

Abstract
Reactivation of the human polyomavirus JC (JCV) in the CNS results in a fatal demyelinating disease, progressive multifocal leukoencephalopathy (PML). The lytic destruction of oligodendrocytes, which occurs at the terminal stage of the viral infection cycle, is considered a critical factor in the development of demyelination and the pathogenesis of PML. However, knowledge is limited about interaction of JCV with oligodendrocytes and its impact on the denudation of axons at the early stage of viral reactivation and prior to the destruction of the infected cells. We have developed an in vitro neuroprogenitor cell culture using human fetal brain that can be differentiated to the oligodendrocyte lineage to investigate interactions of JCV with its host cells. Results show that infection with JCV delays oligodendrocyte maturation as shown by reduced levels of oligodendrocytic markers, including myelin basic protein, proteolipid protein, and platelet-derived growth factor receptor-α. Furthermore, replication of JCV in these cells caused substantial dysregulation of several chemokines, including CCL5/RANTES, GRO, CXCL1/GROα, CXCL16, CXCL8/IL-8, CXCL5/ENA-78, and CXCL10/IP-10, all of which play a role in cell growth and differentiation.
Keywords: neural progenitor, polyomavirus, progressive multifocal leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML), which is caused by the human polyomavirus JC (JCV), remains a fatal disease despite recent advances. JCV infection occurs during early childhood, and then the virus enters a state of latency, during which JCV DNA can be detected but viral proteins cannot (White and Khalili, 2011). Latent virus has been found in several different tissues, including kidney, lymphoid tissue, bone marrow, and brain of healthy and immunosuppressed individuals without PML (for reviews see Berger, 2010; Major, 2010; White and Khalili, 2011). Reactivation of the latent virus, which can occur in patients with impaired immune function, including HIV-1/AIDS, lymphoproliferative disorders, malignancies, and treatment with immunosuppressive dugs, results in the destruction of infected oligodendrocytes in the CNS and the onset of PML (Carson et al., 2009; Clifford et al., 2010; Mateen et al., 2011; Tavazzi et al., 2012).
In PML, demyelination results from the damage of oligodendrocytes by replicating JCV (Del Valle and Piña-Oviedo, 2006; Khalili et al., 2006; Moll et al., 2008). Oligodendrocytes are seen with enlarged nuclei that contain inclusion bodies consisting of crystalline arrays of JCV particles, and virions have also been demonstrated among lamellae of the myelin sheath of viable axons (Mázló et al., 2001).
Because reactivation of JCV occurs mainly in immunocompromised individuals, it is thought that the immune system controls viral latency, especially cellular immune repsonses (Tan and Koralnik, 2010; Gheuens et al., 2011). The chemokine system is a critical part of immune serveillance. Chemokines (chemoattractant cytokines) regulate many important biological processes, including cell adhesion, proliferation, apoptosis, angiogenesis, phagocytosis, and cellular response to viral replication. Chemokines are expressed constitutively in the brain and are implicated in the brain physiology, migration of neuronal progenitor cells in the developing brain, and glial proliferation (Cartier et al., 2005). Although chemokine-induced immune responses can act to eliminate pathogens, they may also be responsible for neuronal damage and are involved in the pathogenesis of a number of diseases of the CNS that are associated with inflammation and neurodegeneration diseases (Bajetto et al., 2002; Miller et al., 2008). The occurence of PML in patients receiving therapies that target leukocyte trafficking into inflamed tissue (for reviews see Berger, 2010; Major, 2010; Carson et al., 2009, Clifford, 2010, 2011) suggests a role for inflammatory chemokines in JCV reactivation and PML progression. A correlation between cytokine expression in HIV-1/AIDS patients and the development of PML was previously suggested (Marzocchetti et al., 2005). Furthermore, a link between cytokine/chemokine gene transcription and JCV infection has been proposed (Manley et al., 2007), and there is evidence that proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) activate JCV gene expression and are present in PML lesions (Wollebo et al., 2011). Thus, it is of interest to explore the mechanisms whereby JCV infection affects chemokine balance and intercellular interactions in the brain.
The mature oligodendrocyte, the cell type that provides myelination and trophic support to neurons (Nave, 2010; Piaton et al., 2010), is the primary target for JCV infection. Oligodendrocytes modify axonal structure via myelination, influence the formation of nodes of Ranvier, control axon extension, and preserve axonal integrity (Dupree et al., 2004; Nave and Trapp, 2008, Rasband et al., 2001).
Studies of the effect of JCV on oligodendrocyte function have been limited by the difficulty of preparing primary oligodendrocyte cultures. Previously, CNS progenitor cells prepared from human fetal brain have been reported to be able to be infected by JCV (Messam et al., 2003). Here, we describe the preparation of primary oligodendrocytic progenitors and immature oligodendrocytes from human fetal brain and their infection by JCV. We found that JCV infection perturbed the differentiation of these cells and expression of chemokines. The importance of these findings in advancing our understanding the pathogenesis of PML is discussed.
MATERIALS AND METHODS
Derivation, Expansion, and Maintenance of Primary Human Embryonic Neural Progenitor Cells in Chemically Defined Media
Cultures of human embryonic neural progenitor cells (hNPC) were prepared from human fetal brain tissue (embryonic age 16 weeks) based on protocols of Espinosa-Jeffrey at al. (2009), with slight modifications. hNPC were isolated from the cerebrum of human fetal brain (16 weeks of gestation) obtained from Advanced Bioscience Resources (ABR), Inc., in accordance with NIH guidelines. After removal of meninges and dissociation of brain tissue in the presence of Tryple Express (Gibco, Grand Island, NY; catalog No. 12605) and DNase I (10 U/ml; Sigma, St. Louis, MO), cells were triturated through a fire-polished Pasteur pipette and plated onto nontissue-culture-grade Petri dishes coated with anti-PSA-NCAM antibody (Developmental Studies Hybridoma Bank, University of Iowa [DSHB]; 5A5) in neural stem cell medium (NSCM: Neurobasal medium; Invitrogen, Carlsbad, CA; catalog No. 10888-022), N2 supplement (Invitrogen; catalog No. 17502048), B27 supplement without vitamin A (Invitrogen; catalog No. 12587-010), GlutaMAX (Invitrogen), 10 ng/ml basic fibroblast growth factor (bFGF; Invitrogen), 10 ng/ml recombinant human epidermal growth factor (EGF; Invitrogen), penicillin/streptomycin, and fungizone. Cells were incubated overnight at 37°C with 5% CO2. Unattached cells were cultured either as two- or as three-dimensional cultures of “neurospheres.” Neurospheres, which grow at a slower pace than attached cells, serve as a renewable source of neural stem cells. Unattached cells were dissociated on the next day and resuspended in 5 ml of their own medium and in 5 ml NSCM. For propagation of hNPC, floating neurospheres were fed with fresh NSCM and one-third conditioned NSCM every other day supplemented with 10 ng/ml human bFGF and 10 ng/ml recombinant human EGF. Neurosphere cultures were grown in flasks and were dissociated routinely to keep the spheres uniformly small (approximately 2 mm in diameter).
