, J20 + 1C22 vs. J20 + 46-4, p < 0.05). Qualitatively, clearance of Aβ by 1C22 was most evident in the subiculum and the ventral region of the dentate gyrus. Moreover, qualitative observations showed smaller diffuse amyloid deposits seemed to be the most reduced, with larger cored plaques remaining, a finding consistent with the observation that 1C22 treatment did not alter 2G3 immunoreactivity or ThS load (Figure 5A, M). Furthermore, we found that a small but significant number of plaques were decorated with 1C22 (Spplementary Figure 3). In contrast, m266 treatment did not reduce Aβ immunoreactivity in the hippocampus of J20 mice (Figure 5), and m266 was not found associated with plaques (Supplementary Figure 5). Much less Aβ is deposited in frontal cortex of J20 mice than in the hippocampus. As in the hippocampus, 1C22 treatment significantly decreased AW7 and 21F12 immunoreactivity and again m266 had no effect (data not shown).
Figure 5. Chronic immunization with 1C22 modestly reduces amyloid pathology in the hippocampus of J20 mice.

Following chronic passive immunization the presence of Aβ deposits in 12 mo J20 mice was investigated using the amyloid binding dye thioflavin S (ThS); the Aβ40-specific monoclonal antibody 2G3; the Aβ42-specific monoclonal antibody, 21F12; and the purified polyclonal anti-Aβ antibody, AW7. Representative micrographs are displayed. (A – D). Levels of ThS positive amyloid were unchanged by anti-Aβ immunotherapy. (E – H) None of the anti-Aβ treatments significantly changed 2G3 immunoreactivity. (I – L) Chronic treatment with 1C22 resulted in a ~31 % decrease in 21F12 immunoreactivity in the hippocampus of J20 mice, compared to both 46-4- and m266-treated J20 mice (1-way ANOVA, p = 0.0006; post tests, J20 + 1C22 vs. J20 + 46-4, p < 0.01; J20 + 1C22 vs. J20 + m266, p < 0.01). (M - P) 1C22 treatment also resulted in a ~23 % decrease in AW7 immunoreactive Aβ pathology in the hippocampus of J20 mice, compared to 46-4-treated J20 mice (1-way ANOVA, p = 0.033; post test, J20 + 1C22 vs. J20 + 46-4, p < 0.05). (D, H, L, P) Percentage positive pixels from 6 sections across 3 equidistant planes were recorded and the average value for each mouse was used to calculate the group mean ± SEM. 1C22 (J20 n = 11), m266 (J20 n = 9), 46-4 (J20 n = 11), Wt n = 2. The magnifying lens was 4 x, and the scale bars are 200 μm.
Anti-Aβ immunotherapy causes unexplained deaths in J20 mice, but not PDAPP mice
Over the course of the 85 days of immunization 3 of 15 1C22-immunized J20 mice and 3 of 13 m266-immunized J20 mice died. Over the same time period and immunization schedule there were no deaths among J20 mice that received 46-4 or Wt mice that received either m266 or 1C22. Microhemorrhage following anti-Aβ immunization is a common reported negative side-effect in hAPP transgenic mice (Burbach et al., 2007; DeMattos et al., 2004; Pfeifer et al., 2002; Racke et al., 2005; Wilcock et al., 2004b), and it has been speculated that deleterious side-effects of anti-Aβ antibodies might counteract the otherwise positive effects of Aβ engagement and clearing (Selkoe, 2013). For this reason, and due to the elevated mortality in anti-Aβ antibody treated J20 mice, we assessed the presence and (i) extent of neuritic degeneration and inflammation, and (ii) microhemorrhage in J20 mice. After 12 weeks of anti-Aβ antibody administration, neither inflammation (Supplementary Figure 6) nor microhemorrhage was exacerbated in J20 mice (Supplementary Figure 7). Indeed, microhemorrhage profiles were similar in 1C22-, m266- and 46-4-treated J20 mice, with no evidence of microhemorrhage in Wt mice. Thus it is unlikely that the increased death we observed in 1C22- and m266-treated mice was a result of increased microhemorrhage.
