

Abstract
Western countries are seeing a constant decline in the incidence of Helicobacter pylori-associated gastritis, coupled with a rising epidemiological and clinical impact of autoimmune gastritis. This latter gastropathy is due to autoimmune aggression targeting parietal cells through a complex interaction of auto-antibodies against the parietal cell proton pump and intrinsic factor, and sensitized T cells. Given the specific target of this aggression, autoimmune gastritis is typically restricted to the gastric corpus-fundus mucosa. In advanced cases, the oxyntic epithelia are replaced by atrophic (and metaplastic) mucosa, creating the phenotypic background in which both gastric neuroendocrine tumors and (intestinal-type) adenocarcinomas may develop. Despite improvements in our understanding of the phenotypic changes or cascades occurring in this autoimmune setting, no reliable biomarkers are available for identifying patients at higher risk of developing a gastric neoplasm. The standardization of autoimmune gastritis histology reports and classifications in diagnostic practice is a prerequisite for implementing definitive secondary prevention strategies based on multidisciplinary diagnostic approaches integrating endoscopy, serology, histology and molecular profiling.
Keywords: Autoimmune gastritis, Metaplasia, Carcinoids, Operative link for gastritis assessment staging
Core tip: Autoimmune gastritis (AIG) is an emerging gastropathy with a significant epidemiological and clinical impact on Western populations. Despite a better understanding of the phenotypic changes or cascades occurring in this autoimmune setting, the etiopathogenic mechanisms behind the disease are still poorly understood, histology reporting is not standardized, and both the AIG-associated cancer risk and its secondary prevention strategies remain confusing.
INTRODUCTION
The clinical relationship between gastric inflammatory diseases and pernicious anemia was foreseen early in the last century. In 1935, in “Gastritis and its consequences”, Knud Faber wrote: “…many observations agree with the hypothesis of gastritis as the cause of the anacidity, which is generally found in patients with pernicious anaemia. All these problems that are associated with pernicious anaemia, and the other forms of anaemia we find in patients with anacidity, must for the moment be approached with a certain going. …It is not improbable that similar conditions as in pernicious anaemia are to be regarded as the cause of simple microcytic anacid anaemia…. In 1913, I mentioned a typical case of this disease (i.e., pernicious anemia) where the microscopical examination post mortem showed a diffuse follicular gastritis”[1]. In 1935, all that was known about the bacterium now called Helicobacter pylori (H. pylori) came from Bizzozero’s microscopic observations in the stomach of dogs[2], but it is safe to assume that most, if not all of Faber’s patients had H. pylori-associated gastritis.
In Western populations the declining incidence of infectious diseases coincides with an increasing prevalence of autoimmune conditions[3-5]. This situation also applies to the gastric setting, where the declining incidence/prevalence of H. pylori infection (in the West, at least) parallels a rising clinical impact of autoimmune gastritis (AIG)[6].
While the natural history of AIG is generally well defined, several crucial aspects of this condition remain unclear: the pathogenesis of autoimmune aggression is poorly understood, the clinical course of the disease in individual patients is difficult to predict, and no specific therapy is available[7].
Regarding the pathogenesis of the autoimmune response, two - not necessarily exclusive - models have been proposed. In one, considered most likely to be seen in areas where the prevalence of H. pylori infection is high, immune responses mounted against H. pylori antigens start cross-reacting with antigens that may be within the proton pump proteins or produced by the host’s gastric mucosa (the intrinsic factor). This leads to a cascade of cellular responses that damage and eventually destroy the oxyntic mucosa, which stops producing acid and thus becomes both functionally and morphologically atrophic. In the second model these same events would occur irrespective of any presence of H. pylori infection, as a “primary” or “pure” autoimmune disorder[8].
From the clinical point of view, in addition to an impaired absorption of vitamin B12, which eventually results in the clinical picture of pernicious anemia, AIG has been associated with an increased risk of two different neoplastic diseases: gastric neuroendocrine tumors (NETs; previously referred as gastric carcinoids), and adenocarcinoma[9].
This review focuses on how and when pathologists can contribute to a multidisciplinary approach to the diagnosis and clinical management of patients with AIG.
EPIDEMIOLOGY
Several studies suggest that AIG is underdiagnosed. Pernicious anemia - the most readily recognizable clinical sign of AIG - is only seen in advanced disease, and microcytic anemia (possibly an earlier sign of gastric autoimmunity) is frequently treated without thoroughly investigating its underlying cause[10].
AIG is more common in women and older people. In the general population, its prevalence has been estimated to vary between 2% and 5%, but - for the reasons outlined above - these estimates may be largely biased by the epidemiological context, comorbidities and patient selection[10]. AIG has traditionally been associated with Northern European descent, but recent studies do not support such a racial clustering. In the US, for instance, a higher prevalence of pernicious anemia has been observed among African American and Hispanic women, and an earlier age of onset has been reported in these ethnic groups too[6]. In short, the available data do not provide solid information on the incidence/prevalence of AIG in the general populations of different countries[11].
CLINICAL PRESENTATION
The clinical presentation of AIG is not associated with any specific gastrointestinal signs or symptoms[12]. In a recent study, Miceli et al[13] examined the clinical and pathological features leading to a diagnosis of AIG in 99 patients: the most common initial findings were hematological disorders (various forms of anemia accounted for 37% of cases), followed by a histology positive for gastritis (34%). In less than 10% of the patients, a clinical suspicion of AIG was aroused by the concomitant presence of other autoimmune diseases, celiac disease, neurological symptoms, or a positive family history.
Microcytic anemia, the expression of iron deficiency, is a common presenting sign and is caused by achlorhydria, which impairs iron absorption[13]. In a study by Hershko et al[14], up to 30% of patients with iron-deficiency anemia and no clinical evidence of blood loss were found to have AIG. Advanced disease is clinically more obvious, when progressive parietal cell loss results in severe cobalamin deficiency and ultimately leads to macrocytic-megaloblastic anemia coexisting with neurological symptoms and atrophic glossitis[15].
The association between AIG and other autoimmune diseases is well recognized. Several reports have highlighted a significant association with type I diabetes mellitus[16]. AIG may also coexist with polyglandular autoimmune (PGA) syndromes. Pernicious anemia occurs in 10% to 15% of patients with PGA type 1 syndrome (hypo-parathyroidism, Addison’s disease, diabetes mellitus, and mucocutaneous candidiasis), and in 15% of PGA type 3 patients (with diabetes mellitus, and autoimmune thyroid diseases)[17,18]. The most common association, however, is with autoimmune thyroiditis (“thyrogastric autoimmunity”): more than 50% of AIG patients have circulating anti-thyroperoxidase antibodies[10]. Significant associations have also been reported with vitiligo, alopecia, celiac disease, myasthenia gravis, and autoimmune hepatitis[17].
