

Abstract
The major type of human liver cancer is hepatocellular carcinoma (HCC), and there are currently many risk factors that contribute to this deadly disease. The majority of HCC occurrences are associated with chronic hepatitis viral infection, and hepatitis B viral (HBV) infection is currently a major health problem in Eastern Asia. Elucidating the genetic mechanisms associated with HBV-induced HCC has been difficult due to the heterogeneity and genetic complexity associated with this disease. A repertoire of animal models has been broadly used to study the pathophysiology and to develop potential treatment regimens for HBV-associated HCC. The use of these animal models has provided valuable genetic information and has been an important contributor to uncovering the factors involved in liver malignant transformation, invasion and metastasis. Recently, transposon-based mouse models are becoming more widely used in liver cancer research to interrogate the genome by forward genetics and also used to validate genes rapidly in a reverse genetic manner. Importantly, these transposon-based rapid reverse genetic mouse models could become crucial in testing potential therapeutic agents before proceeding to clinical trials in human. Therefore, this review will cover the use of transposon-based mouse models to address the problems of liver cancer, especially HBV-associated HCC occurrences in Asia.
Keywords: Hepatocellular carcinoma, Hepatitis B virus, Transposable elements, Sleeping Beauty, Forward and reverse genetic screens
Core tip: Hepatocellular carcinoma (HCC) is the major type of primary liver cancer and the risk factors that contribute to its formation are hepatitis viral infection, alcohol consumption, aflatoxin exposure, hemochromatosis, and tyrosinemia. In vivo forward and reverse genetic transposon models have been used to study the genetic mechanisms of HCC, including hepatitis B viral-induced HCC. These animal models provide valuable genetic information and are important contributors to uncovering the factors involved in liver malignant transformation, invasion and metastasis. They could also be used to test potential therapeutic agents before proceeding to clinical trials in humans.
EPIDEMIOLOGY OF LIVER CANCER
Liver cancer is the second most common cause of cancer related death worldwide, accounting for about 700000 deaths annually. Liver cancer has a poor prognosis, with less than 20% of advanced staged patients surviving for more than one year after initial detection[1]. The major type of primary liver cancer is hepatocellular carcinoma (HCC), which accounts for 85% to 90% of total liver cancer cases[2-4]. In 2012, 83% of 782000 new liver cancer cases worldwide occurred in Eastern Asian countries like Korea, China, Japan, as well as sub-Saharan, middle and Eastern Africa, with a high incidence rate of more than 20 per 100000 individuals. The incidence rate is particularly high in China, with 50% of the total new liver cancer cases diagnosed there[2-6]. Middle Africa, Central America, North America, and Southern Europe have a moderately high incidence rate of about 10 to 20 per 100000 individuals, while a low incidence rate has been reported in South-Central Asia and Northern Europe with less than 5 liver cancer cases per 100000 individuals[2-4,6].
HCC most often is diagnosed between the ages of 55 to 59 years old in China, and between 63 to 65 years old in Europe and North America[3,4]. HCC is a gender-biased disease more commonly found in males than in females with the ratio of about 3:1 in high incidence rate countries. According to long historic studies, hormonal differences between males and females are one of the reasons for this gender bias phenomenon. Researchers have suggested the androgen receptor (AR), which can bind to testosterone and other male hormones and is more highly expressed in males than in females, can promote hepatitis B virus (HBV)-related HCC, while estrogen receptor 1 (ESR1) may work antagonistically[7-10]. The activation of Esr1 has been shown to dampen hepatitis B viral replication[7], and estrogen can also protect female mice by repressing the expression of interleukin 6 (Il6) in Kupffer cells and resident macrophages in liver to prevent liver inflammation[7,8].
HBV INFECTION
HBV infection is a major global health problem and, according to the World Health Organization, more than 300 million people suffer from chronic HBV infection and about 780000 people die every year due to acute or chronic consequences of the disease[4,11]. Since 1970, chronic HBV infection has been shown to be closely related to the development of liver cirrhosis and HCC[12]. The median survival rate for HBV-associated HCC is less than 16 mo, and the five year survival rate is only 15% to 26%[4].
In developing countries, chronic HBV infection accounts for most HCC cases with HBV mostly being transmitted from mother to infant and approximately 90% of infants born to chronic HBV-infected mothers eventually developing the disease[4]. In contrast, HCV infection-associated cirrhosis and alcoholic cirrhosis account for the majority of HCC cases in developed countries that have access to HBV vaccination programs[4]. In countries with these vaccination programs, HBV is usually transmitted sexually between adults and 90% of the virus can be spontaneously cleared by the infected host’s immune system[4]. Although vaccines for HBV have been introduced to reduce the prevalence of HCC, HCC occurrence has not declined in the last two decades due to high chronic HBV infection prevalence and prolonged HCC development latency[4].
HBV GENOTYPES
As of 2011, 10 HBV genotypes (A to J) have been identified and classified by their infectious geographical distribution. Genotype A is widely distributed in South and West Africa and in Western and Northern Europe. Genotypes B and C are commonly found in East and Southeast Asia, including China, while genotype D is distributed mainly in the Mediterranean area[4]. Any genomic variations of HBV, such as genotypes, sub-genotypes, and HBV gene mutations can further contribute to the development of liver cirrhosis and HCC in patients with different HBV replication and hepatitis B surface antigen (HBsAg) status[4,5]. Prolonged expression of HBV correlates with the development of HCC, likely due to the presence of its viral proteins such as the X gene (HBx) and/or large envelope proteins. These viral proteins can activate cellular cancer-related genes, induce oxidative stress, and promote genetic instability that further contributes to hepatocyte transformation[13]. HCC tumorigenesis is also influenced by the patient’s viral status such as the HBV genotype, hepatitis B e antigen (HBeAg) serostatus and mutational status of the HBV genome[4]. There is a higher occurrence of HBV genotypes B and C, and an increasing prevalence of HBsAg serostatus in China. Genotype C seems to have a stronger causative effect than genotype B on HCC development[4,14]. Therefore, it is hypothesized that pathogenicity may be associated with the HBV genotype.
HBV GENOME
HBV belongs to the hepadnaviridae family of enveloped viruses and has an incomplete double-stranded DNA genome of 3.2 kb (Figure 1). The double-stranded HBV genome consists of four open reading frames (ORFs): PreS1/PreS2/S ORF encodes the three large-, middle-, and small HBsAg proteins; C ORF encodes the hepatitis core protein (HBcAg) and HBeAg, a soluble small molecular weight protein produced by the viral core protein gene (PreC/C region) with an alternative start codon and post-translational modification[15]; P ORF encodes the four viral polymerases that consists of the RNase H, viral DNA polymerase/reverse transcriptase, spacer, and terminal protein; X ORF encodes the multi-functional non-structural hepatitis X protein, which can function as a transcriptional transactivator and promote HBV replication (Figure 1)[4,16,17]. The effects of HBV surface proteins and HBx protein on tumorigenesis have been widely studied but their molecular mechanisms remain elusive.
Figure 1.
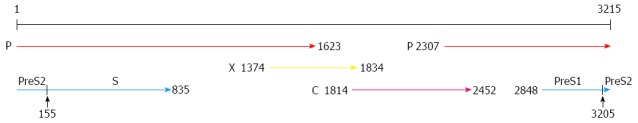
Structure of the hepatitis B virus genome. The hepatitis B virus genome consists of an incomplete double stranded DNA with four open reading frames: surface proteins encoding gene (PreS1/S2/S), polymerase encoding gene (P), X protein encoding gene (X), and core protein encoding gene (C)[17].