Preparation of Anti-PSA-NCAM-Coated Dishes
The surface of 100-mm nontissue-culture-grade Petri dishes was coated with 4 ml of the immunopanning cocktail (50 mM Tris-Cl, pH 9.5, 1% BSA [w/v]/50 μg/ml anti-PSA-NCAM antibody). Cocktail was removed after 40 min, and Petri dishes were washed three times with PBS, then once with 5 ml PBS/1% BSA before use.
Differentiation of hNSCs Into the Oligodendrocyte Lineage
Like hNPC, human oligodendrocyte progenitors (hOPC) can be propagated in two- and three- dimensional cultures (oligospheres). As with hNPC, attached hOPC grow faster. Neurospheres were dissociated and plated onto poly-D-lysine (100 ng/ml final; Sigma)-coated dishes at a density of approximately 1.5 × 106 per 60-mm dish in a mixture of 1 part self-conditioned NSCM and 1 part of an oligodendrocyte specification medium (OSM) containing 1 part NSCM and 1 part glia defined medium with growth factors (GDM+; DMEM/F12 high glucose, N2 supplement, GlutaMAX, 20 ng/ml bFGF, 20 ng/ml EGF, 20 ng/ml PDGF-AA, and penicillin/streptomycin). The transition of hNPC to commit to the OL lineage is achieved by gradual switching of the culture medium to GDM+. To enhance maturation of OL, cells were cultured in GDM without bFGF, EGF, and PDGF-AA supplementation for 9 days.
Infection With JCV
Cells were infected with JCV Mad-1 strain at MOI 10. Quantitative polymerase chain reaction (qPCR) for viral genome copy number was used to determine virus titer. Standards for qPCR were prepared using serial dilutions of pBluescript IIKS + JCV Mad1 plasmid. Primers and probe were generated for VP1 gene from JCV Mad1 strain: JVP-f 5′-AGTTG ATGGGCAGCCTATGTA-3′, JVP-r 5′-TCATGTCTGGGT CCCCTGGA-3′, JCVVPprobe 5′-/5HEX/CATGGATGCT CAAGTAGAGGAGGTTAGAGTTT/3BHQ_1/-3′. qPCR was performed with a LightCycler480 machine (Roche, Indianapolis, IN) using TaqMan Fast Universal PCR Master Mix (23; Applied Biosystems, Foster City, CA). The PCR conditions were activation 95°C 100 sec, PCR 45 cycles: 95°C 15 sec, 60°C 60 sec, cool to 40°C 40 sec. For JCV infection, viral stock with a concentration 1 × 106 viral genome copies/μl was used. Cells were incubated with virus in a minimal volume of Optimem medium for 1 hr (1 ml/60-mm dish/300 cells/7.5 μl viral stock), and then fresh medium was added (3 ml) for overnight incubation. After 24 hr, cells were washed three times in 13 PBS. Medium was changed every 3 days.
Preparation of Protein Extracts and Immunoblot Analysis
For preparation of whole-cell protein, cells were lysed for 30 min on ice in TNN buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP40) containing protease inhibitor cocktail (Sigma; catalog No. P8340) and 0.2 mM Na-orthovana-date. Cell debris was removed by centrifugation at 14,000 rpm for 15 min at 4°C. The supernatant was assayed for protein content by Bradford analysis (Bio-Rad, Hercules, CA) and was either used immediately or stored at −80°C. For immunoblots, 50 μg total cell protein was run on SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with antibody. Bound antibody was detected using an enhanced chemiluminescence (ECL) detection kit (Amersham, Arlington Heights, IL) according to the manufacturer’s recommendations.
RNA Preparation and qRT-PCR
Total RNA was isolated using the RNeasy kit (Qiagen, Valencia, CA) with on-column DNA digestion. The RT reaction was performed with 1 μg total RNA, oligo(dT)-12–18 primer (P/N 58862; Invitrogen), and M-MuLV RT enzyme as described previously (Merabova et al., 2012). cDNA samples were diluted 5×, and PCR was performed using Roche Applied Sciences Sybr Green mix (catalog No. 04707516001) on a LightCycler480 machine (Roche). PCR conditions were activation 95°C 5 min, PCR 45 cycles: 95°C 10 sec, 60°C 20 sec, 72°C 30 sec, melting curve (95–65°C), cool to 40°C 30 sec. The following primers were used: ENA-78/CXCL5 S 5′-CTGTGTTGAGAGAGCTGCGTTGC-3′, AS 5′-GTTTTC CTTGTTTCCACCGTCC-3′; GCP-2/CXCL6, S 5′-GATT GGTAAACTGCAGGTGTTCC-3′, AS 5′-TCCGG GTCC AGACAAACTTGCTTCCC-3′; IL-8/CXCL8, S 5′-ATTTC TGCAGCTCTGTGTGAA-3′, AS 5′-TGAATTCTCAGCCC TCTTCAA-3′; GROα/CXCL1, S 5′-ATGGCCCGCGCTG CTCTCTCC-3′, AS 5′-GTTGGATTTGTCACTGTTCAG-3′; CXCR2, S 5′-GCTCTAGAGCTGGGCAACAA TACAG CAAACT-3′, AS 5′-CCATCGATGGGCACTTAGGCAG-GAGGTCTTA-3′; PDGFRA, S 5′-TCAGCT ACAGATGG CTTGATCC-3′, AS 5′-GCCAAAGTCACAGATCTTCA-CAAT-3′; Olig1 S 5′-CTAAAATAGGTAACCAGGCGTC TCA-3′, AS 5′-CCCGGTACTCCTGCGTGTT-3′; Olig2 S 5′-TGCGCAAGCTTTCCAAGAT-3′, AS 5′-CAGCGAGTT GGTGAGCATGA-3′; Nkx2.2 S 5′-TCTACGACAGCAGC-GACAAC-3′, AS 5′-CTTGGAGCTTGAGTCCTGAG-3′; PLP1, S 5′-AGTCAGGCAGATCTTTGGCG-3′, AS 5′-GAC ACACCCGCTCCAAAGAA-3′; MBP, S 5′-ACTATCTCT TCCTCCCAGCTTAAAAA-3′, AS 5′-TCCGACTATAAAT CGGCTCACA-3′, β-actin, S 5′-CTACAATGAGCTGCGT GTGGC-3′, AS 5′-CAGGTCCAGACGCAGGATGGC-3′, Nanog, S 5′-ATGCCTCACACGGAGACTGT-3′, AS 5′-AAGTGGGTTGTTTGCCTTTG-3′, OCT4B, S 5′-GTTAG GTGGGCAGCTTGGAA-3′, AS 5′-TGTGGCCCCAAGGA ATAGTC-3′; and 5HT2AR, S 5′-GCCCTGCTCAATGT GTTTGTT-3′, AS 5′-GCCAAAGCCGGTATTGTGT-T-3′. For relative quantification, the expression level of genes was normalized to the housekeeping gene β-actin. Results were in arbitrary units.