PATHOGENESIS
Autoimmune gastritis is the result of a complex interaction between host-related factors (genetic susceptibility) and environmental factors (both endogenous and exogenous: the so-called “exposome”[19]). The resulting immunological dysregulations involve sensitized T lymphocytes[20] and autoantibodies against the parietal cell proton pump, and intrinsic factor.
The molecular grounds for the pathogenesis of AIG, and particularly the initial events that precipitate the autoimmune response, have yet to be fully elucidated. The tissue damage results from an antibody-mediated destruction of the parietal cells due to a selective targeting of the H+/K+ ATPase proton pump. This non-self-limiting process also induces a progressive loss of zymogenic chief cells, possibly mediated by sensitized lymphocytes[21]. Eventually, the vanishing oxyntic mucosa cells are replaced by mucous cells and metaplastic glands (of both intestinal and pseudo-pyloric type). In the advanced stages of the disease, the mucosa of the gastric corpus is completely replaced by atrophic and metaplastic epithelium, with no oxyntic glands remaining, so acid production may be completely lacking.
Several studies have addressed the role of H. pylori infection in AIG pathogenesis, and there is solid evidence to support a mechanism of molecular mimicry between H. pylori antigens and the proton pump[22]. Epidemiological studies suggest that a significant number of AIG patients have had, or still have H. pylori infection, and anti-proton pump auto-antibodies have consistently been demonstrated in H. pylori-infected patients. Healthy individuals may have H+/K+ ATPase-autoreactive CD4+ve T cells that have escaped negative thymic selection. Given a particular HLA background (i.e., HLA-DR2/HLA-DR4 or HLADR4/HLADR5), H. pylori infection may abrogate the CD4+ve CD25+ve regulatory T cells that usually suppress the activation of autoantigen-presenting dendritic cells[23,24].
The Japanese scientific literature strongly supports the theory that AIG never occurs in the absence of a triggering H. pylori infection[25] (Sato, personal communication), but this conviction is at odds with the well-established evidence of AIG occurring in H. pylori-naïve patients too[8]. A study by Zhang et al[26] on 9684 patients (53% H. pylori-positive and 55% female) documented that an age-dependent prevalence of anti-parietal cell autoantibodies (APCAs) was only detectable among H. pylori-negative subjects. More importantly, the association observed between APCAs and atrophic gastritis was stronger among H. pylori-negative (OR = 11.3; 95%CI: 7-17) than among H. pylori-positive patients (OR = 2.6; 95%CI: 2-3).
DIAGNOSIS
Serological biopsy
The clinical value of combinations of serological tests for assessing the morphological and functional status of the gastric mucosa has been extensively addressed[6]. In AIG, the typical serological profile includes autoantibodies against intrinsic factor and parietal cells. APCA levels and hypergastrinemia correlate significantly with the inflammatory involvement of the oxyntic mucosa and serum levels of anti-intrinsic factor autoantibodies correlate with mucosal atrophy[21].
Loss of parietal cells results in significant functional changes. The higher gastric pH leads to both antral G cell hyperplasia and hypergastrinemia. The loss of chief and mucous neck cells from the oxyntic glands induces a progressive decrease in serum Pg I, while the levels of Pg II (sustained by the normal secretion of the unaffected antral glands) do not change significantly. This situation leads to a declining Pg I/Pg II ratio, which gives rise to the typical serological profile of gastric autoimmune disease (hypergastrinemia and gradually lower Pg I/Pg II ratios)[18-20]. Several studies have reported on the diagnostic usefulness of the Gastropanel test (Biohit Oy, Finland) in the assessment of AIG patients[27-29].
Histology
The gastric mucosa gradually reveals a spectrum of lesions ranging from minimal inflammatory disease to severe corpus-restricted atrophic gastritis[30]. Inflammation restricted to the oxyntic mucosa is generally considered the most reliable phenotypic marker of AIG, but such an unequivocal topographical picture is rare, particularly when concurrent H. pylori infection induces antral gastritis.
Long-term follow-up studies suggest that early AIG exhibits no distinctive gross changes. In the absence of H. pylori infection, the antral mucosa is essentially devoid of inflammation and the epithelium is either normal or shows foveolar hyperplasia (“reactive gastropathy”), probably related to the trophic effects of hypergastrinemia[31]. As the oxyntic atrophy progresses, the corpus and fundus mucosa becomes irregularly flattened. The uneven loss of oxyntic glands gives rise to a grossly pseudo-polypoid appearance, with lesions consisting of foci of spared oxyntic native mucosa amidst the atrophic areas. This macroscopic appearance emphasizes the need to obtain biopsies from both non-polypoid and polypoid mucosa during endoscopic sampling procedures.
Without wishing to oversimplify the matter, it may be useful for explanatory purposes to separate the distinctive histological features of the oxyntic mucosa in AIG into four basic, sometimes overlapping phases.
Firstly, the earliest phase features an uneven infiltration of plasma cells and lymphocytes involving the full thickness of the lamina propria, with a mainly top-down gradient. The lymphocytes may be organized in nodular or follicular structures. Eosinophils and rare neutrophils can occasionally be seen. Mononuclear cells may be found intimately connected with glandular epithelial cells (intra-epithelial mononuclear infiltrate). There may be a patchy destruction of oxyntic glands.
Secondly, in a later phase of the disease, lymphocytes and plasma cells form a dense infiltrate in the lamina propria, while atrophic glandular alterations become clearly visible. In the oxyntic tubules, the native population of parietal and chief cells alternates with a new phenotype of clear, mucosecreting epithelia, with the phenotype of antral glands (“oxyntic antralization”). This metaplastic process is known as pseudo-pyloric metaplasia, or (as more recently proposed) as spasmolytic polypeptide-expressing metaplasia (SPEM). The etiological specificity of these metaplastic cells is questioned. It has been suggested that they may be pathogenically linked to any longstanding oxyntic inflammation; and this interpretation is consistent with their occurrence in inflammatory gastric polyps and Crohn-associated gastritis. On the other hand, recent experimental studies suggest that SPEM may also result from toxic damage to parietal cells (caused, for example, by DMP-777, an elastase inhibitor that ablates parietal cells without eliciting an inflammatory response)[32]. It has been demonstrated that parietal cell disruption also results in the dysregulation of parietal cell-derived signaling molecules involved in the proper differentiation of zymogen-secreting chief cells[33]. Whatever its pathogenic pathway, this SPEM population is believed to derive either from the transdifferentiation of mature chief cells[34], or from progenitor cells in the neck of the oxyntic glands[32]. SPEM cells are characterized by newly-expressed immunohistochemical markers, such as HE4 and Tff2, neither of which are expressed in native oxyntic glands. On the other hand, the original oxyntic commitment of pseudopyloric metaplastic epithelia may be disclosed by their chimeric expression of Pg I, as documented by immunohistochemistry.