HBV PreS1/S2/S ORF
Three main HBsAg proteins are synthesized by three individual promoters and start sites. The PreS1 promoter drives the transcription of PreS1/S2/S ORF to form the large protein (L); the PreS2/S promoter drives the transcription of PreS2/S ORF to form the middle protein (M); while transcription of S ORF only forms the small protein (S) (Figure 1). All these HBsAg proteins have the same carboxyl (C)-terminus but different amino (N)-terminal extensions: the S protein forms the HBsAg with only 226 amino acids (aa), the M protein has an additional 55 aa N-terminal extension, while the L protein has a further N-terminal extension of 108, 118, or 119 aa, depending on the HBV genotype[5,18,19]. A central major hydrophilic region formed by residues 103-173 is exposed to the surface of the viral particles[5]. HBsAg is the main target for viral neutralization by either natural or vaccine-induced anti-HBs antibodies[5,18]. The PreS1 ORF plays a key role in HBV infectivity through its role in nucleocapsid binding during virus envelopment and in receptor binding during hepatocyte infection[18,20,21].
The PreS1 and PreS2 ORFs of HBV can initiate host immune responses as both contain several epitopes for B cell and T cell recognition. Therefore, mutations in these two ORFs can lead to a host immune response defect, resulting in chronic HBV infection[4,22]. Numerous clinical studies have shown deletions in the PreS1 3’-end and PreS2 5’-end are frequently found in chronic HBV infected patients[22-25]. Additional types of mutations that can occur in the PreS ORFs include deletions, insertions and substitutions. Importantly, presence of these mutations has been found to correlate closely with HCC tumorigenesis[4,22]. Deletion mutations that commonly occur can be classified into four categories: PreS1 start codon deletion, internal deletion of PreS1, PreS2 start codon deletion, and internal deletion of PreS2[23]. Generally, the PreS deletion sites are commonly located at the PreS1 and PreS2 region of the HBV genome (Figure 2)[23,25]. In addition, insertions and substitutions have also been reported in chronic HBV infected patient samples[25]. Most neutralizing antibodies target the immunodominant “a” determinant region that is formed by residues 124-147, whereas glycine to arginine substitution at position 145 (G145R) in the “a” determinant region of the S ORF occurs commonly in immune evasive mutant variants[5,18,26]. Other mutations occurring outside the “a” determinant region but within the S ORF, such as the P120S/T mutation, can also exhibit immune evasive effects. These mutant variants are associated with the clinical detection of HBV DNA in patient serum despite the absence of detectable HBsAg[5]. Substitution mutations can result in premature termination during gene transcription, resulting in the production of truncated HBsAg. These truncated HBsAgs can accumulate in the hepatic endoplasmic reticulum (ER), increase oxidative stress of the cell and eventually accelerate liver cell damage[23,25].
Figure 2.
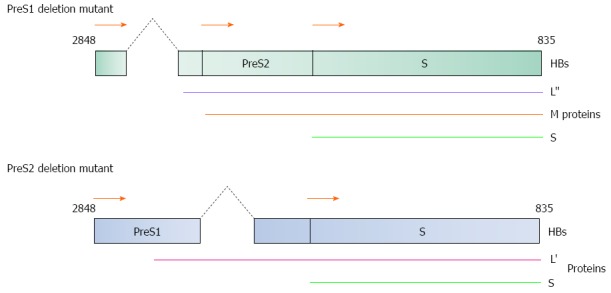
Diagrammatical representation of the PreS1/S2 deletion mutants. (Top) 3’-end PreS1 deletion mutant and (Bottom) 5’-end PreS2 deletion mutant are two common mutation forms found in HBV-induced HCC patients. Both mutants produce truncated large surface proteins (L’ and L”). M: Middle protein; S: Small protein. HBs gene nucleotide position 2848 to 835 as shown in Figure 1.
PreS deletion mutants can induce liver tumor formation by (1) altering the PreS1 mRNA to PreS2/S mRNA ratio; (2) inducing ER oxidative stress; and (3) allowing escape from host immune system surveillance, whereas the 3’-end deletion in the PreS2 domain and removal of the CCAAT element in the S promoter domain is also thought to reduce transcription of the 2.1 kb PreS2/S mRNA[23]. The CCAAT element increases the transcription of PreS2/S ORF and decrease transcription of PreS1 ORF, thus the deletion of this CCAAT element would alter the PreS1 mRNA to PreS2/S mRNA ratio. This decrease in transcription of PreS2/S ORF therefore reduces synthesis of S protein and eliminates synthesis of M protein. Normally, L protein can only be secreted as sub-viral particles or mature virions by forming a complex with the M and S proteins. Insufficient amounts of both M and S proteins would therefore result in the accumulation of L protein in hepatic ER that induces oxidative stress by generating high levels of reactive oxygen species (ROSs) that can cause oxidative DNA damage, induce genetic mutations, and ultimately lead to HCC development[23,27].
PreS deletion mutants have also been implicated in the prevention of apoptosis in hepatocytes by activating the nuclear factor of kappa light polypeptide gene enhancer in B-cell 1 (NFKB1) and v-akt murine thymoma viral oncogene homolog 1 (AKT1)/mechanistic target of rapamycin (serine/threonine kinase) (MTOR) signaling pathways by upregulating the prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) (PTGS2) and vascular endothelial growth factor (VEGF) protein production, respectively[23,27]. In addition, hepatocytes transfected with the PreS2 deletion mutant can also evade the host immune system surveillance due to altered epitopes sites (b10, t5, t6) for B-cell and T-cell recognition and/or reduction in binding affinity of the viral proteins to major histocompatibility complex I molecule[23,27,28].
X ORF
The X ORF encodes the multifunctional HBx protein consisting of 154 aa[16,29]. The genetic mechanism(s) by which HBx induces and/or contributes to HCC development is still not well understood. However, it has been shown that HBx protein plays a critical role in hepatocyte transformation in three ways: (1) changing the epigenetic status; (2) inducing genomic instability; and (3) modulating signaling pathways[13,30].
HBx can change the epigenetic status of hepatocytes, leading to the inactivation of host tumor suppressor genes and/or activation of host oncogenes through induction of various DNA methyltransferases[30,31]. HBx is able to bind to histone acetyltransferase complex and CREB binding protein, promoting transactivation reactions and leading to histone hyperacetylation[13,29-31]. In addition, HBx can promote the production of H3-K4-specific methyltransferase by upregulating SET and MYND domain containing 3 (SMYD3) gene expression that increases H3 lysine K4 methylation[31]. HBx can recruit the binding of DNA (cytosine-5-)-methyltransferase 1 and 3A (DNMT1 and DNMT3A) onto tumor suppressor genes and alter their methylation status and expression level. Conversely, HBx can also inhibit the binding of DNMT3A to promoters, releasing the hypomethylated status of tumor-promoting genes and inducing their expression[31]. DNMT3A has been shown to be associated with hepatocarcinogenesis, as a higher expression level of DNMT3A has been reported in HCC patients[30]. The transcriptional transactivation activity of HBx includes the upregulation of DNMT1 and DNMT3A, which induces cytosine-guanine dinucleotide (CpG) island methylation at the carbon-5 position of cytosine. This DNMT1 and DNMT3A upregulation prevents binding of transcription factors and RNA polymerase II complexes to tumor suppressor genes, such as cyclin-dependent kinase inhibitor 2A (CDKN2A) and cadherin 1, type 1, E-cadherin (epithelial) (CDH1), resulting in their inactivation[30,31]. In addition, HBx can also promote binding of histone deacetylases to tumor suppressor genes to suppress their transcription[31].
In addition to inducing epigenetic changes, HBx is also believed to enhance genomic instability. HBx has been associated with the inactivation of various tumor suppressor genes such as tumor protein p53 (TP53), retinoblastoma 1 (RB1), and axin 1 (AXIN1). HBx inhibits major DNA repair pathways by the interaction with tumor suppressor proteins in these pathways, leading to enhanced genomic instability from the accumulation of mutations and deletions[32-35]. HBx can also increase angiogenesis by upregulating VEGF transcription and stabilizing hypoxia inducible factor 1, alpha subunit [basic helix-loop-helix transcription factor (HIF1A)][29]. Recently, several in vivo research studies have demonstrated HBx overexpression upregulates catenin (cadherin-associated protein), beta 1, 88kDa (CTNNB1) in the important canonical wingless-type MMTV integration site family (WNT)/CTNNB1 signaling pathway implicated in hepatocarcinogenesis[29,36,37].