We have previously reported a significantly reduced viability in J20 mice receiving anti-tau passive immunotherapy (Mably et al., 2015). The number of animals that died following 1C22 (3 of 15) and m266 (3 of 13) treatment did not reach statistical significance compared to 46-4-treated J20 mice. However, considering the significant mortality observed following anti-tau immunization (4 of 13) and the results reported here, it seems likely that antibodies that act on the APP/Aβ/tau axis have highly toxic effects in J20 mice. As with the deaths observed with anti-tau immunotherapy, there were no abnormalities of the major peripheral organs, and gross brain anatomy appeared normal when stained with hematoxylin and eosin. Thus the cause of death in 1C22 and m266 mice is as yet unexplained.
Since anti-Aβ treatment has not been previously shown to have adverse effects in hAPP tgs we sought to determine if the effect in J20 mice (which to our knowledge have never previously been used for anti-Aβ passive immunotherapy) was specific to the model. To this end we repeated our immunization study, using PDAPP mice, a model which has previously been used in passive immunization studies without adverse effect. However, since the behavioral phenotype in PDAPP mice is unstable (Mably, Dodart and Walsh, unpublished; and Personal Communication from Drs Ron Demattos and Sophie Dix) we used these animals only to assess the toxicity of m266 and 1C22, and to measure their effects on cerebral Aβ oligomer levels. Importantly, no immunized PDAPP mice died or exhibited any observable adverse effects (46-4-treated, n = 13; 1C22-treated, n = 15; m266-treated, n = 15). To ensure the difference seen in viability between PDAPP and J20 mice was not due to differences in the effective mAb concentrations, we measured the levels of 1C22 and m266 in PDAPP mice. The levels of antibodies in the plasma of PDAPP mice were comparable to those in J20 mice (J20 + 1C22 = 94.8 μg/ml ± 36.9, PDAPP + 1C22 = 63.3 μg/ml ± 20.7; J20 + m266 = 123.1 μg/ml ± 60.8, PDAPP + m266 = 90.1 μg/ml ± 57.9). As in J20’s, m266 treatment resulted in a large increase (~10 fold) in antibody-bound Aβ in the plasma of PDAPP mice (PDAPP + m266 = 0.39 ng/ml, PDAPP + 46-4 = 0.039 ng/ml), and 1C22 was found to decorate amyloid deposits in PDAPP mouse brain (Supplementary Figure 5). These results demonstrate that the toxic effects of 1C22 and m266 are not generalizable across all hAPP tg models. Thus despite m266 and 1C22 being present at similar levels, and engaging with Aβ in a similar manner, in both PDAPP and J20 mice; some aspect of APP/Aβ/tau metabolism that is particularly sensitive in J20 mice results in certain anti-Aβ and anti-tau mAbs exerting a toxic effect. Interestingly, Aβ oligomer levels were significantly lower in PDAPP brains compared with J20 brains (Supplementary Figure 8A and B), and as in J20’s were unaffected by anti-Aβ mAb treatment (Supplementary Figure 8C and D).
DISCUSSION
Both active and passive immunization against Aβ has been reported to reduce amyloid pathology and improve cognitive impairments in certain hAPP transgenic mice (Bard et al., 2000; Basi et al., 2010; Dodart et al., 2002; Karlnoski et al., 2008; Kotilinek et al., 2002; Lee et al., 2006; Levites et al., 2006; Lord et al., 2009; Maier et al., 2006; Morgan et al., 2000; Oddo et al., 2006; Rasool et al., 2013; Schenk et al., 1999; Seabrook et al., 2007; Weiner et al., 2000; Wilcock et al., 2006; Wilcock et al., 2004b; Zago et al., 2012; Zhang et al., 2011). Despite evidence that antibodies engaged Aβ in vivo, trials of anti-Aβ antibodies in humans have had limited success (Blennow et al., 2012; Salloway, 2012; Sperling, 2012). Indeed, Solanezumab, a humanized version of the mouse anti-Aβ antibody, m266, is the only anti-Aβ immunotherapeutic for which there is verifiable data of a beneficial effect when administered to individuals with mild AD (Chen, 2014; Doody et al., 2014). Aβ is pleomorphic with forms ranging from the innocuous monomer all the way to aggregates of insoluble fibrils like those found in plaques. In recent years much attention has focused on soluble Aβ assemblies with sizes intermediate between monomer and fibrils (Benilova et al., 2012; Lannfelt et al., 2014; Walsh and Teplow, 2012). As such, oligomeric forms of Aβ are suggested to be more appropriate targets for anti-Aβ immunotherapy. There are currently five anti-Aβ mAbs in advanced clinical trials, four of which are purported to be aggregate-preferring, and one (Solanezumab) which preferentially (but not exclusively) recognizes Aβ monomer (Adolfsson et al., 2012; Biogen-Idec, 2015; Bohrmann et al., 2012; Bussiere, 2013; Bussiere et al., 2013; Chen, 2014; clinicaltrials.gov, 2014a; clinicaltrials.gov, 2014b; clinicaltrials.gov, 2014c; clinicaltrials.gov, 2014d; Tucker et al., 2015). Several aggregate-preferring antibodies have been shown to decrease Aβ deposition (Bohrmann et al., 2012; Lord et al., 2009; Tucker et al., 2015) and synapse loss (Dorostkar et al., 2014), and in at least 3 different mouse models aggregate-preferring mAbs have been shown to preserve cognition (Lee et al., 2006; Rasool et al., 2013; Zhang et al., 2011). Although the AD field has quickly embraced the use of aggregate-preferring antibodies there has been little published research directly comparing monomer-preferring and aggregate-preferring antibodies. Thus, in this study, we tested m266, the murine precursor of Solanezumab, alongside the aggregate-preferring mAb, 1C22, (O’Nuallain et al., 2014; Yang et al., 2015) and we used the well-characterized J20 hAPP tg mouse line to examine the effects of mAbs on both behavior and Aβ dynamics.