Thirdly, gastric gland intestinalization may develop in native oxyntic glands, or it may follow after an earlier pseudopyloric transformation. This last hypothesis relies on the immunohistochemical neo-expression of HE4 in both SPEM and intestinalized glands[35,36]. According to Goldenring[36], SPEM progression to intestinal metaplasia probably gives rise to a hyperproliferative state prone to genetic instability, potentially leading to gastric malignancy.
Lastly, advanced AIG is characterized by a marked reduction or complete absence of oxyntic glandular units (oxyntic mucosa atrophy), which are replaced by fibrosis of the lamina propria (“oxyntic mucosa desertification”), and pseudo-pyloric and intestinalized metaplastic glands. Other common features include hyperplasia of the muscularis mucosae, oxyntic pseudo-polyps, hyperplastic or inflammatory polyps, and enterochromaffin-like (ECL) cell hyperplasia. As the target of the autoimmune reaction gradually disappears, the inflammatory component becomes less prominent.
As expected, in cases of concurrent H. pylori infection, the antral mucosa may feature the classic spectrum of H. pylori-related lesions. In such cases, AIG can only be diagnosed on the strength of its specific serological profile (anti-parietal cell and anti-intrinsic factor autoantibodies)[6].
THE ELEMENTARY LESIONS OF AIG
Mucosal atrophy
Gastric mucosal atrophy, defined as the loss of “appropriate” glands, may occur in two different, usually coexisting forms. In one, the disappearing glandular units are replaced by fibrotic expansion of the lamina propria, resulting in a reduced glandular mass with no change in the native glandular phenotype. In the other form, native glands are replaced by metaplastic glands featuring a new commitment[37,38]. Metaplastic atrophy does not necessarily imply a numerical reduction in the number of the glandular units, but the metaplastic replacement of native glands ultimately results in a declining population of glandular structures “appropriate” to the compartment concerned. The progressive replacement of functionally-specialized native glands with non-functioning fibrotic tissue or metaplastic cells results in the loss of the specialized functions of the oxyntic mucosa, the most evident of which is acid production.
Within the oxyntic mucosa, there may be both pseudo-pyloric metaplasia and intestinal metaplasia (IM). A third variant, pancreatic acinic cell metaplasia, may also occur. Its detection in the oxyntic mucosa of gastritis patients “should inject a strong suspicion for an autoimmune pathogenesis”, but its clinical impact seems to be negligible[38].
IM is defined as the replacement of glandular or foveolar epithelium by intestinal-type epithelium. In AIG, IM arises in previously-antralized oxyntic glands (i.e., from pseudo-pyloric metaplasia). Based on its histological features on hematoxylin and eosin staining, IM has been divided into two main subtypes: small-intestinal and colonic. Histochemical methods [staining mucins with high-iron diamine (HID)] help to distinguish Type I (incomplete, or small-intestinal type) IM from Types II and III (complete, or colonic-type), depending on the acidity of the mucus and the morphology of the mucosecreting epithelia. Type III features sulfomucins in the columnar epithelium and is generally regarded as the IM subtype associated with the highest risk of neoplastic transformation[39]. Intestinalization of the gastric glands parallels the induction of the intestinal transcription factor Cdx2, which results in both MUC2 upregulation, and a decreased expression of MUC5AC[40].
Neuroendocrine cell hyperplasia
The gastric hypochlorhydia or achlorhydria associated with advanced AIG stimulates sustained gastrin secretion by the antral G cells, and the resulting hypergastrinemia triggers ECL cell proliferation. ECL cells are involved in the synthesis of the molecules responsible for histamine processing (histidine decarboxylase, and vesicular monoamine transporter type 2), and possibly for histamine secretion, which in turn stimulates acid secretion from adjacent parietal cells[6,12].
In advanced AIG, ECL cells may feature a broad spectrum of changes, ranging from “true” hyperplasia to endocrine neoplasia.
The earliest lesion is linear ECL cell hyperplasia, conventionally described as a set of five adjacent chromogranin-expressing ECL cells lining the glandular neck region. ECL cell hyperplasia is also known to develop from the hypergastrinemia caused by the long-term administration of proton pump inhibitors (PPIs), although the use of PPIs has not been associated with gastric neuroendocrine tumors (NETs)[41]. The term “micronodular hyperplasia” is applied to more advanced lesions that form clusters of neuroendocrine cells (not exceeding the diameter of a gastric gland, i.e., < 150 μm) surrounded by basement membrane. Five or more clusters of micronodules are defined as adenomatoid hyperplasia, a confusing term rarely used in practice[6].
ECL cell dysplasia refers to individual nodules (larger than 150 μm in diameter) with no evidence of basal membrane. Dysplastic nodules may progress to microinvasive tumors infiltrating the lamina propria, associated with perinodular fibroplasia. The factors that may contribute to neoplastic progression include mutations in the MEN1 or REG genes, which provide a negative feedback for gastrin secretion[42].
It has been demonstrated in a transgenic mouse model that, under certain conditions, progenitor cells of the oxyntic glands may give rise to a cell population characterized by the loss of native parietal cells marker (i.e., H/K-ATPase) expression, the neo-expression of neuroendocrine antigens (chromogranin A), and a phenotype on electron microscopy consistent with entero-endocrine cells[43]. These findings may point to a possible morphogenetic pathway for neuroendocrine tumors associated with AIG.
Neuroendocrine tumors
Micro endocrine tumors (i.e., micro carcinoids) are nodules of ECL cells (ranging from 0.5-5 mm in diameter) that escape detection at endoscopy. When the diameter of these neuroendocrine proliferations exceeds 5 mm, they become endoscopically detectable and are histologically categorized as NETs.
Gastric NETs are relatively rare, with an annual incidence between 0.4% and 2%. Amongst all NETs, the US SEER database has demonstrated a rising incidence of gastric NETs: from 2.2% between 1950 and 1969 to 6.0% between 2000 and 2007. They account for 0.6%-2% of all gastric polyps identified at endoscopy[44]. The rising incidence/prevalence of gastric NETs may be due to the increasing number of endoscopy procedures performed in AIG patients, and to a greater capacity to recognize these tumors.