Four common mutation sites have been identified in the HBx ORF: nucleotide position T1753C (I127T amino acid substitution), A1762T (K130M amino acid substitution), G1764A (V131I amino acid substitution) and T1768A (F132Y amino acid substitution)[38] (Figure 3). The combinatory effects of these mutations have been studied in vitro using the human liver cell line (CCL13) where both K130M and V131I mutations had the potential to induce cell proliferation[38,39].
Figure 3.
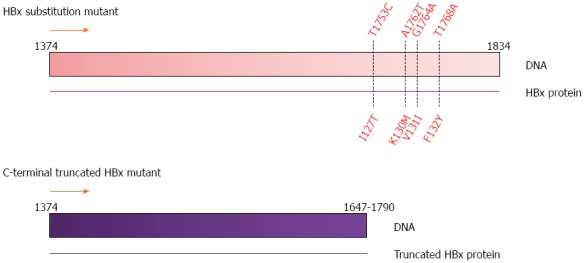
Common mutations in the HBx gene. Substitution mutations at nucleotide positions T1753C, A1762T, G1764A and T1768A; and C-terminal truncations ranging from nucleotide position 1647 to 1790 observed in human HCC tumor samples[43]. HBx gene nucleotide position 1374 to 1834 as shown in Figure 1.
Another form of HBx mutation is the C-terminal truncation (Figure 3). The role of HBx in HBV replication has been carefully studied in HepG2 and Huh7 cell lines[16,40,41]. HBx contains two active domains: a negative regulatory domain in the N-terminus and a transactivation or co-activation domain in the C-terminus that can transactivate viral and cellular promoters[16]. In vitro experiments have shown 52 to 65 aa and 88 to 154 aa in the C-terminus of HBx are necessary for its transactivation activity, cell cycle regulation, and HBx stability[16,40,41]. Upon integration of the HBV genome into the host genome, the 3’-end of HBx is often deleted and produces the C-terminal truncated form of HBx[42,43]. Clinically, C-terminal truncated HBx has been commonly detected in tumors of HCC patients[42,44,45]. Importantly, this C-terminal truncated HBx has been shown to promote tumor cell proliferation and metastasis compared to full-length HBx[42,44,46]. Furthermore, C-terminal truncated HBx has shown to abrogate the growth suppression and apoptotic effect of full-length HBx in cell lines[42-44,46]. However, the genetic mechanism(s) of how this C-terminal truncated HBx affects the tumorigenicity of HBV still remains unclear.
MECHANISMS OF HBV-INDUCED HCC
Increasing evidence has shown HBV infection plays an important role in the development of HCC. Two major mechanisms involved in HBV-associated HCC pathogenesis are: (1) integration of the viral genome into the host chromosome; and (2) the expression of the trans-activating factors derived from the HBV genome[13,15,29,47]. Although, during the normal lifecycle of HBV, the viral genome present in the host nucleus is a covalently closed circular DNA (cccDNA) (Figure 4), about 80% of HBV-related HCC are found to have the HBV genome integrated into the host chromosomes[13]. The integration of the viral genome into the host DNA can disrupt and/or promote cellular gene expression involved with cell growth and differentiation[47]. In addition, the expression of trans-activating factors derived from the expression of viral DNA may influence intracellular signaling pathways and affect host gene expression[47]. Elevated levels of truncated PreS2/S, HBx, and hepatitis B spliced protein have been found in infected tumor tissue[47]. HBx has been shown to play a role in pleiotropic functions and to be involved in the malignant transformation of chronically-infected liver cells[47]. HBx can induce cell proliferation, oxidative stress, and host DNA damage and ultimately contribute to HCC tumorigenesis[13,15]. Recent studies have found HBx can activate CTNNB1 expression and inactivate TP53[15,37]. The activation of the WNT/CTNNB1 signaling pathway is vital in the genetic evolution of HCC[48]. Elevated levels of truncated surface proteins have also been found in HCC patients chronically infected with HBV.
Figure 4.
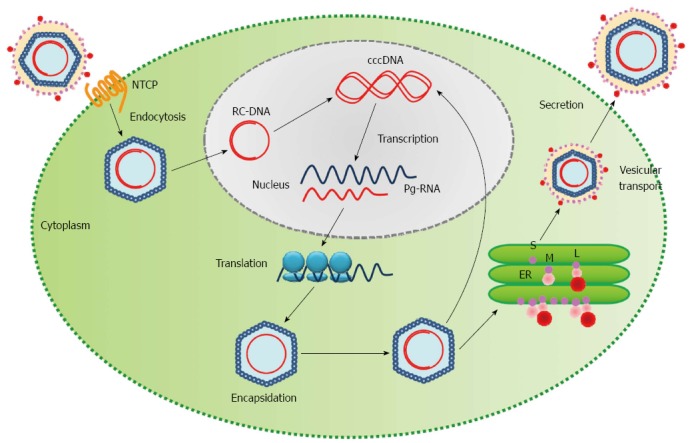
Life cycle of the hepatitis B virus. HBV enters the hepatocyte and is uncoated in the cytoplasm. The relaxed-circular viral DNA (RC-DNA) is released from the nucleocapsid into the host nucleus and is converted into covalently closed circular DNA (cccDNA). The cccDNA acts as a template for transcription of pregenomic virion RNAs (Pg-RNA). Pg-RNA is then transferred into the cytoplasm, bound by viral polymerase and encapsidated. Inside the nucleocapsid, the Pg-RNA is reverse transcribed into RC-DNA, where either the RC-DNA can be released back into the host nucleus for amplification of viral DNA or the entire nucleocapsid can be transferred into the endoplasmic reticulum (ER) for viral surface protein coating and ultimately released from the cell[13]. S: Small surface protein; M: Middle surface protein; L: Large surface protein; HBV: Hepatitis B viral.
MOST FREQUENTLY MUTATED GENES IN HBV-ASSOCIATED HCC
The relationship of somatic mutation of telomerase reverse transcriptase (TERT) to different types of cancer has been highly reported. Recently, the relationship between TERT and HCC is becoming more obvious: the TERT promoter was shown to be mutated in 54% of 469 HCC cases and in 37% of HBV-positive cases, where the mutation site was most commonly located 124 bp upstream the TERT start codon[49]. Moreover, there was a 22% focal amplification rate of TERT in HBV-positive samples[49]. The combined TERT promoter mutation and focal amplification causes higher TERT expression in HCC[49]. Apart from TERT, recent exome sequencing of HCC tumors has identified at least 30 significant putative HCC driver genes involved in 11 signaling pathways: WNT/CTNNB1, mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/AKT/MTOR, TERT, TP53/cell cycle, hepatic differentiation, epigenetic regulation, chromatin remodeling, oxidative stress, IL6/Janus kinase (JAK)-STAT3 and TGFB[49,50]. Amongst these putative driver genes, somatic mutations and copy number changes in CTNNB1, TP53, AT rich interactive domain 1A (SWI-like) (ARID1A) and AXIN1 were significantly altered in 10% of HCC patient samples[49,50]. TP53 mutations are common in most cancer types, so it is not surprising that similar TP53 mutation frequencies in HBV-related HCC have been widely reported by numerous studies[37,49-51]. Furthermore, 16% of the TP53 mutations identified in genotype B HBV-related HCC were found at the R249S site, usually associated with aflatoxin-induced HCC[51-53].
THE NEED FOR MOUSE MODELS OF HBV-INDUCED HCC
Although a few mechanisms by which HBV may promote HCC have been identified, they need to be better characterized in order for the understanding of the genetic mechanisms of HBV-induced HCC to be complete. While it is promising that several common mutations driving HCC in context of HBV infection have been identified, there are many more mutations present in HBV-associated liver tumors that may be important in driving tumor phenotypes[54,55]. Of the many mutations found in HBV-associated liver tumors, the drivers need to be identified and characterized. Mouse models may provide an effective tool for this.