J20 mice develop highly reproducible age-dependent impairments in spatial reference memory (Cheng et al., 2007; Chin et al., 2005; Karl et al., 2012; Mably et al., 2015; Palop et al., 2003; Roberson et al., 2011; Roberson et al., 2007) which are believed to be mediated by pre-plaque forms of Aβ (Cheng et al., 2007; Karl et al., 2012; Roberson et al., 2007), however, neither m266 nor 1C22 were able to attenuate memory deficits in these mice. The lack of effect on behavior, particularly for m266, is somewhat surprising as m266 had previously been shown to alleviate hyperactivity (Bales et al., 2006) and to improve object recognition and spatial memory in PDAPP mice (Dodart et al., 2002). Moreover, the humanized derivative of m266, Solanezumab, produced a small but significantly beneficial effect on cognition in humans (Doody et al., 2014). Why the behavioral deficits in J20 mice are refractive to m266 and 1C22, whereas m266 had benefit in PDAPP mice (Dodart et al. 2002) and aggregate-preferring antibodies were effective in Tg2576 (Lee et al., 2006), 3xTg (Rasool et al., 2013) and SAMP8 (Zhang et al., 2011) mice is unclear, but raises the thorny issue of how well APP tg mice represent the situation in AD brain? Given that Solanezumab had only marginal effects in humans, whereas m266 had dramatic protective and preventative effects in PDAPPs, but no benefit in J20s, one could argue that J20s better model the AD brain than PDAPPs. Since a lack of antibody effect could result due to a number of trivial factors we were careful to confirm that m266 and 1C22 were present and active in the blood and brains of immunized J20 mice, and that m266 engaged with Aβ as had been described previously.
Examination of blood, brain extracts, and brain tissue from J20 mice, revealed that m266 treatment caused a 70-fold increase in the levels of Aβ in blood – the vast majority of which was bound to m266, but m266 had little effect on amyloid load. These findings are consistent with the notion that m266 accentuates the Aβ concentration gradient between the CNS and the periphery, such that Aβ is drawn from the brain to the blood (DeMattos et al., 2001; DeMattos et al., 2002; Dodart et al., 2002). Similarly, our finding that a small amount of m266 is bound to Aβ in the aqueous phase of brain demonstrates that m266 can enter the brain and directly bind to soluble Aβ (Dodart et al., 2002; Schenk, 2002). Thus, in terms of the effects of m266 on Aβ homeostasis our results are entirely consistent with those reported previously (DeMattos et al., 2001; DeMattos et al., 2002). In addition, we used 2 distinct ELISAs to measure the effects of m266 on oligomeric forms of Aβ, and found that m266 did not alter the levels of cerebral Aβ oligomers in J20 mice. Like m266, 1C22 did not affect Aβ oligomer levels, but 1C22 did engage with Aβ in other significant ways. Specifically, 1C22 treatment caused an increase in peripheral Aβ and a reduction in amyloid load. Unlike m266, 1C22 treatment increased free and not antibody-bound Aβ, thus indicating that 1C22 and m266 interact with Aβ in distinct ways. Despite previous studies reporting that aggregate-preferring mAbs decreased water-, detergent- and/or formic acid-soluble pools of Aβ (Lord et al., 2009; Rasool et al., 2013; Tucker et al., 2015), we found 1C22 had no effects on these Aβ pools in J20 mice. That 1C22 did not alter the levels of formic acid-soluble – which is thought to represent plaque Aβ, suggests that this antibody may have caused a shift in the size of aggregates i.e. preventing the accumulation of visible plaques, but allowing formation of aggregates not readily detected by IHC. Detection of 1C22 bound to amyloid plaques demonstrates that at least a portion of mAb made it to the brain and that the observed reduction in macroscopically detectable amyloid deposits likely resulted due to 1C22 directly binding to and disrupting plaques.