Antibodies
Rabbit polyclonal antibodies against JCV agnoprotein R7903 (rabbit polyclonal) and VP1 pAB 597 (mouse) were previously described (Darbinyan et al., 2007). SV40 T-antigen Ab-2 PAb416 (mouse) DP02 was obtained from Calbiochem (La Jolla, CA). Antigalactocerebroside (catalog No. MAB342) and anti-A2B5 monoclonal antibody clone A2B5-105 (catalog No. MAB312R) were purchased from EMD Millipore (Bedford, MA). Neuronal class III β-tubulin (TUJ1) monoclonal antibody, Alexa Fluor-labeled (catalog No. A488-435L), was obtained from Covance (Berkeley, CA). Anti-GFAP(C19) goat polyclonal IgG (catalog No. sc-6170) and anti-GAPDH (6C5, sc-32233) antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antinestin antibody (catalog No. 611658) was from BD Pharmingen (San Jose, CA).
Immunocytochemistry
hNPC or hOPC were seeded on poly-D-lysine (0.1 mg/ml final concentration)-coated glass chamber slides. Cells were fixed with 4% paraformaldehyde at designated time points according to the experimental design and permeabilized for 5 min in 0.25% Triton X-100/1× PBS solution. Fixed cells were blocked with 5% normal horse, or goat, or rabbit serum (depending on type of primary antibody) in 0.1% PBS-BSA for 1 hr following incubation with specific antibodies. For immunolabeling, control cells were processed without primary antibody. Cells were then washed three times with PBS-0.01% Triton X-100 and incubated with FITC-conjugated anti-mouse, or anti-goat, or anti-rabbit secondary antibody for 1 hr. Slides were washed three times with PBS and blocked a second time with 5% normal horse, or goat, or rabbit serum (depending on type of second primary antibody) for 1 hr following incubation with second set of specific antibodies. After 16 hr, cells were washed, incubated with rhodamine-conjugated secondary antibodies, and mounted using mounting medium for fluorescence from Vectashield (Vector, Burlingame, CA; catalog No. H-1400). Nuclear DNA was labeled with DAPI, and slides were examined via fluorescence microscopy.
Methylthiazoletetrazolium Assay
The Cell Proliferation Kit I (methylthiazoletetrazolium; MTT) was used according to the manufacturer’s protocol (Roche). Cells were plated onto poly-D-lysine-coated six-well plates in triplicate in two sets and induced to differentiate into OL lineage. After 2 days in GDM, they were either infected with JCV or treated with conditioned medium (CM) from JCV-infected hOPCs after JCV depletion from CM by immunoprecipitation with anti-VP1 antibody or normal mouse serum. Control cells were left uninfected or were treated with CM from uninfected cultures after immunoprecipitation as described above. At designated time points, MTT (5 mg/ml) was added to the wells (final concentration 0.5 mg/ml) for 4 hr, and the reaction was stopped by addition of solubilization solution. The tetrazolium salt MTT is cleaved to form a formazan dye by mitochondrial reductase enzymes that are active only in viable cells and not in dead cells. The amount of formazan generated is directly proportional to the number of metabolically active cells. The spectrophotometric absorbance of cells under each condition, which correlates with cell viability, was measured using a microplate (enzyme-linked immunosorbent assay) reader at 570 nm with a reference wavelength of 650 nm.
Chemokine Array Assay
Levels of chemokines in CM from cells were analyzed using RayBio Human Chemokine Antibody Array 1 according to the manufacturer’s protocol (RayBiotech, Norcross, GA).