FORWARD GENETIC SCREENS FOR DRIVERS OF HCC USING TRANSPOSON INSERTIONAL MUTAGENESIS
The discovery of inducing lymphoma and mammary cancer formation in mouse models through retroviral insertional mutagenesis by murine leukemia virus or mouse mammary tumor virus has accelerated the speed of identifying driver mutations within these tumor types[56]. However, the application of retroviral insertional mutagenesis has been limited to these specific cancers. Therefore, it may be more valuable to apply an insertional mutagenesis method that is applicable in oncogenomic studies for more human cancer types.
Transposons are such genetic tools that can be used for insertional mutagenesis in multiple tissues in most mammalian species. One transposon that has been successfully used for both forward and reverse genetic interogation of the oncogenome is Sleeping Beauty (SB)[57]. The synthetic SB transposon belongs to the Tc1/mariner family of class II transposable elements[57]. The SB transposon system consists of two components: a transposon and transposase. The transposon can be any DNA sequence flanked by inverted repeat/direct terminal repeat (IR/DR) sequences. The transposase binds to the IR/DR sequences and mediates the excision and reintegration of the transposon from one locus to another in a “cut-and-paste” manner[57]. The target integration site for SB is a TA-dinucleotide pair and, with approximately 300 million sites in the mouse genome, allows for ample sites for insertional mutagenesis[58,59]. The process of transposition or mobilization is relatively random, although “local hopping” may occur[60-62].
Tissue-specific mutagenesis with the SB transposon system has been made possible with the use of a conditional system, which has allowed for transposon insertional mutagenesis in mice to recapitulate several important human cancers for genetic analyses, including liver, gastrointestinal tract, skin, blood, bone, prostate and nervous system[63-75]. In this review we focus on the use of the conditional SB insertional mutagenesis system in several forward genetic screens for HCC candidate genes[63-67,75]. Briefly, mice carrying the following transgenes were generated for each of these forward genetic screens for liver cancer genes: conditional SB transposase transgene, mutagenic transposon, hepatocyte-specific Cre recombinase and predisposed genetic background[63,66]. The SB transposase (SB11) carrying a floxed-stop (lsl) cassette knocked into the mouse endogenous Rosa26 locus can only be activated by a tissue-specific Cre recombinase, allowing for expression and mobilization of transposons exclusively in hepatocytes[64]. The mutagenic transposon, T2/Onc, consists of splice acceptor/polyadenylation (SA/pA) sequences in both orientations and a murine stem cell virus (MSCV) long terminal repeat (LTR) containing promoter/enhancer elements followed by a splice donor that can facilitate splicing of transcripts initiated in the MSCV into downstream endogenous exons[68,74]. The mutagenic transposon was designed to allow for both gain-of-function and loss-of-function mutational activity when integrated into the host genome. When the mutagenic transposon is inserted in a tumor suppressor gene, normal splicing event will be disrupted by the SA/pA elements. Alternatively, misexpression of a proto-oncogene by the MSCV LTR element will occur if the mutagenic transposon is inserted within or near these genes[68,74].
Since TP53 is commonly mutated in HBV-related HCC[51-53], and HBx has been found to inactivate TP53[76-78], studying the genetic drivers of HCC in context of transformation related protein 53 (Trp53) mutation in a mouse model may shed light on the genetic alterations required for HBV-induced HCC development. Using the conditional transposon system together with a conditional dominant negative Trp53 transgene as a predisposed genetic background, 19 highly significant common insertional sites (CISs) for HCC-associated genes were identified[66]. The three most significant signaling/disease functional annotations were identified through analyzing CIS genes by Ingenuity Pathway Analysis (IPA): post-translational modification, cancer, and tumor morphology. Epidermal growth factor receptor (EGFR), HIF1A, mitogen-activated protein kinase kinase 4 (MAP2K4), MET proto-oncogene, receptor tyrosine kinase (MET), p21 protein (Cdc42/Rac)-activated kinase 4 (PAK4), vaccinia related kinase 2 (VRK2), transient receptor potential cation channel, subfamily M, member 7 (TRPM7) and TAO kinase 3 (TAOK3) have been shown to be involved in tumor formation and apoptsis[66]. Nuclear factor I/B (NFIB) and HIF1A network pathways were also identified by IPA and have been implicated in the transduction of phosphorylation-signaling cascades from EGFR[66]. PAK4, NFIB, TAOK3, EGFR, MET, MAP2K4, HIF1A, ubiquitin-conjugating enzyme E2H (UBE2H), and QKI, KH domain containing, RNA binding (QKI) were found to potentially interact with tumor necrosis factor (TNF), inducing tyrosine phosphorylation and internalization of EGFR that might activate the nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (NFKB1) pathway that regulates apoptosis during liver tumor formation (Table 1)[66]. In addition, a high frequency of mutagenic transposon insertions were found in intron 24 of the Egfr gene. This insertion results in the production of C-terminal truncated Egfr protein, which transphosphorylates the tyrosine sites of v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian) (Erbb2) and activates other signaling pathways that contribute to HCC tumorigenesis[66]. In addition, this study also revealed 3 strong candidate genes (UBE2H, QKI, MAP2K4) associated with HCC. QKI and MAP2K4 have a significant decrease in DNA copy number and mRNA level and a high proportion of deletion mutations in human HCC samples, suggesting that both are putative HCC tumor-suppressor genes[66,79,80]. The significant increase in DNA copy number, mRNA level, and high mRNA up-regulation rate of UBE2H indicate that it is a putative HCC tumor-promoting gene[66,79,80].
Table 1.
Hepatocellular carcinoma signaling pathways identified by Sleeping Beauty transposon insertional mutagenesis system[63,66]
| Pathways | Wnt/Ctnnb1 | Trp53 | Pi3k-Akt-Mtor | Mapk | Hippo | Tgfb-Bmp | Il6-Stat3 | Tnf-Akt1 |
| Genes involved in corresponding pathways | Lrp1 | Prkag2 | Insr | Egfr | Fat1 | Acvr1 | Il6st (gp130) | Tnf |
| Lrp5 | Ywhaz | Igf1 | Met | Wwc1 | Acvr2a | Jak1 | Egfr | |
| Lrp6 | Crebbp | Pten | Grb2 | Taok1 | Bmp1 | Stat3 | Met | |
| Gsk3b | Mdm2 | Pik3ca | Sos1 | Taok2 | Bmpr1a | Pak4 | ||
| Axin1 | Usp10 | Pik3r1 | Sos2 | Taok3 | Sar1a | Nfib | ||
| Apc | Usp7 | Pik3c2a | Kras | Mobkl2b | Tab2 | Taok3 | ||
| Ctnnb1 | Pias1 | Pik3ap1 | Raf1 | Mobkl1a | Tab3 | Map2k4 | ||
| Tcf7l2 | Trp53inp1 | Akt2 | Map3k1 | Sav1 | Smad3 | Akt | ||
| Csnk1a1 | Ppp2r1a | Rps6kb1 | Map3k2 | Lats1 | Smad2 | Hif1a | ||
| Csnk1d | Ppp2r2a | Foxo1 | Map2k1 | Yap1 | Smad1 | Ube2h | ||
| Csnk1g1 | Ppp2r2d | Mapk1 | Tead1 | Smad4 | Qk | |||
| Csnk1g3 | Ppp2cb | Smad5 | Pi3k | |||||
| Tnks | Ppp2r5e | Jnk | ||||||
| Tnks2 | Trp53 |
Indicates a network.
Incorporating the SB transposon system with a commonly dysregulated human malignancy gene, v-myc avian myelocytomatosis viral oncogene homolog (MYC), 18 CISs in early-developing liver tumors were identified[67]. It was shown that zinc finger protein, X-linked (Zfx) plays a tumor suppressor role in liver tumorigenesis, while nuclear receptor coactivator 2 (Ncoa2) and dystrobrevin, beta (Dtnb) function as putative tumor-suppressors[67]. In addition, expression of NCOA2 and its target gene glucose-6-phosphatase, catalytic subunit (G6PC) were significantly reduced in human tumor samples, and this reduction was strongly associated with the low survival rates of HCC patients[67].