Three different types of gastric NET have been described: (1) NETs arising in corpus-predominant atrophic gastritis (Type 1); (2) NETs associated with gastrinoma and MEN-1 syndrome (Type 2; due to inactivating mutations in the MEN1 and REG genes, which provide a negative feedback for gastrin secretion[15]); and (3) sporadic NETs (Type 3).
AIG-related NETs (i.e., Type 1 NETs) account for 70% to 80% of all gastric neoplasms deriving from ECL cells. Type I carcinoids are significantly associated with pernicious anemia: a 13-fold higher risk of AIG-related NETs emerged among 4000 Swedish patients with pernicious anemia[45].
These tumors are often multiple, small (< 1 cm in diameter), with the endoscopic appearance of a sessile polyp in the oxyntic mucosa. Clinically, they are rather indolent and functionally “mute”, and the prevalence of regional nodal metastases is less than 5%. These tumors are mostly restricted to the mucosa or submucosa and consist of well-differentiated monotonous cells, with a weak proliferative activity. Because of their peculiar phenotype/immuno-phenotype, their consistent expression of neuroendocrine markers (i.e., chromogranin, synaptophysin, and CD56), and their clinical-pathological setting, gastric NETs are usually easy to diagnose. Mib1 staining is needed to identify proliferating cells and establish the tumor’s grade (which can be done by using the Mib1 labeling index or the mitotic count)[46].
The overall survival rate for patients with gastric NETs is similar to that of the general population, i.e., 95% at 5 years and 74% at 10 years[46].
Although the WHO recommends avoiding the historically-used term “carcinoid”, which should only be applied to the clinical syndrome, well-differentiated gastric NETs are still commonly referred to as carcinoids.
Oxyntic pseudo-polyps
Against a background of atrophic AIG, the remnants of unaffected oxyntic mucosa may have the endoscopic features of pseudo-polypoid lesions. These areas are endoscopically similar to their intestinal counterparts in inflammatory bowel diseases. Histologically, they consist of oxyntic mucosa spared by the atrophic and metaplastic process, but often showing moderate chronic inflammation.
Hyperplastic polyps
Hyperplastic polyps are meta-inflammatory proliferations of the gastric foveolar cells. They are characterized by elongated, branching, and dilated hyperplastic foveolae lying in an edematous, hypervascular, inflammatory stroma. Superficial erosions are common, and result in chronic blood loss, further contributing to these patients’ iron-deficiency anemia. While hyperplastic polyps arising in patients with AIG are morphologically similar to those encountered in other types of gastritis, they more often tend to be proximally located and multiple.
Pyloric gland adenoma
Pyloric gland adenomas are relatively rare tumors, usually occurring in females and AIG patients[3]. Pyloric gland adenoma prevails mainly among AIG patients (51% of a German cohort of 373 patients, and 77% of a series of 189 patients in the United States). The most common site of the lesion was the oxyntic mucosa (54%), followed by the cardia region (17%) and gastric bulb (8%)[47].
Pyloric gland adenoma consists of closely-packed pyloric-type glands, lined with cubic or columnar mucus-secreting cells that express both MUC6 and concavalin A. Although the adenomatous cell population is bland, dysplasia may occur, and there have been reports of progression to pyloric-type gastric adenocarcinoma[48-50].
Gastric adenocarcinoma
Longstanding atrophic AIG is considered a precancerous condition[41]. In a Swedish cohort of 4000 patients with pernicious anemia, a 20-year follow-up documented a three-fold risk of gastric cancer (GC)[45]. The most important risk factors include clinical evidence of pernicious anemia, severity of atrophy, the presence of intestinal metaplasia, the duration of the disease, and age over 50 years[9].
In a T-cell receptor transgenic mouse model, Nguyen et al[51] recently demonstrated that an immunological attack against a peptide from the parietal cell antigen H/K ATPase may result in the whole spectrum of histological lesions associated with AIG. By 2-4 mo of age, these transgenic mice develop severe oxyntic atrophic/metaplastic lesions (including SPEM), higher levels of mRNA for cancer biomarkers (HE4, OLFM4, TFF2) and of phosphorylated STAT3, and high-grade intra-epithelial neoplasia.
Few studies have addressed the difference in the cancer risk between H. pylori-associated (i.e., “secondary”) AIG and “primary” AIG. This is not surprising, given the difficulty of reliably distinguishing between the two. While some studies suggested a higher cancer risk associated with corpus gastritis, they all contained significant confounding factors. For instance: (1) most of them involved patients with concomitant/previous H. pylori infection; (2) the atrophic/metaplastic lesions in the antrum and corpus mucosa were not scored separately; and (3) the prevalence of concomitant autoimmunity was not explored. The gastric cancer risk associated with “primary” as opposed to “secondary” AIG consequently remains unknown[52,53].
THE PATHOLOGIST’S ROLE IN ASSESSING AIG: THE DIAGNOSTIC IMPACT OF BIOPSY MAPPING
The definitive diagnosis of AIG results mainly from a combination of clinical findings (hematological profile, anti-parietal cell antibodies, serum gastrin), and histology (Figure 1). For cases with a presumptive diagnosis of AIG, the histological assessment demands a topographically defined biopsy sampling procedure that is uncommonly performed in most practices, where one or two antral specimens are often all that is taken. Pathologists are therefore rarely provided with an optimal set of samples for the purpose of even suggesting a diagnosis of AIG.
Figure 1.
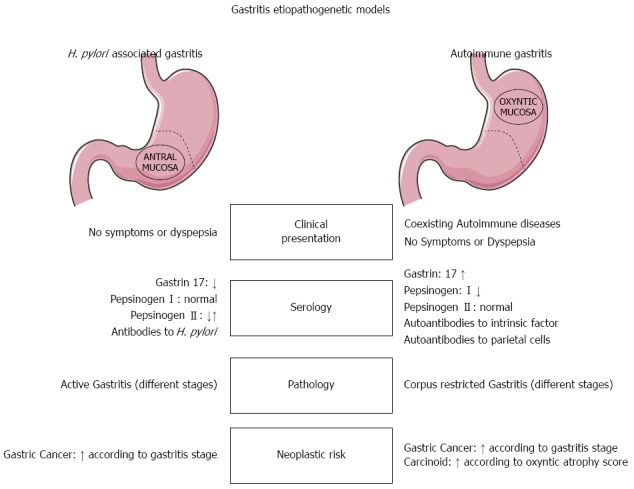
Helicobacter pylori-related vs autoimmune gastritis. Main differences in clinical presentation, serology (immunological and functional profiles), pathology, and neoplastic risk. H. pylori: Helicobacter pylori.