It is important to note that prior to Solanezumab being used in human trials, only one published preclinical study investigated the ability of m266 to reverse cognitive deficits (Dodart et al., 2002). The divergence between the findings reported in that study, and those reported here could arise due to multiple different factors, including, but not limited to differences between the mouse models and behavioral tasks used. With regard to mouse models it is conceivable that the cognitive deficits seen in J20 and PDAPP mice are mediated by different forms of Aβ, or indeed other APP metabolites (Born et al., 2014; Kim et al., 2013a; Rodgers et al., 2012). In this regard it is intriguing that we detected substantial levels of Aβ oligomers in J20 mouse brain, but little or no specific signal in brain extracts from 12 mo PDAPP mice (Supplementary Figure 8), this despite the fact that these PDAPP mice expressed high levels of human APP and had deposits readily detected by IHC. The inability of 1C22 to attenuate impairment of spatial reference memory in J20 mice could also be due to a number of reasons. For instance, plaque-binding antibodies, such as 1C22, have previously been shown to worsen microhemorrhage in PDAPP mice (Racke et al., 2005), but we saw no evidence of 1C22 induced microhemorrhages. Alternatively, the clearance of Aβ deposition by 1C22 may not have been sufficient to elicit a cognitive improvement. However, cognitive improvement in mouse models of AD, following immunotherapy, has occurred both with (Adolfsson et al., 2012; Karlnoski et al., 2008; Oddo et al., 2006; Rasool et al., 2013) and without (Dodart et al., 2002; Kotilinek et al., 2002; Lee et al., 2006) significant clearance of Aβ deposits. One common biochemical factor that was unchanged by both 1C22 and m266 was the level of water-soluble Aβ oligomers, suggesting that cognitive improvement may require engagement with Aβ oligomers. But how can 1C22 be useful to measure Aβ oligomers (Yang et al., 2015), yet be incapable of engaging with the same oligomers in mouse brain? One possibility is that when in solution the affinity of 1C22 for J20 mouse oligomers is relatively poor. In contrast, when 1C22 is immobilized at high density on the surface of a microtitre plate this allows for a massive increase in avidity (O’Nuallain et al., 2011), and hence strong binding and recognition of the repeating structures present in oligomers and aggregates. Moreover, since 1C22 bound to plaques in J20 brain the effective concentration of 1C22 may have been insufficient to bind the smaller Aβ oligomers. The reason why m266 could not alter oligomer levels is obvious when one considers the amount of m266 in brain and the amount bound to Aβ monomer. It is widely accepted that ~0.05% of circulating antibodies penetrate the brain (Pardridge, 2007; Tabrizi et al., 2010) thus the concentration of m266 would be expected to be ~0.25 pM – a level closely similar to the concentration of Aβ1-x found bound to m266 in TBS brain extract (Figure 3B). Thus in J20 brain essentially all of the m266 is bound to Aβ monomer and therefore unable to engage with other forms of Aβ.
The fact that 1C22 was unable to alleviate impairment in spatial reference memory in J20 mice, whereas similar aggregate-preferring mAbs have beneficial effects in other hAPP transgenic mice (Adolfsson et al., 2012; Rasool et al., 2013; Zhang et al., 2011) suggests that the lack of effect of anti-Aβ mAbs seen here may primarily be due to differences between the J20 model and other hAPP transgenic mice. Specifically that Aβ oligomers or certain other APP metabolites mediate the cognitive deficits evident in J20 mice and these are not targeted by either m266 or 1C22. It is not clear which of the available AD mouse models best reflect the human disease; therefore great care should be taken when attempting to extrapolate success in a single preclinical model. Moreover, there is a false impression from published preclinical immunotherapy trials that almost all anti-Aβ antibodies are successful; but anecdotal evidence, and a lack of translation to clinical trials, suggests this is not the case. Some of this misperception likely arises from a strong bias toward publishing positive data and a lack of interest in publishing potentially important negative data. The findings reported here emphasize the need for in-depth characterization of both current and newly emerging AD models. Additionally, they highlight the importance of assessing preferential binding of anti-Aβ antibodies using both in vitro and in vivo paradigms. The use of in vivo paradigms in which an array of Aβ isoforms and assemblies are present is particularly important since only a fraction of these may have pathogenic activity, and the competition between binding to innocuous forms of Aβ versus toxic forms of Aβ will be a critical determinant of efficacy in humans.