Using the SB transposon insertional mutagenesis mouse model incorporating the HBsAg transgene in a forward genetic screen, over 2000 candidate drivers of HBV-associated HCC were identified[63]. In this study, 21 genes with highly significant sequencing read counts and frequencies of occurrence were identified to be candidate HCC genes[63]. Of these candidate genes, 4 genes were identified as putative tumor suppressor genes: adenosine kinase (Adk), dihydropyrimidine dehydrogenase (Dpyd), lysine (K)-specific methyltransferase 2E (Kmt2e) and nuclear factor I/A (Nfia); 6 genes were found to have tumor suppressing effects in HCC: Arid1a, Gsk3b, IG motif containing GTPase activating protein 2 (Iqgap2), membrane associated guanylate kinase, WW and PDZ domain containing 1 (Magi1), phosphatase and tensin homolog (Pten) and salvador homolog 1 (Sav1); 2 genes were found to be involved in hepatocyte differentiation and maturation: zinc finger and BTB domain containing 20 (Zbtb20) and ankyrin repeat domain 17 (Ankrd17); 6 genes were found to regulate hepatic metabolism: Pten, glycogen synthase kinase 3 beta (Gsk3b), growth hormone receptor (Ghr), Adk, Zbtb20, and Dpyd; and 7 genes were found to be associated with HCC transcription modulation: Kmt2e, SET domain containing 2 (Setd2), WW domain containing adaptor with coiled-coil (Wac), Arid1a, Nfia, staphylococcal nuclease and tudor domain containing 1 (Snd1), and Zbtb20[63]. Through CIS gene annotation enrichment analysis, it was found that HBsAg CIS genes drive HCC through conserved cancer signaling pathways, including Wnt/Ctnnb1, Trp53, Pi3k-Akt-Mtor, Mapk, Hippo, transforming growth factor beta (Tgfb)-bone morphogenic protein (Bmp) and Il6-signal transducer and activator of transcription 3 (Stat3) (Table 1)[63]. Moreover, it was revealed that the majority of the HCC CIS genes were involved in cellular metabolic processes[63]. It was suggested that disruption of glycolytic and glutaminolytic pathways provided bioenergetics, biosynthesis and redox regulation benefits for tumor cell division, thus inhibiting glycolysis and/or glutaminolysis might impede liver tumor formation or progression[63]. Genetic alterations, copy number changes, and expression levels of the human orthologs of CIS genes from three separate SB transposon forward genetic screens with different genetic backgrounds were analyzed in TCGA database and presented as a word cloud (Wordle) to view the degree of genetic alteration reported in clinical samples (Figure 5)[63,65,67].
Figure 5.

Word cloud of frequent CIS genes from three separate studies using SB transposon forward genetic screening with different genetic backgrounds[63,65,67]. The roles of these frequent CIS gene human orthologs were analyzed using TCGA database. The font size indicates the frequency of genetic alteration reported in human HCC samples. Color of the font indicates the copy number change or expression profile of the gene: red, amplification; blue, deletion; tomato, tends to upregulation; purple, tends to downregulation; yellow, frequently found in all three studies.
REVERSE GENETIC VALIDATION USING RAPID TRANSPOSON-BASED MOUSE MODELS
The SB transposon system can also be used in a reverse genetic manner to introduce tumorigenic genes into mouse hepatocytes for stable expression when delivered on transposon plasmids to the mouse liver by hydrodynamic tail vein injection[37,59,65,66,75,81]. Hydrodynamic injection is a rapid and high-volume infusion of naked plasmid DNA into the tail vein. It is an effective method for in vivo gene delivery, in which about 40% of the hepatocytes in a test animal take up the transgene and express > 95% of the transgene after hydrodynamic injection[59]. The mechanism of DNA uptake is still poorly understood, but it is suggested that the injected high-volume of DNA solution enters the inferior vena cava and causes over-stretching of myocardial fibers, induces cardiac congestion, resulting in delivery of injected solution into liver[82]. In addition to its use in mice, this method can also be used to transfer naked plasmid DNA into porcine and rabbit livers and into muscles of larger animals[59,82]. The sporadic expression of target genes mimics a more realistic situation in human liver cancer than other conventional conditional transgenic models.
A powerful method to test genes’ oncogenic roles in the liver employs this gene delivery system to deliver tumorigenic genes to the livers of fumarylacetoacetate hydrolase (Fah)-deficient/Rosa26-SB11 (Fah-/-/SB+/-) transgenic mice[37,59,65,66,75,81,83]. This is a selective model allowing for rapid generation of mice in which nearly all hepatocytes express the delivered transgenes. Fah-deficient mice have a defect in the last step of the tyrosine catabolic pathway in which fumarylacetoacetate is hydrolyzed to acetoacetate and fumarate, similar to the human hereditary tyrosinemia type I disease[84]. These mice must be maintained on nitisinone in the drinking water, which blocks this pathway upstream of fumarylacetoacetate production[84]. Oncogenes to be tested are co-delivered on transposon plasmids with a Fah rescue cDNA, and nitisinone is removed after gene delivery. Nitisinone removal causes Fah-deficient hepatocytes to die, and the liver is regenerated by hepatocytes that stably express the delivered transgenes[37,59,65,66,75,81]. This system has been used in a reverse genetic manner to introduce tumorigenic genes into the livers of Fah-/-/SB+/- transgenic mice by hydrodynamic tail vein injection for validation in several studies[37,59,65,66,75,81].
SB-mediated gene delivery to Fah-/-/SB+/- transgenic mice by hydrodynamic tail vein injection has been used to study the role of HBx in promoting liver cancer[37]. In this study, a transposon vector containing the HBx gene and Fah cDNA was delivered with and without a transposon vector containing a short hairpin RNA directed against Trp53 (shp53). HBx expression activated Ctnnb1 expression, and HBx cooperated with shp53 to induce the formation of hyperplastic nodules. This study showed this method could be used to model liver cancer driven by HBV gene components and elucidate mechanisms by which they may promote cancer.
We are currently attempting to use this method to test the roles of other HBV genes in driving liver cancer. Using the Gateway cloning system (Life Technologies), we have constructed transposon plasmids with various individual HBV gene components for tumorigenic analyses in livers of Fah-/-/SB+/- transgenic mice. Hepatocytes that are transgenic for both the Fah cDNA and HBV gene-of-interest will repopulate the Fah-deficient liver, mimicking disease progression. Preliminary unpublished studies have yielded promising development of tumors in certain HBV gene-injected mice compared with empty vector controls (Figure 6). These tumors will be further interrogated for their genetic information such as gene expression profiles and/or pathway analyses. It is envisaged that genetic information from such in vivo studies will provide insight into underlying genetic mechanisms of HBV-induced HCC.
Figure 6.
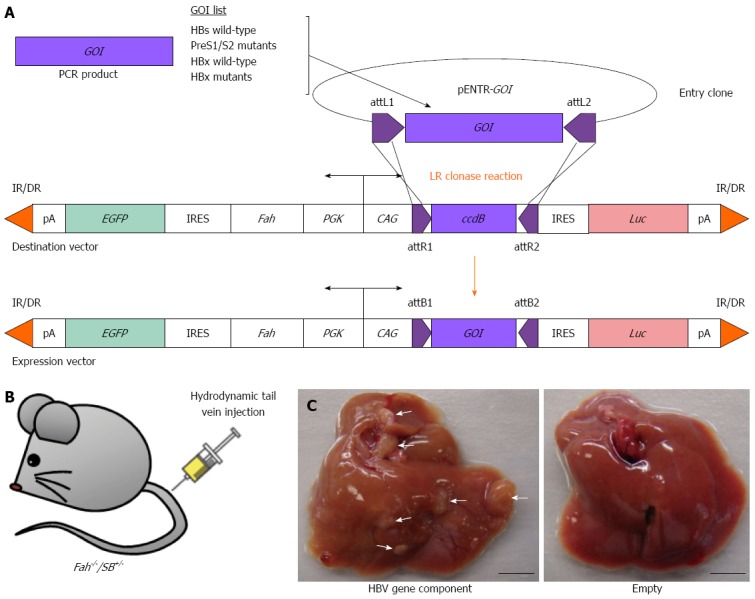
Reverse genetic screening of individual HBV gene components using the Fah-/-/SB+/- transgenic mouse model. A: Construction of gene delivery expression vectors that carry various individual HBV gene-of-interest (GOI) components by PCR amplification and insertion of GOI into an entry clone (pENTR) and followed by LR clonase reaction (Life Technologies); B: Hydrodynamic tail vein injection of gene delivery expression vector into Fah-/-/SB+/- transgenic mouse; C: Liver tumors indicated by white arrowheads observed in Fah-/-/SB+/- mouse injected with a HBV gene component (left) after 160-d post-hydrodynamic injection. Fah-/-/SB+/- transgenic mouse injected with an empty gene delivery expression vector displayed normal liver morphology (right). EGFP: Enhanced green fluorescent protein gene; Luc: Luciferase gene; IR/DR: Inverted repeat/direct repeat sequences for Sleeping Beauty transposase binding and mobilization; pA: Polyadenalytion signal; IRES: Internal ribosome entry site; PGK: Phosphoglycerate kinase 1 promoter; CAG: Cytomegalovirus enhancer fused to the chicken β-actin promoter; attL/R/B sites: Recombination sites used by the Gateway cloning system (Life Technologies); ccdB: Topoisomerase poison from Escherichia coli. Scale bar: 0.5 cm.