RESULTS
Preparation of hNPC and Their Differentiation Into hOPC and hOL
hNPC were prepared from human fetal brain tissue (hfB; embryonic age 16 weeks) based on protocols of Espinosa-Jeffrey at al. (2009), as described in Materials and Methods (Fig. 1A). After dissociation of brain tissue, cells were plated onto nontissue-culture-grade Petri dishes coated with anti-PSA-NCAM antibody. Unattached cells were cultured in neural stem cell medium (NSCM) either as two- or as three-dimensional hNPC cultures. The stage of differentiation of these cultures was verified by immunolabeling of hNPC plated on slides with nestin, glial fibrillary acid protein (GFAP), A2B5, and βIII-tubulin (Fig. 1B). Only 11% were positive for the neuronal marker βIII-tubulin. About 67% and 17% of cells were positive for neural stem cell markers nestin and A2B5, respectively, and 19–20% of the cell population was expressing GFAP, a protein commonly associated with astrocytes, although a large body of evidence indicates that postnatal and adult hNPC express GFAP both in vivo (Doetsch et al., 1999; Garcia et al., 2004) and in vitro (Laywell et al., 2000; Imura et al., 2003). Nuclei were stained with DAPI. As shown schematically in Figure 1A, hNPC were gradually switched to glia defined medium with growth factors (GDM+) to induce OL lineage progression (Fig. 1C). To obtain hOL specification and enhance maturation of hOL, cells were cultured in GDM without growth factors (GDM) for 9 days (Fig. 1D). The hOPC arise from the neural precursors with a characteristic bipolar shape and undergo sequential morphological changes during the course of differentiation as they acquire features specific to OL. Each developmental stage of OL is characterized by the presence of specific markers. Neural stem cells express NKX2.2. PSA-NCAM, nestin, and Pax6. hOPCs, while continuing to express Nkx2.2, become positive for Olig1, Olig2, NG2, A2B5, CD3, Sox9, and PDGFRα. Immature OL express O4, O1, and 2′,3′-cyclic nucleotide 3′phosphohydrolase (CNP), followed by galactocerebroside (GalC). Mature OL in culture are characterized by a highly developed network of cytoplasmic extensions and expression of the myelin-specific proteins proteolipid protein (PLP), myelin basic protein (MBP), myelin-associated glycoprotein (MAG), and myelin oligodendrocyte glycoprotein (MOG). After 2 days in GDM, the hOPC progressively express the markers A2B5 and GalC while extending numerous processes during differentiation. Immunolabeling of hOPC (Fig. 1C) showed that expression of nestin was reduced to 29%, whereas expression of A2B5-positive cells increased to 31–37%. The number of GFAP-expressing cells also was decreased, making up 9–11% of the cell population. In hOL cultures (Fig. 1D), the number of hOPC, which have developed into immature oliodendrocytes, as apparent from their increased morphological complexity and GalC expression, increased significantly and reached 65%, while A2B5 expression was reduced to 18%. The number of cells positive for βIII-tubulin and GFAP remained low in cultures of hOPC and hOL.
Fig. 1.
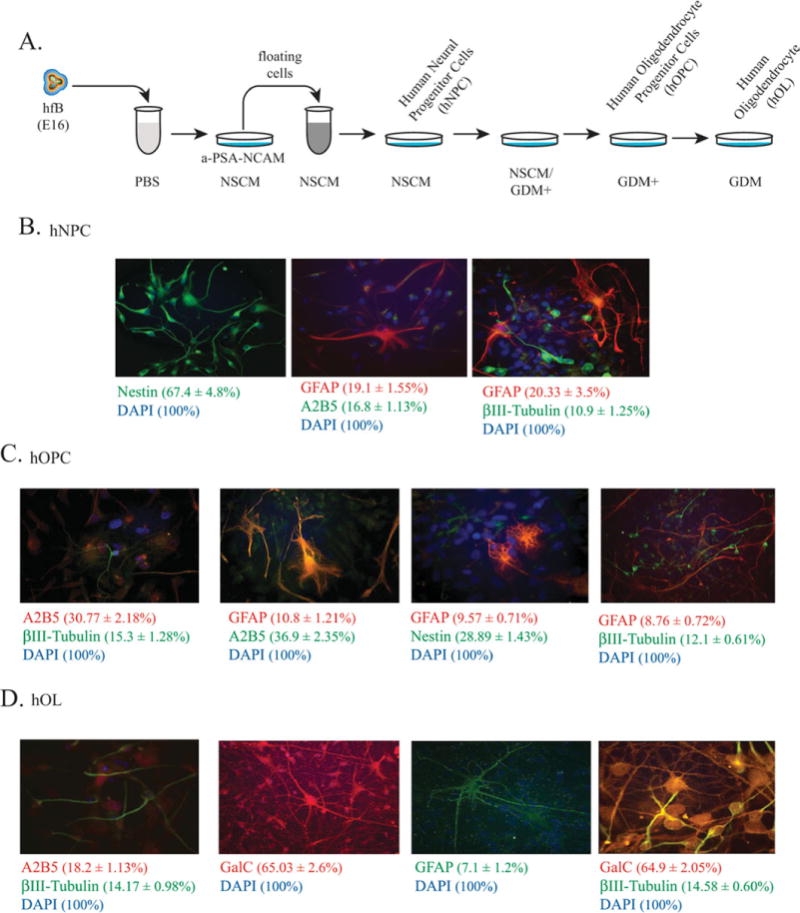
Preparation of human neural progenitor cells (hNPC) and their differentiation into human oligodendrocyte progenitor cells (hOPC) and human oligodendrocytes (hOL). A: Schematic representation of the preparation of hNPC from human fetal brain tissue (hfB, embryonic age 16 weeks) based on protocols of Espinosa-Jeffrey at al. (2009), with slight modifications. After dissociation of brain tissue, cells were plated onto nontissue-culture-grade Petri dishes coated with anti-PSA-NCAM antibody. Unattached cells were cultured in neural stem cell medium (NSCM) either as two- or as three-dimensional hNPC cultures. The hNPC were then gradually switched mixed with glial defined medium plus growth factors (GDM+) to differentiate them into hOPC and then to glial defined medium minus growth factors (GDM) to differentiate them into hOL. B: The hNPC stage of cultures was ascertained by immunolabeling of hNPC plated on slides with antibodies specific to nestin, A2B5, βIII-tubulin, and GFAP. Nuclei were stained with DAPI. C: Immunolabeling of hOPC for expression of nestin, A2B5, βIII-tubulin, and GFAP. Nuclei were stained with DAPI. D: Immunolabeling of hOL cultures for expression of GalC, A2B5, βIII-tubulin, and GFAP. Nuclei were stained with DAPI. Average percentages of cells expressing specific markers were quantified in at least 15 fields of view (~700–800 cells total). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Next, we analyzed the expression of cytokines/chemokines in hNPC and during differentiation to the OL lineage by assaying CM with a chemokine array. As can be seen in Table I, the levels of several cytokines/chemokines, including CXCL16, CCL2/MCP-1, CCL26/eotaxin-3, and CCL27/CTACK, were significantly increased, whereas others, including CXCL1/GROα, GRO, CCL17/TARC, and CCL3/MIP-1α, showed significant decreases. Because there is some heterogeneity of cellular population, it is difficult to determine unambiguously the role of particular cell types in chemokine expression. Nevertheless, the differentiated cultures were predominantly GalC positive.
TABLE I.