Aside from the lack of effect of m266 and 1C22 on cognition in J20’s we also observed a substantial (but not quite statistically significant) number of deaths among m266- and 1C22-immunized J20 mice. Specifically, approximately 20% of m266- and 1C22-treated J20’s died after 4 to 11 immunizations. That this effect is highly specific for anti-Aβ mAbs and J20 mice is evident from the findings that: (1) there were no deaths among 14 J20 mice immunized with the isotype control IgG, 46-4, or 14 wt littermate controls that received 1C22, and (2) there were no loses among 15 PDAPP mice that received a total of 14 immunizations with m266 and 15 PDAPP mice that received a total of 14 immunizations with 1C22. As yet the cause of the sudden death of three of fifteen 1C22-immunized, and three of thirteen m266-immunized J20s remains unexplained. New studies will be required to shed light on the molecular mechanisms that led to these sudden deaths, but we speculate that these mAbs enhance aberrant excitatory neuronal activity (Lalonde et al., 2012; Palop et al., 2007; Roberson et al., 2011; Sanchez et al., 2012) and that this leads to fatal convulsive seizures. These studies follow on from our recent report that a mid-region anti-tau mAb also caused death in J20 mice (Mably et al., 2015). The toxicity of anti-Aβ antibodies under the conditions used was clearly specific to J20s and suggest that J20 mice are particularly susceptible to targeting of the APP/Aβ/tau axis. That the murine precursor of a lead experimental therapeutic, Solanezumab, did not engage with putatively pathogenic Aβ oligomers, while at the same time was associated with death in three out of thirteen immunized animals merits further investigation.
Supplementary Material
Highlights.
m266 caused a dramatic increase in blood and aqueous brain Aβ in J20 mice.
1C22 increased the levels of free Aβ in blood and bound to parenchymal amyloid in J20 mice.
Neither antibody attenuated cognitive deficits or altered brain Aβ oligomers in J20 mice.
Anti-Aβ antibody treatment increased mortality in J20, but not PDAPP mice.
The adverse side-effects and lack of engagement with Aβ oligomers is worrying.
Acknowledgments
We thank Dr. Roderick Bronson for technical assistance. Monoclonal antibodies 8E5, m266, 21F12 and 2G3 were a kind gift from Drs Peter Seubert and Dale Schenk, Elan pharmaceuticals. This research was funded by a grant to DMW from the NIH (R01 AG046275), the Foundation for Neurologic Diseases, and portions of this study were supported by the Harvard NeuroDiscovery Center’s NeuroBehavior Laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolfsson O, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique Abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. 2012;32:9677–89. doi: 10.1523/JNEUROSCI.4742-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, et al. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-Abeta antibody. J Clin Invest. 2006;116:825–32. doi: 10.1172/JCI27120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Basi GS, et al. Structural correlates of antibodies associated with acute reversal of amyloid beta-related behavioral deficits in a mouse model of Alzheimer disease. J Biol Chem. 2010;285:3417–27. doi: 10.1074/jbc.M109.045187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, et al. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–57. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Biogen-Idec. Biogen Idec 2014 revenues increase 40% to $9.7 billion. 2015 http://www.biogenidec.com/press_release_details.aspx?ID=14712&M=News&PID=61997&NewsID=2459, accessed February 12th 2015.