SB-mediated gene delivery for reverse genetic studies in mice, in addition to its use in studying the oncogenic roles of viral genes, could be used to rapidly validate candidate genetic drivers of HBV-associated HCC discovered in forward genetic screens as described above or genes found to be commonly altered in human HBV-associated HCC. Candidate drivers could be tested in context of expression of HBV viral genes using this system. In addition, hydrodynamic delivery of plasmid DNA expressing HBV genome components has been used to model HBV infection in mice[85-87]. Candidate HBV-associated HCC drivers could be added on separate plasmids to test their roles in HCC in context of a model of HBV infection in vivo.
DISCUSSION
Based on the limited existing treatments and the poor prognosis of HCC, continuous efforts should be put into uncovering the mechanisms of drug resistance and tumor progression and on the development of biomarkers for more sensitive diagnosis and as molecular targets for new therapies. HBV infection is a major risk factor for HCC development; understanding its interactions with cancer-driving signaling pathways and its contributions to tumor initiation, promotion and progression will be critical for developing biomarkers for diagnosis and therapy.
In addition, recent exome sequencing of HCC patient samples have identified multiple cellular signaling pathways, including WNT/CTNNB1, MAPK, PI3K/AKT/MTOR, TERT, TP53/cell cycle, hepatic differentiation, epigenetic regulation, chromatin remodeling, oxidative stress, IL6/JAK-STAT3 and TGFB[49,50]. In addition, numerous significant HCC driver genes, such as TERT, TP53, CTNNB1, AXIN1 and ARID1A have also been identified[49,50]. Additionally, TP53 mutation was frequently found in HBV-related HCC patient samples[50]. A large number of components are involved in each of these pathways, many of which may pose effects on the tumor initiation, promotion and progression, which challenges the screening of potential therapeutic drugs for treatment. Insertional mutagenesis by the SB transposon system is a powerful tool for studying HBV-induced HCC through forward genetic screens, and SB-mediated gene delivery is a powerful tool for reverse genetic studies. Snd1, an oncogene that promotes HCC angiogenesis, was a recurrent CIS gene in liver tumor samples from various studies using the SB transposon system forward genetic approach including a study done in the context of transgenic HBsAg expression[63,65-67]. Many other candidate genes likely to play a role in promoting HBV-associated HCC have been identified in these studies. There is also ample data on genes commonly mutated in HBV-associated liver tumors, and genes whose expression or function may be altered by HBV proteins. We have developed a unique rapid in vivo model to validate candidate liver cancer genes that can be used to study potential drivers of HBV-associated HCC. It relies on the use of the Fah-deficient mouse that is transgenic for the SB transposase gene (Fah-/-/SB+/-)[81,84]. As described earlier, candidate genes can be easily cloned into transposon-based delivery gene vectors and introduced specifically into the livers of these mice using hydrodynamic tail vein injection[59]. This system can be used to study both cellular and viral genes. Importantly, these models could be used in pre-clinical trials to test novel therapeutic drugs. Therefore, we propose to use this rapid system to generate many genetic mouse models of HBV-induced HCC to further investigate the tumorigenic mechanisms. Currently, we have generated mouse models that recapitulate several molecular subclasses of human HCC: neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS), CTNNB1, Poly7, and HBV-induced[37,65,81]. The heterogeneity and complexity of HBV-induced HCC has thus far precluded the full understanding of the genetic mechanisms associated with this deadly disease needed to develop effective treatments and preclinical models. We believe the SB transposon system may allow the discovery of drug targets and development of preclinical models desperately needed to advance this field.
Footnotes
Supported by Health Medical Research Fund No. 11122171, the Food and Health Bureau, and the Hong Kong SAR Government; the Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong SAR (1-ZVAG, G-YBAY, G-UA94 and 1-ZE19); and the NIH IMVTP grant No. T32 AI083196-04 to Tschida BR.
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 13, 2015
First decision: September 11, 2015
Article in press: September 30, 2015
P- Reviewer: Cao GW, Mizuguchi T S- Editor: Qi Y L- Editor: A E- Editor: Ma S
References
- 1.Scudellari M. Drug development: try and try again. Nature. 2014;516:S4–S6. doi: 10.1038/516S4a. [DOI] [PubMed] [Google Scholar]
- 2.Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist. 2010;15 Suppl 4:5–13. doi: 10.1634/theoncologist.2010-S4-05. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273.e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q, Cao G. Genotypes, mutations, and viral load of hepatitis B virus and the risk of hepatocellular carcinoma: HBV properties and hepatocarcinogenesis. Hepat Mon. 2011;11:86–91. [PMC free article] [PubMed] [Google Scholar]
- 5.Cao GW. Clinical relevance and public health significance of hepatitis B virus genomic variations. World J Gastroenterol. 2009;15:5761–5769. doi: 10.3748/wjg.15.5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Tuteja G, Schug J, Kaestner KH. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell. 2012;148:72–83. doi: 10.1016/j.cell.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 9.Liu WC, Liu QY. Molecular mechanisms of gender disparity in hepatitis B virus-associated hepatocellular carcinoma. World J Gastroenterol. 2014;20:6252–6261. doi: 10.3748/wjg.v20.i20.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu MH, Ma WL, Hsu CL, Chen YL, Ou JH, Ryan CK, Hung YC, Yeh S, Chang C. Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med. 2010;2:32ra35. doi: 10.1126/scitranslmed.3001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerlich WH. Medical virology of hepatitis B: how it began and where we are now. Virol J. 2013;10:239. doi: 10.1186/1743-422X-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Bisceglie AM. Hepatitis B and hepatocellular carcinoma. Hepatology. 2009;49:S56–S60. doi: 10.1002/hep.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. 2010;52:594–604. doi: 10.1016/j.jhep.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 14.Chan HL, Hui AY, Wong ML, Tse AM, Hung LC, Wong VW, Sung JJ. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut. 2004;53:1494–1498. doi: 10.1136/gut.2003.033324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayub A, Ashfaq UA, Haque A. HBV induced HCC: major risk factors from genetic to molecular level. Biomed Res Int. 2013;2013:810461. doi: 10.1155/2013/810461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gong DY, Chen EQ, Huang FJ, Leng XH, Cheng X, Tang H. Role and functional domain of hepatitis B virus X protein in regulating HBV transcription and replication in vitro and in vivo. Viruses. 2013;5:1261–1271. doi: 10.3390/v5051261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghany M, Liang TJ. Drug targets and molecular mechanisms of drug resistance in chronic hepatitis B. Gastroenterology. 2007;132:1574–1585. doi: 10.1053/j.gastro.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 18.Churin Y, Roderfeld M, Roeb E. Hepatitis B virus large surface protein: function and fame. Hepatobiliary Surg Nutr. 2015;4:1–10. doi: 10.3978/j.issn.2304-3881.2014.12.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarocchi M, Polvani S, Marroncini G, Galli A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J Gastroenterol. 2014;20:11630–11640. doi: 10.3748/wjg.v20.i33.11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meier A, Mehrle S, Weiss TS, Mier W, Urban S. Myristoylated PreS1-domain of the hepatitis B virus L-protein mediates specific binding to differentiated hepatocytes. Hepatology. 2013;58:31–42. doi: 10.1002/hep.26181. [DOI] [PubMed] [Google Scholar]