Differential Expression of Cytokines in Human Neuroprogenitor Cell (hNPC) and During Oligodendrocyte Lineage Progression*
| Cytokine/chemokine | hNPC | hOPC | hOL(early) | hOL(late) |
|---|---|---|---|---|
| CCL1/I-309 | 4.0±0.7 | 10.0±0.17 | 11.2±0.09 | 12.9±0.35 |
| CCL2/MCP-1 | 6.0±0.006 | 109.3±3.3 | 83.7±0.2 | 73.7±0.5 |
| CCL3/MIP-1α | 9.0±0.4 | 4.8±1.6 | 3.1±0.1 | 3.3±1.37 |
| CCL4/MIP-1β | 10.0±0.48 | 20.6±1.0 | 22.7±0.86 | 22.1±3.6 |
| CCL5/RANTES | 12.0±2.0 | 11.5±2.7 | 17.2±0.6 | 12.7±1.77 |
| CCL13/MCP-4 | 7.0±0.7 | 0.0 | 0.3±0.67 | 0.1±0.2 |
| CCL17/TARC | 12.0±0.9 | 5.7±1.6 | 5.2±0.67 | 2.7±0.65 |
| CCL22/MDC | 8.0±4.4 | 7.5±0.06 | 5.3±0.5 | 4.7±0.4 |
| CCL26/eotaxin-3 | 1.0±1.3 | 10.4±1.6 | 9.9±2.2 | 6.0±1.2 |
| CCL27/CTACK | 1.9±0.5 | 13.6±0.8 | 16.1±2.1 | 15.1±1.8 |
| GRO | 102.3±0.8 | 66.2±6.06 | 62.5±0.78 | 18.1±1.89 |
| CXCL1/GROα | 49.3±2.9 | 4.8±1.8 | 2.0±0.7 | 1.0±0.2 |
| CXCL5/ENA-78 | 9.9±1.6 | 7.3±1.7 | 8.2±0.5 | 5.5±1.4 |
| CXCL6/GCP-2 | 3.0±1.8 | 7.7±1.8 | 8.5±1.0 | 6.1±2.7 |
| CXCL7/NAP2 | 11.0±3.78 | 18.2±0.99 | 21.8±0.97 | 19.6±3.0 |
| CXCL8/IL-8 | 18.6±0.7 | 14.7±1.3 | 15.2±1.6 | 12.1±0.5 |
| CXCL10/IP-10 | 12.3±2.2 | 13.8±2.78 | 25.3±2.0 | 14.2±3.0 |
| CXCL16 | 0.0 | 14.5±1.9 | 16.9±1.2 | 8.0±1.8 |
| XCL1/lymphoactin | 5.0±0.17 | 10.4±1.2 | 7.0±1.6 | 7.2±1.6 |
| CX3CL1/fractalkine | 2.0±2.29 | 4.5±0.9 | 4.03±1.4 | 2.5±1.25 |
The intensities of signals corresponding to each cytokine/chemokine were analyzed in NIH Image, normalized to background and positive controls, and presented as optic density units.
Productive JCV Infection Can Occur in Undifferentiated hNPC
hNPC were plated onto poly-D-lysine-coated dishes and slides in two sets and were cultured in NSCM. After 2 days, one set of cells was infected with the Mad-1 strain of JC virus, and the second set served as an uninfected control. After 24 hr, the culture medium was removed, and fresh NSCM was added. Cells were cultured in NSCM for 7 days, and then the CM was collected, total protein lysates were prepared, and cells on slides were fixed. As shown in Figure 2A, infection of hNPC was detected by immunocytochemistry for VP1; by Western blot for JCV T-antigen, VP1, and agnoprotein; and by detection of a viral titer of 1.1 ± 0.43 × 103 viral copies/μl in CM by qPCR.
Fig. 2.
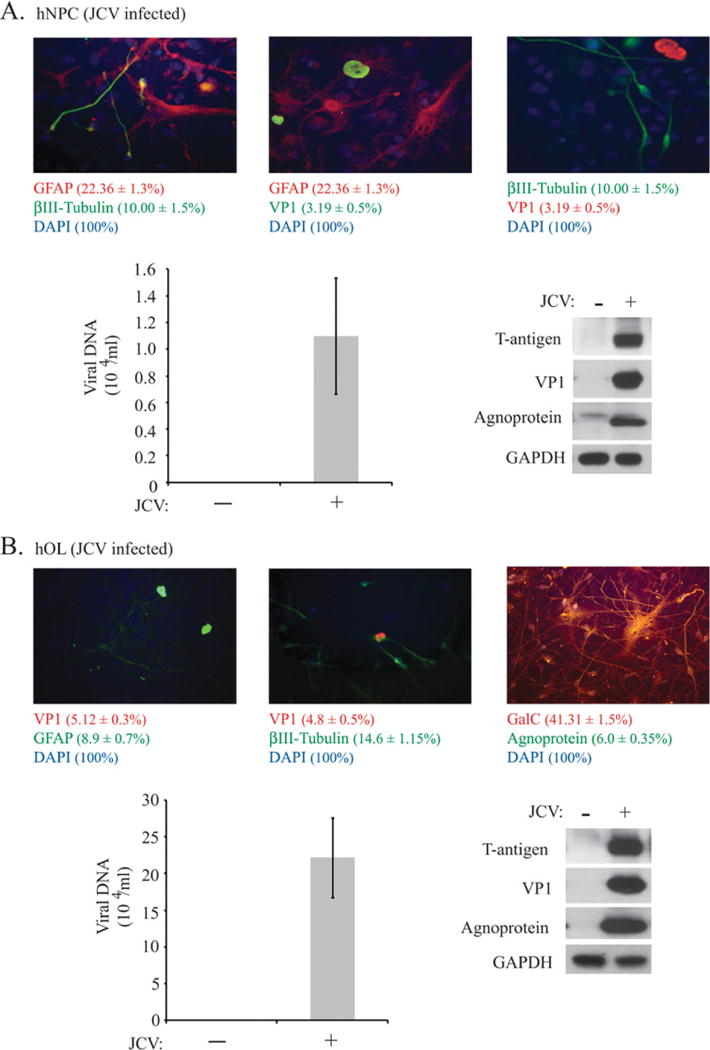
JCV infection of undifferentiated hNPC and hNPC induced to differentiate into hOL. A: Cultures of hNPC on poly-D-lysinecoated dishes and slides in two sets were left uninfected or were infected with the Mad-1 strain of JC virus for 7 days, and CM was collected, total protein lysates were prepared, and cells on slides were fixed. Immunocytochemistry was performed for βIII-tubulin, GFAP, GalC, and the viral proteins VP1 and agnoprotein. Expression of JCV T-antigen, VP1, and agnoprotein was analyzed by Western blot assay. Viral DNA content in CM was determined by qPCR. B: Cultures of hNPC were grown in NSCM for 3 days, and then differentiation of hNPC into the OL lineage was initiated. Cells were kept in GDM+ with growth factors for 4 days, and then culture medium was switched to the GDM without growth factors. Two days later, cells were uninfected or infected with the Mad-1 strain of JC virus. At day 7 of JCV infection (9 days in GDM without growth factors), CM was collected, protein lysates were prepared, and cells on slides were fixed. Immunocytochemistry was performed for βIII-tubulin, GFAP, GalC, and the viral proteins VP1 and agnoprotein. Expression of JCV T-antigen, VP1, and agnoprotein was analyzed by Western blot assay. Viral DNA content in CM was determined by qPCR. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Productive JCV Infection Can Occur in hNPC Induced To Differentiate Into OL