The assessment of a case relies on effective communications between gastroenterologist and pathologist. Whenever significant inflammation, atrophy, or metaplasia are detected in the oxyntic samples in a representative gastric biopsy set, and the antral mucosa only shows minimal or reactive changes, it is incumbent on the histopathologist to report a strong suspicion of an autoimmune component, even in the absence of other clinical information to support this impression.
Early AIG is essentially impossible to diagnose and even difficult to suspect. According to the Sydney system, the diagnosis of gastritis should be based on the separate assessment of at least three samples from the antrum (including the incisura angularis), and two from the gastric body; and any focal lesions should be biopsied too[54,55]. When such topographically well-defined samples are available, the presence of chronic gastritis with minimal or no active inflammation in the corpus, and a relatively normal antral mucosa in the absence of H. pylori infection should elicit the suspicion of an early autoimmune phenomenon.
A histological underestimation of AIG may stem from three (possibly concurrent) technical situations: (1) inconsistencies in the number of biopsy samples available (e.g., only 2 specimens, 1 of antral and the other of oxyntic mucosa); (2) inconsistent mapping of the gastric mucosa (e.g., four biopsy samples, all from the antrum); and (3) inappropriate sample submission procedures (e.g., biopsy samples obtained from different sites, but all submitted in the same vial). Metaplastic changes can affect the oxyntic mucosa (i.e., pseudo-pyloric metaplasia), so biopsy samples obtained from the corpus and fundus may be misinterpreted as coming from a naturally mucosecreting (antral) mucosa. Hence the need to submit two biopsy samples from the oxyntic mucosa, two from the antrum and one from the angularis mucosa, all in different vials. In exceptional cases, the immunohistochemical detection of the chimeric expression of Pg I, and the rarity/absence of G cells may nonetheless support the oxyntic origin of a biopsy sample[54,55].
A schematic representation of the main diagnostic scenarios occurring in AIG histopathology is shown in Figure 2.
Figure 2.
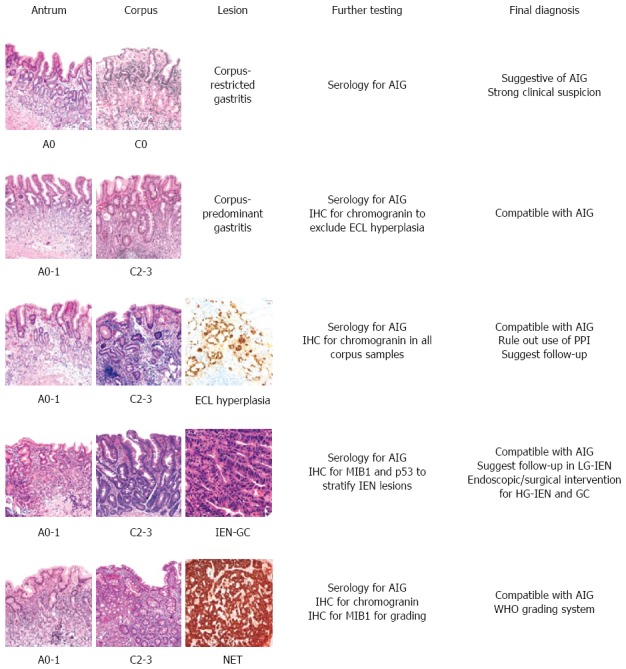
Schematic representation of the main diagnostic histological pictures seen in autoimmune gastritis. Metaplastic changes in the antrum and corpus are staged using the OLGA system. IHC: Immunohistochemistry; ECL: Enterochromaffin-like; IEN: Intraepithelial neoplasia; LG-IEN: Low-grade IEN; HG-IEN: High-grade IEN; GC: Gastric carcinoma; OLGA: Operative link for gastritis assessment; AIG: Autoimmune gastritis; PPI: Proton pump inhibitor.
THE PATHOLOGIST’S ROLE IN SECONDARY PREVENTION STRATEGIES
Both pernicious anemia and atrophic gastritis are considered precancerous conditions[56]: the real GC risk associated with AIG is still debated, and concomitant risk factors (particularly H. pylori infection) are believed to act as cancer co-promoters, significantly increasing the magnitude of the basic risk associated with a pure autoimmune etiology. Based on this rationale, AIG patients are eligible for GC secondary prevention strategies. No evidence-based protocols are currently available, however, for stratifying AIG patients in different risk classes.
The American Society for Gastrointestinal Endoscopy 2006 guidelines for endoscopy of premalignant conditions recommend a single endoscopic assessment to identify neoplastic lesions, but no routine follow-up surveillance[8,57].
The European MAPS (Management of Precancerous Conditions and Lesions in the Stomach) guidelines recommend a 3-yearly endoscopic and bioptic follow-up for all patients with “extensive atrophy or intestinal metaplasia”[58]. This recommendation is based on solid evidence of the risk of cancer developing in parallel with the extent of mucosal atrophy[37]. It is important to bear in mind, however, that the greater risk associated with concomitant H. pylori infection has not been separately weighted.
A more objective and consistent assessment of the extent and location of atrophy has been achieved by replacing the descriptive histology report with a staging approach. The operative link for gastritis assessment (OLGA) staging system ranks gastritis in different classes of cancer risk. As in H. pylori infection, so too in the autoimmune setting, OLGA stages III-IV have been significantly associated with a higher GC risk[36]. In a retrospective, single-institution study on a consecutive series of 562 AIG biopsy sets, low atrophy stages clearly prevailed (91.8%), and incidental gastric neoplasia (both intra-epithelial and invasive) only occurred in high-risk stages (III-IV) when atrophy involved the antral mucosa as well, and was associated with H. pylori infection[8]. As expected, when gastritis stage was matched with endocrine cell hyperplasia/neoplasia (linear and micronodular hyperplasia and Type I NETs) the vast majority of the hyperplastic/neoplastic cases were in the most advanced stages (III-IV) of gastritis[8].
Another gastritis staging system derived from OLGA, called OLGIM, relies on the histological scoring of IM alone. Because it excludes both non-metaplastic atrophy and pseudo-pyloric metaplasia from the atrophy score, the OLGIM disregards the atrophy phenotypes specifically occurring in AIG, and this results in a down-staging of AIG patients who would be considered at high risk if a global atrophy score were applied[8,59,60]. The OLGIM may therefore be less than optimal for assessing the risk of gastric cancer in AIG.
Despite the widespread availability of functional serological tests (Pg I, Pg II, and gastric 17, as stand-alone tests or integrated in a panel (Gastropanel, Biohit), serology is still notably absent from the international recommendations on the initial assessment and clinical monitoring of AIG. Pepsinogens and the pepsinogen ratio have been extensively validated as reliable markers of atrophy, and their low cost, accessibility, and high negative predictive value could be usefully exploited in the AIG setting. Pepsinogen I is consistently considered a surrogate for loss of zymogenic chief cells from the oxyntic mucosa, but large studies are now available that have correlated the organic with the functional disease in AIG. Low Pg I levels (particularly with a low Pg I/Pg II ratio) are consistently associated with oxyntic atrophy. Gastrin 17 is an elective marker of proton pump efficiency and is considered a reliable marker of oxyntic atrophy.