- 22.Yeung P, Wong DK, Lai CL, Fung J, Seto WK, Yuen MF. Association of hepatitis B virus pre-S deletions with the development of hepatocellular carcinoma in chronic hepatitis B. J Infect Dis. 2011;203:646–654. doi: 10.1093/infdis/jiq096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CM, Wang GM, Jow GM, Chen BF. Functional analysis of hepatitis B virus pre-s deletion variants associated with hepatocellular carcinoma. J Biomed Sci. 2012;19:17. doi: 10.1186/1423-0127-19-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Na B, Huang Z, Wang Q, Qi Z, Tian Y, Lu CC, Yu J, Hanes MA, Kakar S, Huang EJ, et al. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PLoS One. 2011;6:e26240. doi: 10.1371/journal.pone.0026240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen T, Yan XM, Zhang JP, Wang JL, Zuo RX, Li L, Wang LP. Evolution of Hepatitis B Virus in a Chronic HBV-Infected Patient over 2 Years. Hepat Res Treat. 2011;2011:939148. doi: 10.1155/2011/939148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Datta S, Chatterjee S, Veer V, Chakravarty R. Molecular biology of the hepatitis B virus for clinicians. J Clin Exp Hepatol. 2012;2:353–365. doi: 10.1016/j.jceh.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu L, Kuai X, Liu T, Chen T, Ni Z, Shen X. Pre-S deletion and complex mutations of hepatitis B virus related to young age hepatocellular carcinoma in Qidong, China. PLoS One. 2013;8:e59583. doi: 10.1371/journal.pone.0059583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D, Dong P, Zhang K, Deng L, Bach C, Chen W, Li F, Protzer U, Ding H, Zeng C. Whole genome HBV deletion profiles and the accumulation of preS deletion mutant during antiviral treatment. BMC Microbiol. 2012;12:307. doi: 10.1186/1471-2180-12-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brechot C, Kremsdorf D, Soussan P, Pineau P, Dejean A, Paterlini-Brechot P, Tiollais P. Hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC): molecular mechanisms and novel paradigms. Pathol Biol (Paris) 2010;58:278–287. doi: 10.1016/j.patbio.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Lu JW, Hsia Y, Tu HC, Hsiao YC, Yang WY, Wang HD, Yuh CH. Liver development and cancer formation in zebrafish. Birth Defects Res C Embryo Today. 2011;93:157–172. doi: 10.1002/bdrc.20205. [DOI] [PubMed] [Google Scholar]
- 31.Tian Y, Yang W, Song J, Wu Y, Ni B. Hepatitis B virus X protein-induced aberrant epigenetic modifications contributing to human hepatocellular carcinoma pathogenesis. Mol Cell Biol. 2013;33:2810–2816. doi: 10.1128/MCB.00205-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van de Klundert MA, van Hemert FJ, Zaaijer HL, Kootstra NA. The hepatitis B virus x protein inhibits thymine DNA glycosylase initiated base excision repair. PLoS One. 2012;7:e48940. doi: 10.1371/journal.pone.0048940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qadri I, Fatima K, AbdeL-Hafiz H. Hepatitis B virus X protein impedes the DNA repair via its association with transcription factor, TFIIH. BMC Microbiol. 2011;11:48. doi: 10.1186/1471-2180-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuda Y, Ichida T. Impact of hepatitis B virus X protein on the DNA damage response during hepatocarcinogenesis. Med Mol Morphol. 2009;42:138–142. doi: 10.1007/s00795-009-0457-8. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh YH, Hsu JL, Su IJ, Huang W. Genomic instability caused by hepatitis B virus: into the hepatoma inferno. Front Biosci (Landmark Ed) 2011;16:2586–2597. doi: 10.2741/3874. [DOI] [PubMed] [Google Scholar]
- 36.Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus X protein is essential for the activation of Wnt/beta-catenin signaling in hepatoma cells. Hepatology. 2004;39:1683–1693. doi: 10.1002/hep.20245. [DOI] [PubMed] [Google Scholar]
- 37.Keng VW, Tschida BR, Bell JB, Largaespada DA. Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the Sleeping Beauty transposon system. Hepatology. 2011;53:781–790. doi: 10.1002/hep.24091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin X, Xu X, Huang QL, Liu YQ, Zheng DL, Chen WN, Lin JY. Biological impacts of “hot-spot” mutations of hepatitis B virus X proteins are genotype B and C differentiated. World J Gastroenterol. 2005;11:4703–4708. doi: 10.3748/wjg.v11.i30.4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindh M, Gustavson C, Mårdberg K, Norkrans G, Dhillon AP, Horal P. Mutation of nucleotide 1,762 in the core promoter region during hepatitis B e seroconversion and its relation to liver damage in hepatitis B e antigen carriers. J Med Virol. 1998;55:185–190. doi: 10.1002/(sici)1096-9071(199807)55:3<185::aid-jmv1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 40.Lizzano RA, Yang B, Clippinger AJ, Bouchard MJ. The C-terminal region of the hepatitis B virus X protein is essential for its stability and function. Virus Res. 2011;155:231–239. doi: 10.1016/j.virusres.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo N, Cai Y, Zhang J, Tang W, Slagle BL, Wu X, He S. The C-terminal region of the hepatitis B virus X protein is required for its stimulation of HBV replication in primary mouse hepatocytes. Virus Res. 2012;165:170–178. doi: 10.1016/j.virusres.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Ma NF, Lau SH, Hu L, Xie D, Wu J, Yang J, Wang Y, Wu MC, Fung J, Bai X, et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin Cancer Res. 2008;14:5061–5068. doi: 10.1158/1078-0432.CCR-07-5082. [DOI] [PubMed] [Google Scholar]
- 43.Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013;34:787–798. doi: 10.1093/carcin/bgs406. [DOI] [PubMed] [Google Scholar]
- 44.Sze KM, Chu GK, Lee JM, Ng IO. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology. 2013;57:131–139. doi: 10.1002/hep.25979. [DOI] [PubMed] [Google Scholar]
- 45.Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26 Suppl 1:144–152. doi: 10.1111/j.1440-1746.2010.06546.x. [DOI] [PubMed] [Google Scholar]
- 46.Yip WK, Cheng AS, Zhu R, Lung RW, Tsang DP, Lau SS, Chen Y, Sung JG, Lai PB, Ng EK, et al. Carboxyl-terminal truncated HBx regulates a distinct microRNA transcription program in hepatocellular carcinoma development. PLoS One. 2011;6:e22888. doi: 10.1371/journal.pone.0022888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chemin I, Zoulim F. Hepatitis B virus induced hepatocellular carcinoma. Cancer Lett. 2009;286:52–59. doi: 10.1016/j.canlet.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Thompson MD, Wickline ED, Bowen WB, Lu A, Singh S, Misse A, Monga SP. Spontaneous repopulation of β-catenin null livers with β-catenin-positive hepatocytes after chronic murine liver injury. Hepatology. 2011;54:1333–1343. doi: 10.1002/hep.24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 50.Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D, Gaston Mathe Y, Laurent C, Laurent A, Bioulac-Sage P, et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut. 2015;64:820–829. doi: 10.1136/gutjnl-2013-306228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chittmittrapap S, Chieochansin T, Chaiteerakij R, Treeprasertsuk S, Klaikaew N, Tangkijvanich P, Komolmit P, Poovorawan Y. Prevalence of aflatoxin induced p53 mutation at codon 249 (R249s) in hepatocellular carcinoma patients with and without hepatitis B surface antigen (HBsAg) Asian Pac J Cancer Prev. 2013;14:7675–7679. doi: 10.7314/apjcp.2013.14.12.7675. [DOI] [PubMed] [Google Scholar]