In the next set of experiments, hNPC were plated onto poly-D-lysine-coated dishes and slides in two sets and cultured in NSCM. After 3 days, the differentiation of hNPC into the OL lineage was initiated: cells in both sets were kept in GDM+ with growth factors for 4 days, and then culture medium was switched to the GDM without growth factors. Two days later, cells in one set were infected with the Mad-1 strain of JC virus, and the second set was left uninfected. After 24 hr, the culture medium was removed, and cells were cultured in GDM without growth factors for another 6 days. At day 7 of JCV infection (9 days in GDM without growth factors), CM was collected, protein lysates and total RNA were prepared, and cells on slides were fixed. As shown in Figure 2B, infection of hOL was detected by immunocytochemistry for VP1, by Western blot for JCV T antigen, VP1, and agnoprotein; and by detection of a viral titer of 2.2 ± 0.54 × 104 viral copies/μl in the CM by qPCR. The increased number of VP1-positive hOL compared with hNPC in the previous experiment (5% vs. 3%) and the higher viral titer for hOL indicate that hOL support viral infection better than hNPC. We also observed a decreased number of GalC-positive cells in JCV-infected hOL cultures compared with uninfected (42% vs. 65% respectively).
JCV Infection Interferes With the Expression of Oligodendroglial Lineage-Specific Markers in hNPC Undergoing OL Lineage Progression
hNPC were induced to differentiate into the oligodendroglial lineage and infected or uninfected with JCV as described in the previous section. At 7 days following infection (9 days in GDM), RNA was isolated, and the levels of mRNAs for oligodendroglial lineage markers were measured by qRT-PCR (Fig. 3). All values in the histograms were normalized to the housekeeping gene β-actin and compared with the expression levels of mRNAs in hOPC cultures that were neither induced to differentiate nor infected with JCV (1.0). All values are shown as fold change. The expression of the oligodendroglial lineage markers was increased in cells induced to differentiate, but this was reduced by JCV infection. For example, PDGFRα was increased about sixfold by differentiation but was reduced to 0.3-fold in cells that had undergone differentiation but were infected by JCV (Fig. 3A). Similarly, MBP was increased almost tenfold by differentiation but only 1.5-fold in cells that had undergone differentiation but were infected by JCV (Fig. 3B). It was also of interest to study dedifferentiation by measuring expression of hOPC markers. We found that expression of Oct4B and Nanog is reduced on differentiation but shows increased expression upon JCV infection (Fig. 3C). Expression of A2B5 (a cell surface carbohydrate antigen) was also decreased by differentiation and increased by JCV infection as measured by immunocytochemistry (Fig. 3D).
Fig. 3.
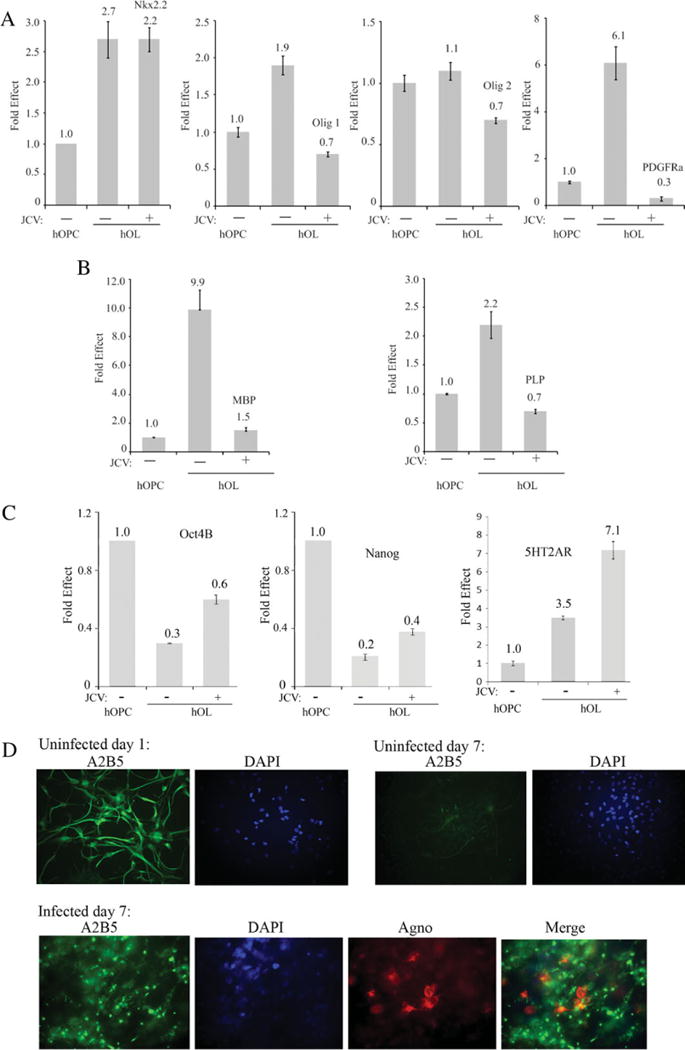
Effect of JCV infection on the expression of OL lineage-specific markers in hNPC undergoing OL lineage progression. hNPC were induced to differentiate into the oligodendroglial lineage and infected or uninfected with JCV as described in the legend to Figure 2. Seven days following infection (9 days in GDM), RNA was isolated, and the levels of mRNAs for the oligodendroglial lineage markers Nkx2.2, Olig1, Olig2, and PDGFRα (A); MBP and PLP (B); and Oct4B, Nanog, and 5HT2AR (C) were measured by qRT-PCR. All values in the histograms are normalized relative to hOPC cultures that were neither induced to differentiate nor infected with JCV (1.0). Finally, expression of A2B5 was examined in these cells by immunocytochemistry (D). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
JCV Infection Increases the Growth and/or Survival of hNPC Undergoing OL Lineage Progression
hNPC were induced to differentiate into the oligodendroglial lineage and infected or uninfected with JCV as described above. At 7 days following infection (9 days in GDM), the number of viable cells was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Viable cells with active mitochondria cleave the tetrazolium ring of MTT into a visible dark blue formazan reaction product, which was quantified by spectrophotometry as absorbance and correlates with cell viability. As shown in Figure 4A (left), JCV infection increased the number of viable cells by 25–30% (absorbance 0.36 ± 0.004 vs. 0.27 ± 0.004). Next, we collected the CM from these cultures and after depletion of viral particles by immunoprecipitation with VP1 antibody (data not shown). We used the CM to treat hOPC that were undergoing OL lineage progression. As shown in Figure 4A (right), CM from JCV-infected cells caused a nearly 30% increase in the number of viable cells compared with CM from uninfected cells (absorbance 0.44 ± 0.005 vs. 0.32 ± 0.001).