CONCLUSION
There are gaps in the information available on the epidemiology of gastric autoimmunity, and the incidence of AIG is probably underestimated. Secondary autoimmune gastric disease may be triggered by longstanding H. pylori infection, but “primary” autoimmune gastritis exists as a separate clinical entity as well. The clinical signs of AIG include a broad spectrum of non-hematological and hematological disorders, but megaloblastic anemia is mostly associated with advanced gastric disease.
From a clinical viewpoint, AIG can be diagnosed reliably by means of: (1) specific autoantibody assays (anti-parietal cells, and anti-intrinsic factor autoantibodies); (2) functional serology of the gastric mucosa (Pg I/Pg II ratio, gastrin 17); and (3) histology (applying a standard diagnostic biopsy sampling protocol).
There is clear evidence of a link between longstanding AIG and a spectrum of ECL cell changes ranging from hyperplastic to neoplastic lesions (gastric NETs).
The magnitude of the AIG-associated GC risk probably differs in “primary” vs H. pylori-associated, “secondary” autoimmune disease (and is presumably higher in the latter, but also in advanced “primary” AIG). Both gastric “functional serology” and histological staging are reliable in monitoring disease progression, and for stratifying patients in different prognostic classes.
No comprehensive molecular profiling is available for AIG, and no differential molecular profiling is capable of distinguishing primary from secondary autoimmune gastritis. In clinical practice, such information could make AIG assessment and prognostication more reliable, as well as supporting the rationale for biologically-targeted therapies.
Footnotes
Supported by A grant from the Italian Association for Cancer Research (partly, AIRC Regional grant 2008 No. 6421); and published under the auspices of the Healthy Stomach Initiative (HIS)
Conflict-of-interest statement: Dr. Graham is an unpaid consultant for Novartis in relation to vaccine development for treatment or prevention of H. pylori infection. Dr. Graham is a paid consultant for RedHill Biopharma regarding novel H. pylori therapies and has received research support for culture of H. pylori. He is a consultant for Otsuka Pharmaceuticals regarding diagnostic breath testing. Dr. Graham has received royalties from Baylor College of Medicine patents covering materials related to 13C-urea breath test.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 26, 2015
First decision: June 2, 2015
Article in press: September 2, 2015
P- Reviewer: Huang ZH S- Editor: Yu J L- Editor: A E- Editor: Liu XM
References
- 1.Faber K. Gastritis and Its Consequences. JAMA. 1935;105:2098. [Google Scholar]
- 2.Bizzozero G. "Ueber die schlauchförmigen Drüsen des Magendarmkanals und die Beziehungen ihres Epitheles zu dem Oberflächenepithel der Schleimhaut". Archiv für mikroskopische Anatomie. 1893;42:82–152. [Google Scholar]
- 3.Vojdani A. A Potential Link between Environmental Triggers and Autoimmunity. Autoimmune Dis. 2014;2014:437231. doi: 10.1155/2014/437231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamradt T, Göggel R, Erb KJ. Induction, exacerbation and inhibition of allergic and autoimmune diseases by infection. Trends Immunol. 2005;26:260–267. doi: 10.1016/j.it.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Kondrashova A, Seiskari T, Ilonen J, Knip M, Hyöty H. The ‘Hygiene hypothesis’ and the sharp gradient in the incidence of autoimmune and allergic diseases between Russian Karelia and Finland. APMIS. 2013;121:478–493. doi: 10.1111/apm.12023. [DOI] [PubMed] [Google Scholar]
- 6.Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis--pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. 2013;10:529–541. doi: 10.1038/nrgastro.2013.101. [DOI] [PubMed] [Google Scholar]
- 7.Baggett RT, Welsh JD. Observations on the effects of glucocorticoid administration in pernicious anemia. Am J Dig Dis. 1970;15:871–881. doi: 10.1007/BF02236052. [DOI] [PubMed] [Google Scholar]
- 8.Rugge M, Fassan M, Pizzi M, Zorzetto V, Maddalo G, Realdon S, De Bernard M, Betterle C, Cappellesso R, Pennelli G, et al. Autoimmune gastritis: histology phenotype and OLGA staging. Aliment Pharmacol Ther. 2012;35:1460–1466. doi: 10.1111/j.1365-2036.2012.05101.x. [DOI] [PubMed] [Google Scholar]
- 9.Vannella L, Lahner E, Annibale B. Risk for gastric neoplasias in patients with chronic atrophic gastritis: a critical reappraisal. World J Gastroenterol. 2012;18:1279–1285. doi: 10.3748/wjg.v18.i12.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massironi S, Cavalcoli F, Rossi RE, Conte D, Spampatti MP, Ciafardini C, Verga U, Beck-Peccoz P, Peracchi M. Chronic autoimmune atrophic gastritis associated with primary hyperparathyroidism: a transversal prospective study. Eur J Endocrinol. 2013;168:755–761. doi: 10.1530/EJE-12-1067. [DOI] [PubMed] [Google Scholar]
- 11.Carmel R. Prevalence of undiagnosed pernicious anemia in the elderly. Arch Intern Med. 1996;156:1097–1100. [PubMed] [Google Scholar]
- 12.Rugge M, Correa P, Dixon MF, Fiocca R, Hattori T, Lechago J, Leandro G, Price AB, Sipponen P, Solcia E, et al. Gastric mucosal atrophy: interobserver consistency using new criteria for classification and grading. Aliment Pharmacol Ther. 2002;16:1249–1259. doi: 10.1046/j.1365-2036.2002.01301.x. [DOI] [PubMed] [Google Scholar]
- 13.Miceli E, Lenti MV, Padula D, Luinetti O, Vattiato C, Monti CM, Di Stefano M, Corazza GR. Common features of patients with autoimmune atrophic gastritis. Clin Gastroenterol Hepatol. 2012;10:812–814. doi: 10.1016/j.cgh.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 14.Hershko C, Ronson A, Souroujon M, Maschler I, Heyd J, Patz J. Variable hematologic presentation of autoimmune gastritis: age-related progression from iron deficiency to cobalamin depletion. Blood. 2006;107:1673–1679. doi: 10.1182/blood-2005-09-3534. [DOI] [PubMed] [Google Scholar]