- Blennow K, et al. Effect of immunotherapy with bapineuzumab on cerebrospinal fluid biomarker levels in patients with mild to moderate Alzheimer disease. Arch Neurol. 2012;69:1002–10. doi: 10.1001/archneurol.2012.90. [DOI] [PubMed] [Google Scholar]
- Bohrmann B, et al. Gantenerumab: a novel human anti-Abeta antibody demonstrates sustained cerebral amyloid-beta binding and elicits cell-mediated removal of human amyloid-beta. J Alzheimers Dis. 2012;28:49–69. doi: 10.3233/JAD-2011-110977. [DOI] [PubMed] [Google Scholar]
- Born HA, et al. Genetic suppression of transgenic APP rescues hypersynchronous network activity in a mouse model of Alzeimer’s disease. J Neurosci. 2014;34:3826–40. doi: 10.1523/JNEUROSCI.5171-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach GJ, et al. Vessel ultrastructure in APP23 transgenic mice after passive anti-Abeta immunotherapy and subsequent intracerebral hemorrhage. Neurobiol Aging. 2007;28:202–12. doi: 10.1016/j.neurobiolaging.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Bussiere T. BIIB037: A fully-human monoclonal antibody that binds specifically to fibrillar abeta, and reduces amyloid burden without affecting vascular amyloid; 11th International Conference on Alzheimer’s and Parkinson’s Diseases.2013. [Google Scholar]
- Bussiere T, et al. Differential in vitro and in vivo binding profiles of BIIB037 and other anti-abeta clinical antibody cadidates; 11th International Conference on Alzheimer’s and Parkinson’s Diseases.2013. [Google Scholar]
- Chen C. Biogen Will Push Alzheimer’s Drug to Phase 3. 2014 http://www.bloomberg.com/news/articles/2014-12-02/biogen-rises-on-results-of-early-stage-alzheimer-s-drug-trial, accessed February 12th 2015.
- Cheng IH, et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–28. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Chin J, et al. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2005;25:9694–703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- clinicaltrials.gov. Clinical Trial of Solanezumab for Older Individuals Who May be at Risk for Memory Loss (A4) 2014a https://clinicaltrials.gov/ct2/show/NCT02008357, accessed February 12th 2015.
- clinicaltrials.gov. Dominantly Inherited Alzheimer Network Trial: An Opportunity to Prevent Dementia. A Study of Potential Disease Modifying Treatments in Individuals at Risk for or With a Type of Early Onset Alzheimer’s Disease Caused by a Genetic Mutation. 2014b (DIAN-TU) https://clinicaltrials.gov/ct2/show/NCT01760005, accessed February 12th 2015.
- clinicaltrials.gov. A Study of Crenezumab Versus Placebo in Preclinical PSEN1 E280A Mutation Carriers to Evaluate Efficacy and Safety in the Treatment of Autosomal-Dominant Alzheimer Disease, Including a Placebo-Treated Noncarrier Cohort. 2014c doi: 10.1016/j.trci.2018.02.002. https://clinicaltrials.gov/ct2/show/NCT01998841, accessed February 12th 2015. [DOI] [PMC free article] [PubMed]
- clinicaltrials.gov. A Study to Evaluate Safety, Tolerability, and Efficacy of BAN2401 in Subjects With Early Alzheimer’s Disease. 2014d https://clinicaltrials.gov/ct2/show/NCT01767311, accessed February 12th 2015.
- Colgin LL, Moser EI. Gamma oscillations in the hippocampus. Physiology (Bethesda) 2010;25:319–29. doi: 10.1152/physiol.00021.2010. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, et al. Peripheral anti-Abeta antibody alters CNS and plasma Abeta clearance and decreases brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2001;98:8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, et al. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science. 2002;295:2264–7. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, et al. In vitro and in vivo characterization of beta-amyloid antibodies binding to cerebral amyloid angiopathy (CAA) and selective exacerbation of CAA-associated micohemorrhage. Neurobiol Aging. 2004;25(S2):577. [Google Scholar]
- Dillon GM, et al. Excitotoxic lesions restricted to the dorsal CA1 field of the hippocampus impair spatial memory and extinction learning in C57BL/6 mice. Neurobiol Learn Mem. 2008;90:426–33. doi: 10.1016/j.nlm.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Dodart JC, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5:452–7. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Doody RS, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311–21. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- Dorostkar MM, et al. Immunotherapy alleviates amyloid-associated synaptic pathology in an Alzheimer’s disease mouse model. Brain. 2014;137:3319–26. doi: 10.1093/brain/awu280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow M, et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012;8:261–71. doi: 10.1016/j.jalz.2011.09.224. [DOI] [PubMed] [Google Scholar]
- Games D, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Hilbich C, et al. Amyloid-like properties of peptides flanking the epitope of amyloid precursor protein-specific monoclonal antibody 22C11. J Biol Chem. 1993;268:26571–7. [PubMed] [Google Scholar]
- Hodges H. Maze procedures: the radial-arm and water maze compared. Brain Res Cogn Brain Res. 1996;3:167–81. doi: 10.1016/0926-6410(96)00004-3. [DOI] [PubMed] [Google Scholar]