Fig. 4.
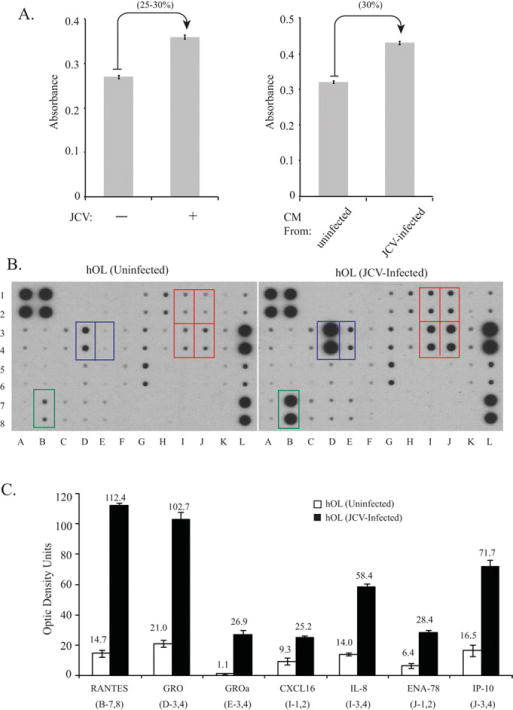
Effect of JCV infection on growth/survival and secretion of specific cytokines by hNPC undergoing OL lineage progression. hNPC were induced to differentiate into the oligodendroglial lineage and infected or uninfected with JCV as described in the legend to Figure 2. A: Seven days following infection (9 days in GDM), the number of viable cells was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide (MTT) assay. CM from JCV-infected and uninfected hOL was subjected to immunoprecipitation with anti-VP1 antibody to deplete viral particles and for treatment of differentiating hOPC. Cell viability was measured by MTT assay (right). B: A chemokine array assay was performed using CM from the JCV-infected or uninfected cells. Chemokines that were significantly upregulated by JCV infection are indicated by colored boxes. C: Expression of chemokines that were significantly upregulated by JCV infection in the chemokine array assay was quantitated by densitometry: CCL5/RANTES, GRO, CXCL1/GROα, CXCL16, CXCL8/IL-8, CXCL5/ENA-78, and CXCL10/IP-10. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
JCV Infection Increases the Secretion of Specific Cytokines/Chemokines by hNPC Undergoing OL Lineage Progression
In light of the data from the CM, we hypothesized that differential release of soluble factors may be involved in the regulation of OL growth and survival. We next performed a chemokine array assay using CM from the JCV-infected or uninfected cells (Fig. 4B). We found that seven cytokines/chemokines (marked by colored boxes) were significantly upregulated by JCV infection. These were RANTES, GRO, GROα, CXCL16, IL-8, ENA-78, and IP-10 (Fig. 4C).
JCV Infection Augments the Expression of mRNAs for Specific Cytokines/Chemokines in hNPC Undergoing OL Lineage Progression
Because the CM from the JCV-infected hOL showed increased levels of specific cytokines compared with CM from uninfected cells, we next examined cytokine mRNA expression levels in these hOL cultures by qRT-PCR using the same RNA preparations that were used to evaluate expression of the oligodendroglial lineage-specific markers described above. We found increased mRNA levels for ENA78, GCP-2, GROα, and IL-8 (Fig. 5). Interestingly, the CXC chemokine receptor CXCR2 was also upregulated.
Fig. 5.
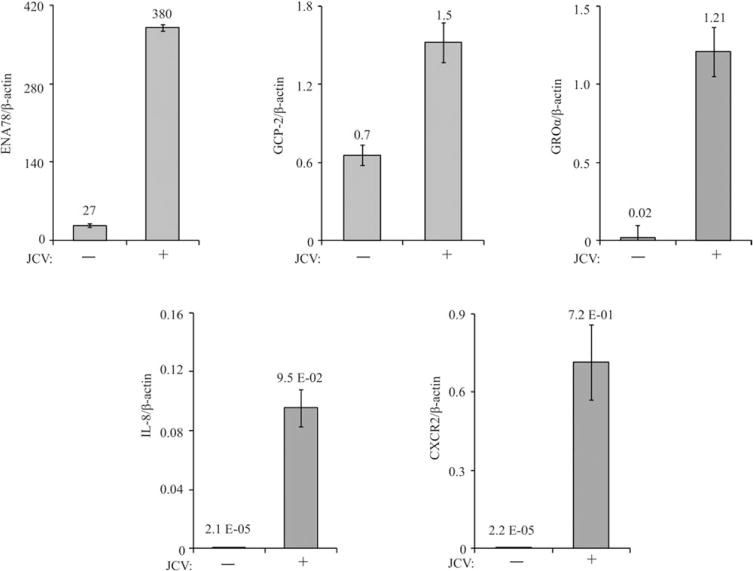
Effect of JCV infection on the expression of specific cytokines/chemokines in hNPC undergoing OL lineage progression. Chemokine mRNA expression levels in the hOL cultures were measured by qRT-PCR using the same RNA preparations that were analyzed for Figure 3. The levels of mRNAs for CXCL1/GROα, CXCL8/IL-8, CXCL5/ENA-78, CXCL6/GCP-2, and the CXC cytokine receptor CXCR2 are shown. ENA78, GCP-2, GROα, and IL-8 cytokines are shown, along with the mRNA for the CXC cytokine receptor CXCR2.