- 15.Vannella L, Lahner E, Osborn J, Annibale B. Systematic review: gastric cancer incidence in pernicious anaemia. Aliment Pharmacol Ther. 2013;37:375–382. doi: 10.1111/apt.12177. [DOI] [PubMed] [Google Scholar]
- 16.Dengue in the South Pacific. Wkly Epidemiol Rec. 1990;65:255–256. [PubMed] [Google Scholar]
- 17.Amerio P, Tracanna M, De Remigis P, Betterle C, Vianale L, Marra ME, Di Rollo D, Capizzi R, Feliciani C, Tulli A. Vitiligo associated with other autoimmune diseases: polyglandular autoimmune syndrome types 3B+C and 4. Clin Exp Dermatol. 2006;31:746–749. doi: 10.1111/j.1365-2230.2006.02171.x. [DOI] [PubMed] [Google Scholar]
- 18.Betterle C, Zanchetta R. Update on autoimmune polyendocrine syndromes (APS) Acta Biomed. 2003;74:9–33. [PubMed] [Google Scholar]
- 19.Bogdanos DP, Smyk DS, Invernizzi P, Rigopoulou EI, Blank M, Pouria S, Shoenfeld Y. Infectome: a platform to trace infectious triggers of autoimmunity. Autoimmun Rev. 2013;12:726–740. doi: 10.1016/j.autrev.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toh BH. Diagnosis and classification of autoimmune gastritis. Autoimmun Rev. 2014;13:459–462. doi: 10.1016/j.autrev.2014.01.048. [DOI] [PubMed] [Google Scholar]
- 21.Park JY, Lam-Himlin D, Vemulapalli R. Review of autoimmune metaplastic atrophic gastritis. Gastrointest Endosc. 2013;77:284–292. doi: 10.1016/j.gie.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 22.Bergman MP, Faller G, D'Elios MM, Del Prete G, Vandenbroucke-Grauls CMJE, Appelmelk BJ. Gastric automminity. In: Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington DC: ASM Press. 2001:chapter 36. [Google Scholar]
- 23.Bergman MP, D’Elios MM. Cytotoxic T cells in H. pylori-related gastric autoimmunity and gastric lymphoma. J Biomed Biotechnol. 2010;2010:104918. doi: 10.1155/2010/104918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach EM. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- 25.Yamada T, Miwa H, Fujino T, Hirai S, Yokoyama T, Sato N. Improvement of gastric atrophy after Helicobacter pylori eradication therapy. J Clin Gastroenterol. 2003;36:405–410. doi: 10.1097/00004836-200305000-00009. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Weck MN, Schöttker B, Rothenbacher D, Brenner H. Gastric parietal cell antibodies, Helicobacter pylori infection, and chronic atrophic gastritis: evidence from a large population-based study in Germany. Cancer Epidemiol Biomarkers Prev. 2013;22:821–826. doi: 10.1158/1055-9965.EPI-12-1343. [DOI] [PubMed] [Google Scholar]
- 27.Lombardo L, Leto R, Molinaro G, Migliardi M, Ravarino N, Rocca R, Torchio B. Prevalence of atrophic gastritis in dyspeptic patients in Piedmont. A survey using the GastroPanel test. Clin Chem Lab Med. 2010;48:1327–1332. doi: 10.1515/CCLM.2010.256. [DOI] [PubMed] [Google Scholar]
- 28.Telaranta-Keerie A, Kara R, Paloheimo L, Härkönen M, Sipponen P. Prevalence of undiagnosed advanced atrophic corpus gastritis in Finland: an observational study among 4,256 volunteers without specific complaints. Scand J Gastroenterol. 2010;45:1036–1041. doi: 10.3109/00365521.2010.487918. [DOI] [PubMed] [Google Scholar]
- 29.McNicholl AG, Forné M, Barrio J, De la Coba C, González B, Rivera R, Esteve M, Fernandez-Bañares F, Madrigal B, Gras-Miralles B, et al. Accuracy of GastroPanel for the diagnosis of atrophic gastritis. Eur J Gastroenterol Hepatol. 2014;26:941–948. doi: 10.1097/MEG.0000000000000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strickland RG, Mackay IR. A reappraisal of the nature and significance of chronic atrophic gastritis. Am J Dig Dis. 1973;18:426–440. doi: 10.1007/BF01071995. [DOI] [PubMed] [Google Scholar]
- 31.Torbenson M, Abraham SC, Boitnott J, Yardley JH, Wu TT. Autoimmune gastritis: distinct histological and immunohistochemical findings before complete loss of oxyntic glands. Mod Pathol. 2002;15:102–109. doi: 10.1038/modpathol.3880499. [DOI] [PubMed] [Google Scholar]
- 32.Weis VG, Goldenring JR. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer. 2009;12:189–197. doi: 10.1007/s10120-009-0527-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, Karam SM, Gordon JI. Diphtheria toxin-mediated ablation of parietal cells in the stomach of transgenic mice. J Biol Chem. 1996;271:3671–3676. [PubMed] [Google Scholar]
- 34.Nam KT, Lee HJ, Sousa JF, Weis VG, O’Neal RL, Finke PE, Romero-Gallo J, Shi G, Mills JC, Peek RM, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology. 2010;139:2028–2037.e9. doi: 10.1053/j.gastro.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sousa JF, Ham AJ, Whitwell C, Nam KT, Lee HJ, Yang HK, Kim WH, Zhang B, Li M, LaFleur B, et al. Proteomic profiling of paraffin-embedded samples identifies metaplasia-specific and early-stage gastric cancer biomarkers. Am J Pathol. 2012;181:1560–1572. doi: 10.1016/j.ajpath.2012.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldenring JR, Nam KT, Wang TC, Mills JC, Wright NA. Spasmolytic polypeptide-expressing metaplasia and intestinal metaplasia: time for reevaluation of metaplasias and the origins of gastric cancer. Gastroenterology. 2010;138:2207–2210, 2210.e1. doi: 10.1053/j.gastro.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jhala NC, Montemor M, Jhala D, Lu L, Talley L, Haber MM, Lechago J. Pancreatic acinar cell metaplasia in autoimmune gastritis. Arch Pathol Lab Med. 2003;127:854–857. doi: 10.5858/2003-127-854-PACMIA. [DOI] [PubMed] [Google Scholar]