- Hsia AY, et al. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–33. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Wood K, et al. Amyloid precursor protein processing and Abeta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–5. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl T, et al. Cognitive phenotyping of amyloid precursor protein transgenic J20 mice. Behav Brain Res. 2012;228:392–7. doi: 10.1016/j.bbr.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Karlnoski RA, et al. Deglycosylated anti-Abeta antibody dose-response effects on pathology and memory in APP transgenic mice. J Neuroimmune Pharmacol. 2008;3:187–97. doi: 10.1007/s11481-008-9114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. Normal cognition in transgenic BRI2-Abeta mice. Mol Neurodegener. 2013a;8:15. doi: 10.1186/1750-1326-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WS, et al. Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci. 2013b;33:4387–94. doi: 10.1523/JNEUROSCI.4165-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornecook TJ, et al. Isoform-specific effects of apolipoprotein E on cognitive performance in targeted-replacement mice overexpressing human APP. Genes Brain Behav. 2010;9:182–92. doi: 10.1111/j.1601-183X.2009.00545.x. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, et al. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22:6331–5. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, et al. Neurologic and motor dysfunctions in APP transgenic mice. Rev Neurosci. 2012;23:363–79. doi: 10.1515/revneuro-2012-0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannfelt L, et al. Perspectives on future Alzheimer therapies: amyloid-beta protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther. 2014;6:16. doi: 10.1186/alzrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, et al. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–9. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- Levites Y, et al. Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord A, et al. An amyloid-beta protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2009;36:425–34. doi: 10.1016/j.nbd.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Mably AJ, et al. Tau immunization: a cautionary tale? Neurobiol Aging. 2015;36:1316–32. doi: 10.1016/j.neurobiolaging.2014.11.022. [DOI] [PubMed] [Google Scholar]
- Maier M, et al. Short amyloid-beta (Abeta) immunogens reduce cerebral Abeta load and learning deficits in an Alzheimer’s disease mouse model in the absence of an Abeta-specific cellular immune response. J Neurosci. 2006;26:4717–28. doi: 10.1523/JNEUROSCI.0381-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews PM, et al. Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of calpains enhances endosomal generation of beta-cleaved C-terminal APP fragments. J Biol Chem. 2002;277:36415–24. doi: 10.1074/jbc.M205208200. [DOI] [PubMed] [Google Scholar]
- Mc Donald JM, et al. The aqueous phase of Alzheimer’s disease brain contains assemblies built from approximately 4 and approximately 7 kDa Abeta species. Alzheimers Dement. 2015 doi: 10.1016/j.jalz.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JM, et al. The levels of water-soluble and triton-soluble Abeta are increased in Alzheimer’s disease brain. Brain Res. 2012;1450:138–47. doi: 10.1016/j.brainres.2012.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercken M, et al. Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 1992;84:265–72. doi: 10.1007/BF00227819. [DOI] [PubMed] [Google Scholar]
- Morgan D, et al. Abeta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–5. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Mucke L, et al. High-level neuronal expression of Abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Nuallain B, et al. Are anti-Abeta aggregate-preferring antibodies the furture for AD immunotherapy? Alzheimers Dement. 2014;10:P881–P882. [Google Scholar]
- O’Nuallain B, et al. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–9. doi: 10.1523/JNEUROSCI.3537-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Nuallain B, et al. A monoclonal antibody against synthetic Abeta dimer assemblies neutralizes brain-derived synaptic plasticity-disrupting Abeta. J Neurochem. 2011;119:189–201. doi: 10.1111/j.1471-4159.2011.07389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, et al. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–23. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Palop JJ, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, et al. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–7. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM. Drug targeting to the brain. Pharm Res. 2007;24:1733–44. doi: 10.1007/s11095-007-9324-2. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, et al. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–36. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S, et al. Systemic vaccination with anti-oligomeric monoclonal antibodies improves cognitive function by reducing Abeta deposition and tau pathology in 3xTg-AD mice. J Neurochem. 2013;126:473–82. doi: 10.1111/jnc.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves JP, et al. Mouse monoclonal antibodies to human immunodeficiency virus glycoprotein 120 generated by repeated immunization with glycoprotein 120 from a single isolate, or by sequential immunization with glycoprotein 120 from three isolates. Hybridoma. 1995;14:235–42. doi: 10.1089/hyb.1995.14.235. [DOI] [PubMed] [Google Scholar]
- Roberson ED, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31:700–11. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rodgers SP, et al. Transgenic APP expression during postnatal development causes persistent locomotor hyperactivity in the adult. Mol Neurodegener. 2012;7:28. doi: 10.1186/1750-1326-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–33. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, et al. A randomized, double-blind, placebo-controlled clinical trial of intravenous bapineuzumab in patients with mild to moderate Alzheimer’s disease who are apolipoprotein E ɛ4 non-carriers. 2012 http://www2.kenes.com/efns/info/Documents/Salloway_Bapineuzumab%20IV%20Study%20301_EFNS%20Presentation%20Slides_9-11-2012.pdf.