- 53.Jiang W, Wang XW, Unger T, Forgues M, Kim JW, Hussain SP, Bowman E, Spillare EA, Lipsky MM, Meck JM, et al. Cooperation of tumor-derived HBx mutants and p53-249(ser) mutant in regulating cell proliferation, anchorage-independent growth and aneuploidy in a telomerase-immortalized normal human hepatocyte-derived cell line. Int J Cancer. 2010;127:1011–1020. doi: 10.1002/ijc.25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woo HG, Kim SS, Cho H, Kwon SM, Cho HJ, Ahn SJ, Park ES, Lee JS, Cho SW, Cheong JY. Profiling of exome mutations associated with progression of HBV-related hepatocellular carcinoma. PLoS One. 2014;9:e115152. doi: 10.1371/journal.pone.0115152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uren AG, Kool J, Berns A, van Lohuizen M. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656–7672. doi: 10.1038/sj.onc.1209043. [DOI] [PubMed] [Google Scholar]
- 57.Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 58.Dupuy AJ. Transposon-based screens for cancer gene discovery in mouse models. Semin Cancer Biol. 2010;20:261–268. doi: 10.1016/j.semcancer.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bell JB, Podetz-Pedersen KM, Aronovich EL, Belur LR, McIvor RS, Hackett PB. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc. 2007;2:3153–3165. doi: 10.1038/nprot.2007.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Keng VW, Yae K, Hayakawa T, Mizuno S, Uno Y, Yusa K, Kokubu C, Kinoshita T, Akagi K, Jenkins NA, et al. Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat Methods. 2005;2:763–769. doi: 10.1038/nmeth795. [DOI] [PubMed] [Google Scholar]
- 61.Dupuy AJ, Fritz S, Largaespada DA. Transposition and gene disruption in the male germline of the mouse. Genesis. 2001;30:82–88. doi: 10.1002/gene.1037. [DOI] [PubMed] [Google Scholar]
- 62.Luo G, Ivics Z, Izsvák Z, Bradley A. Chromosomal transposition of a Tc1/mariner-like element in mouse embryonic stem cells. Proc Natl Acad Sci USA. 1998;95:10769–10773. doi: 10.1073/pnas.95.18.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bard-Chapeau EA, Nguyen AT, Rust AG, Sayadi A, Lee P, Chua BQ, New LS, de Jong J, Ward JM, Chin CK, et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat Genet. 2014;46:24–32. doi: 10.1038/ng.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, Liu P, Largaespada DA, Scheetz TE, Jenkins NA, Copeland NG. A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res. 2009;69:8150–8156. doi: 10.1158/0008-5472.CAN-09-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keng VW, Sia D, Sarver AL, Tschida BR, Fan D, Alsinet C, Solé M, Lee WL, Kuka TP, Moriarity BS, et al. Sex bias occurrence of hepatocellular carcinoma in Poly7 molecular subclass is associated with EGFR. Hepatology. 2013;57:120–130. doi: 10.1002/hep.26004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keng VW, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I, Silverstein KA, Sarver A, Starr TK, Akagi K, et al. A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma. Nat Biotechnol. 2009;27:264–274. doi: 10.1038/nbt.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Donnell KA, Keng VW, York B, Reineke EL, Seo D, Fan D, Silverstein KA, Schrum CT, Xie WR, Mularoni L, et al. A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci USA. 2012;109:E1377–E1386. doi: 10.1073/pnas.1115433109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 69.Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, Gupta M, O’Sullivan MG, Matise I, Dupuy AJ, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–1750. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moriarity BS, Otto GM, Rahrmann EP, Rathe SK, Wolf NK, Weg MT, Manlove LA, LaRue RS, Temiz NA, Molyneux SD, et al. A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis. Nat Genet. 2015;47:615–624. doi: 10.1038/ng.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rahrmann EP, Collier LS, Knutson TP, Doyal ME, Kuslak SL, Green LE, Malinowski RL, Roethe L, Akagi K, Waknitz M, et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a Sleeping Beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009;69:4388–4397. doi: 10.1158/0008-5472.CAN-08-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rahrmann EP, Watson AL, Keng VW, Choi K, Moriarity BS, Beckmann DA, Wolf NK, Sarver A, Collins MH, Moertel CL, et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat Genet. 2013;45:756–766. doi: 10.1038/ng.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mann MB, Black MA, Jones DJ, Ward JM, Yew CC, Newberg JY, Dupuy AJ, Rust AG, Bosenberg MW, McMahon M, et al. Transposon mutagenesis identifies genetic drivers of Braf(V600E) melanoma. Nat Genet. 2015;47:486–495. doi: 10.1038/ng.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 75.Riordan JD, Keng VW, Tschida BR, Scheetz TE, Bell JB, Podetz-Pedersen KM, Moser CD, Copeland NG, Jenkins NA, Roberts LR, et al. Identification of rtl1, a retrotransposon-derived imprinted gene, as a novel driver of hepatocarcinogenesis. PLoS Genet. 2013;9:e1003441. doi: 10.1371/journal.pgen.1003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huo TI, Wang XW, Forgues M, Wu CG, Spillare EA, Giannini C, Brechot C, Harris CC. Hepatitis B virus X mutants derived from human hepatocellular carcinoma retain the ability to abrogate p53-induced apoptosis. Oncogene. 2001;20:3620–3628. doi: 10.1038/sj.onc.1204495. [DOI] [PubMed] [Google Scholar]
- 77.Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci USA. 1994;91:2230–2234. doi: 10.1073/pnas.91.6.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang XW, Gibson MK, Vermeulen W, Yeh H, Forrester K, Stürzbecher HW, Hoeijmakers JH, Harris CC. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995;55:6012–6016. [PubMed] [Google Scholar]
- 79.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wangensteen KJ, Wilber A, Keng VW, He Z, Matise I, Wangensteen L, Carson CM, Chen Y, Steer CJ, McIvor RS, et al. A facile method for somatic, lifelong manipulation of multiple genes in the mouse liver. Hepatology. 2008;47:1714–1724. doi: 10.1002/hep.22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suda T, Liu D. Hydrodynamic gene delivery: its principles and applications. Mol Ther. 2007;15:2063–2069. doi: 10.1038/sj.mt.6300314. [DOI] [PubMed] [Google Scholar]
- 83.Montini E, Held PK, Noll M, Morcinek N, Al-Dhalimy M, Finegold M, Yant SR, Kay MA, Grompe M. In vivo correction of murine tyrosinemia type I by DNA-mediated transposition. Mol Ther. 2002;6:759–769. doi: 10.1006/mthe.2002.0812. [DOI] [PubMed] [Google Scholar]
- 84.Grompe M, al-Dhalimy M, Finegold M, Ou CN, Burlingame T, Kennaway NG, Soriano P. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 1993;7:2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- 85.Li L, Shen H, Li A, Zhang Z, Wang B, Wang J, Zheng X, Wu J, Yang D, Lu M, et al. Inhibition of hepatitis B virus (HBV) gene expression and replication by HBx gene silencing in a hydrodynamic injection mouse model with a new clone of HBV genotype B. Virol J. 2013;10:214. doi: 10.1186/1743-422X-10-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peng XH, Ren XN, Chen LX, Shi BS, Xu CH, Fang Z, Liu X, Chen JL, Zhang XN, Hu YW, et al. High persistence rate of hepatitis B virus in a hydrodynamic injection-based transfection model in C3H/HeN mice. World J Gastroenterol. 2015;21:3527–3536. doi: 10.3748/wjg.v21.i12.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu J, Huang S, Zhao X, Chen M, Lin Y, Xia Y, Sun C, Yang X, Wang J, Guo Y, et al. Poly(I: C) treatment leads to interferon-dependent clearance of hepatitis B virus in a hydrodynamic injection mouse model. J Virol. 2014;88:10421–10431. doi: 10.1128/JVI.00996-14. [DOI] [PMC free article] [PubMed] [Google Scholar]