- 38.Rugge M, Fassan M, Pizzi M, Pennelli G, Nitti D, Farinati F. Operative Link for Gastritis Assessment gastritis staging incorporates intestinal metaplasia subtyping. Hum Pathol. 2011;42:1539–1544. doi: 10.1016/j.humpath.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 39.Rugge M, Pennelli G, Pilozzi E, Fassan M, Ingravallo G, Russo VM, Di Mario F. Gastritis: the histology report. Dig Liver Dis. 2011;43 Suppl 4:S373–S384. doi: 10.1016/S1590-8658(11)60593-8. [DOI] [PubMed] [Google Scholar]
- 40.Rugge M, Fassan M, Graham DY. Clinical guidelines: Secondary prevention of gastric cancer. Nat Rev Gastroenterol Hepatol. 2012;9:128–129. doi: 10.1038/nrgastro.2012.19. [DOI] [PubMed] [Google Scholar]
- 41.Cockburn AN, Morgan CJ, Genta RM. Neuroendocrine proliferations of the stomach: a pragmatic approach for the perplexed pathologist. Adv Anat Pathol. 2013;20:148–157. doi: 10.1097/PAP.0b013e31828d185d. [DOI] [PubMed] [Google Scholar]
- 42.Basuroy R, Srirajaskanthan R, Prachalias A, Quaglia A, Ramage JK. Review article: the investigation and management of gastric neuroendocrine tumours. Aliment Pharmacol Ther. 2014;39:1071–1084. doi: 10.1111/apt.12698. [DOI] [PubMed] [Google Scholar]
- 43.Syder AJ, Karam SM, Mills JC, Ippolito JE, Ansari HR, Farook V, Gordon JI. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc Natl Acad Sci USA. 2004;101:4471–4476. doi: 10.1073/pnas.0307983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lahner E, Esposito G, Pilozzi E, Galli G, Corleto VD, Di Giulio E, Annibale B. Gastric cancer in patients with type I gastric carcinoids. Gastric Cancer. 2015;18:564–570. doi: 10.1007/s10120-014-0393-8. [DOI] [PubMed] [Google Scholar]
- 45.Hsing AW, Hansson LE, McLaughlin JK, Nyren O, Blot WJ, Ekbom A, Fraumeni JF. Pernicious anemia and subsequent cancer. A population-based cohort study. Cancer. 1993;71:745–750. doi: 10.1002/1097-0142(19930201)71:3<745::aid-cncr2820710316>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 46.Borch K, Ahrén B, Ahlman H, Falkmer S, Granérus G, Grimelius L. Gastric carcinoids: biologic behavior and prognosis after differentiated treatment in relation to type. Ann Surg. 2005;242:64–73. doi: 10.1097/01.sla.0000167862.52309.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vieth M, Montgomery EA. Some observations on pyloric gland adenoma: an uncommon and long ignored entity! J Clin Pathol. 2014;67:883–890. doi: 10.1136/jclinpath-2014-202553. [DOI] [PubMed] [Google Scholar]
- 48.Chen ZM, Scudiere JR, Abraham SC, Montgomery E. Pyloric gland adenoma: an entity distinct from gastric foveolar type adenoma. Am J Surg Pathol. 2009;33:186–193. doi: 10.1097/PAS.0b013e31817d7ff4. [DOI] [PubMed] [Google Scholar]
- 49.Arslan Pagnini C, Rugge M. Gastric cancer: problems in histological diagnosis. Histopathology. 1982;6:391–398. doi: 10.1111/j.1365-2559.1982.tb02736.x. [DOI] [PubMed] [Google Scholar]
- 50.Rugge M, Correa P, Dixon MF, Hattori T, Leandro G, Lewin K, Riddell RH, Sipponen P, Watanabe H. Gastric dysplasia: the Padova international classification. Am J Surg Pathol. 2000;24:167–176. doi: 10.1097/00000478-200002000-00001. [DOI] [PubMed] [Google Scholar]
- 51.Nguyen TL, Khurana SS, Bellone CJ, Capoccia BJ, Sagartz JE, Kesman RA, Mills JC, DiPaolo RJ. Autoimmune gastritis mediated by CD4+ T cells promotes the development of gastric cancer. Cancer Res. 2013;73:2117–2126. doi: 10.1158/0008-5472.CAN-12-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Norcross JW, Monroe SE, Griffin BG. The development of gastric carcinoma in pernicious anemia. Ann Intern Med. 1952;37:338–343. doi: 10.7326/0003-4819-37-2-338. [DOI] [PubMed] [Google Scholar]
- 53.Vannella L, Lahner E, Osborn J, Bordi C, Miglione M, Delle Fave G, Annibale B. Risk factors for progression to gastric neoplastic lesions in patients with atrophic gastritis. Aliment Pharmacol Ther. 2010;31:1042–1050. doi: 10.1111/j.1365-2036.2010.04268.x. [DOI] [PubMed] [Google Scholar]
- 54.Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20:1161–1181. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- 55.Price AB. The Sydney System: histological division. J Gastroenterol Hepatol. 1991;6:209–222. doi: 10.1111/j.1440-1746.1991.tb01468.x. [DOI] [PubMed] [Google Scholar]
- 56.Morson BC, Sobin LH, Grundmann E, Johansen A, Nagayo T, Serck-Hanssen A. Precancerous conditions and epithelial dysplasia in the stomach. J Clin Pathol. 1980;33:711–721. doi: 10.1136/jcp.33.8.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirota WK, Zuckerman MJ, Adler DG, Davila RE, Egan J, Leighton JA, Qureshi WA, Rajan E, Fanelli R, Wheeler-Harbaugh J, et al. ASGE guideline: the role of endoscopy in the surveillance of premalignant conditions of the upper GI tract. Gastrointest Endosc. 2006;63:570–580. doi: 10.1016/j.gie.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Dinis-Ribeiro M, Areia M, de Vries AC, Marcos-Pinto R, Monteiro-Soares M, O’Connor A, Pereira C, Pimentel-Nunes P, Correia R, Ensari A, et al. Management of precancerous conditions and lesions in the stomach (MAPS): guideline from the European Society of Gastrointestinal Endoscopy (ESGE), European Helicobacter Study Group (EHSG), European Society of Pathology (ESP), and the Sociedade Portuguesa de Endoscopia Digestiva (SPED) Virchows Arch. 2012;460:19–46. doi: 10.1007/s00428-011-1177-8. [DOI] [PubMed] [Google Scholar]
- 59.Rugge M, Capelle LG, Cappellesso R, Nitti D, Kuipers EJ. Precancerous lesions in the stomach: from biology to clinical patient management. Best Pract Res Clin Gastroenterol. 2013;27:205–223. doi: 10.1016/j.bpg.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 60.Rugge M, Capelle LG, Fassan M. Individual risk stratification of gastric cancer: evolving concepts and their impact on clinical practice. Best Pract Res Clin Gastroenterol. 2014;28:1043–1053. doi: 10.1016/j.bpg.2014.09.002. [DOI] [PubMed] [Google Scholar]