- Sanchez PE, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–8. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- Schenk D, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–7. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schroeter S, et al. Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J Neurosci. 2008;28:6787–93. doi: 10.1523/JNEUROSCI.2377-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabrook TJ, et al. Dendrimeric Abeta1–15 is an effective immunogen in wildtype and APP-tg mice. Neurobiol Aging. 2007;28:813–23. doi: 10.1016/j.neurobiolaging.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann Neurol. 2013;74:328–336. doi: 10.1002/ana.24001. [DOI] [PubMed] [Google Scholar]
- Seubert P, et al. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature. 1992;359:325–7. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- Shankar GM, et al. Isolation of low-n amyloid beta-protein oligomers from cultured cells, CSF, and brain. Methods Mol Biol. 2011;670:33–44. doi: 10.1007/978-1-60761-744-0_3. [DOI] [PubMed] [Google Scholar]
- Sperling R, et al. A randomized, double-blind, placebo-controlled clinical trial of intravenous bapineuzumab in patients with mild to moderate Alzheimer’s disease who are apolipoprotein E ɛ4 carriers. 2012 http://www2.kenes.com/efns/info/Documents/Sperling_Bapineuzumab%20IV%20Study%20302_EFNS%20Presentation%20Slides_9-11-2012.pdf.
- Tabrizi M, et al. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 2010;12:33–43. doi: 10.1208/s12248-009-9157-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker S, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J Alzheimers Dis. 2015;43:575–88. doi: 10.3233/JAD-140741. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Teplow DB. Alzheimer’s disease and the amyloid beta-protein. Prog Mol Biol Transl Sci. 2012;107:101–24. doi: 10.1016/B978-0-12-385883-2.00012-6. [DOI] [PubMed] [Google Scholar]
- Weidemann A, et al. Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell. 1989;57:115–26. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- Weiner HL, et al. Nasal administration of amyloid-beta peptide decreases cerebral amyloid burden in a mouse model of Alzheimer’s disease. Ann Neurol. 2000;48:567–79. [PubMed] [Google Scholar]
- Wenk GL. Assessment of spatial memory using the radial arm maze and Morris water maze. Curr Protoc Neurosci. 2004 doi: 10.1002/0471142301.ns0805as26. Chapter 8, Unit 8 5A. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, et al. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–6. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004a;24:6144–51. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004b;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AL, et al. Neuroinflammation and Neuronal Loss Precede Abeta Plaque Deposition in the hAPP-J20 Mouse Model of Alzheimer’s Disease. PLoS One. 2013;8:e59586. doi: 10.1371/journal.pone.0059586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, et al. Abeta immunotherapy: intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J Neurosci. 2009;29:11393–8. doi: 10.1523/JNEUROSCI.2021-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, et al. New ELISAs with high specificity for soluble oligomers of amyloid beta-protein detect natural Abeta oligomers in human brain but not CSF. Alzheimers Dement. 2013 doi: 10.1016/j.jalz.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, et al. A highly sensitive novel immunoassay specifically detects low levels of soluble Abeta oligomers in human cerebrospinal fluid. Alzheimers Res Ther. 2015;7:14. doi: 10.1186/s13195-015-0100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zago W, et al. Neutralization of soluble, synaptotoxic amyloid beta species by antibodies is epitope specific. J Neurosci. 2012;32:2696–702. doi: 10.1523/JNEUROSCI.1676-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Administration of amyloid-beta42 oligomer-specific monoclonal antibody improved memory performance in SAMP8 mice. J Alzheimers Dis. 2011;23:551–61. doi: 10.3233/JAD-2010-091195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.