DISCUSSION
Development of new therapeutic strategies aimed at the treatment of PML requires advancement in the of understanding complex pathogenetic mechanisms underlying JCV-induced damage in CNS, which involve not only the cytolytic destruction of JCV-infected oligodendrocytes but also dysfunction of neighboring cells, including astrocytes, microglial cells, and neurons. Existing in vitro models of JCV infection are limited to transformed astrocytic cell lines and primary human fetal astrocytes (Radhakrishnan et al., 2003, 2004) and thus cannot provide a complete platform for understanding the complexity of cellular interactions in the JCV-infected brain. In particular, studies have been limited by the lack of a model for JCV infection of the OL, a cell type central to the pathogenesis of PML. Here we report the preparation of primary hNPC from fetal brain and their differentiation into hOPC and hOL by manipulation of the growth medium and that these cell types support productive infection by JCV. We believe that this is a novel approach to the study of the interactions of JCV with cells of the oligodendroglial lineage, because the differentiated cultures are predominately GalC positive. However, it should be noted that these cultures are not totally pure, so the possibility of occurrence of effects of other cell types cannot be excluded. For example, the low numbers of βIII-tubulin- and GFAP-positive cells indicate that some neurons and astrocytes are present, which may influence effects such as those with regard to chemokines.
Our data demonstrate that susceptibility to JCV infection is a property of hNPC but increases as cells differentiate toward OL. Our results show that OL at a late stage of differentiation can also be efficiently infected with JCV (unpublished data). We have found that JCV infection of hOPC delays progression of the oligodendroglial lineage, suggesting that JCV induces a block of OPC differentiation, which is significant in that it may play a crucial role in failure of remyelination in PML. Thus the dysregulation events that occur early in JCV infection may be important in pathogenesis long before the OL undergoes cytolytic destruction.
Another interesting observation is that the JCV-infected cultures show enhanced viability, suggesting perhaps that the deficit in differentiation is associated with an increased capacity to proliferate. It is possible that secretion of these chemokines also has pathological effects on other cells. For example, we previously reported that changes in differentiation and chemokine secretion by rat OL that express JCV agnoprotein are associated with activation of apoptotic signaling in the differentiation into OL of rat O2A cells and neurons (Merabova et al., 2008, 2012).
Endogenous remyelination, partial or complete, has been observed in multiple sclerosis (MS; Lassman, 1983; Raine and Wu, 1993; Waxman 1996; Patrikios et al., 2006; Bramow et al., 2010), and numerous studies have shown that the adult brain retains the ability to generate oligodendroglial progenitors (Zhang et al., 1999; Menn et al., 2006; Alvarez-Buylla et al., 2008). Repair of demyelinated lesions in the CNS relies on the availability of OPC and their potential to differentiate into mature OL. Recent studies have reported that chemokines are involved in the development of the OL lineage and myelination. There is evidence that the chemokine GROa regulates the proliferation of OL precursor cells (Robinson et al., 1998; Wu et al., 2000; Robinson and Franic, 2001). A critical role of CXCR2 receptor in OL development, which is a receptor for a number of CXC ligands, including GRO and IL-8, was demonstrated in CXCR2 knockout studies (Tsai et al., 2002). Animals with disruption of the CXCR2 gene exhibited abnormal positioning of OL precursors in the developing spinal cord. It has also been reported that GROα and IL-8 stimulate MBP synthesis in primary myelinating cultures and proliferation of A2B5 precursor-like cell line (Kadi et al., 2006). Several studies have reported increased expression of a large number of chemokines in CNS demyelinating diseases, including MS (Ransohoff and Trebst, 2002; Bartosik-Psujek and Stelmasiak, 2005). It was also shown that inhibition of CXCR2 signaling can enhance myelin repair in models of MS (Kerstetter et al., 2009). It is now evident that chemokines and their receptors play important roles not only in recruitment of inflammatory cells and the initiation of immune-mediated demyelination but also in remyelination and repair of lesions (Arnett et al., 2003; Omari et al., 2005a,b; Liu et al., 2010a,b).
In conclusion, the development of a primary cell culture model for cells of the oligodendroglial lineage has revealed new facets of JCV infection that may contribute to the pathogenesis of PML. Understanding the mechanisms of failure of remyelination is important for developing strategies to promote myelin repair and prevent axonal injury.
Acknowledgments
We thank past and present members of the Department of Neuroscience and Center for Neurovirology for their continued support, insightful discussions, and sharing of reagents and ideas. We also thank C. Papaleo for editorial assistance. The authors declare that there is no conflict of interest, finacial or otherwise, concerning the studies described in this article.
Contract grant sponsor: NIH; Contract grant number: R01 NS35000 (to K.K.).
References
- Alvarez-Buylla A, Kohwi M, Nguyen TM, Merkle FT. The heterogeneity of adult neural stem cells and the emerging complexity of their niche. Cold Spring Harbor Symp Quant Biol. 2008;73:357–365. doi: 10.1101/sqb.2008.73.019. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Wang Y, Matsushima GK, Suzuki K, Ting JP. Functional genomic analysis of remyelination reveals importance of inflammation in oligodendrocyte regeneration. J Neurosci. 2003;23:9824–9832. doi: 10.1523/JNEUROSCI.23-30-09824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem. 2002;82:1311–1329. doi: 10.1046/j.1471-4159.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- Bartosik-Psujek H, Stelmasiak Z. The levels of chemokines CXCL8, CCL2 and CCL5 in multiple sclerosis patients are linked to the activity of the disease. Eur J Neurol. 2005;12:49–54. doi: 10.1111/j.1468-1331.2004.00951.x. [DOI] [PubMed] [Google Scholar]
- Berger JR. Progressive multifocal leukoencephalopathy and newer biological agents. Drug Saf. 2010;33:969–983. doi: 10.2165/11537510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Bramow S, Frischer JM, Lassmann H, Koch-Henriksen N, Lucchinetti CF, Sørensen PS, Laursen H. Demyelination vs. remyelination in progressive multiple sclerosis. Brain. 2010;133:2983–2998. doi: 10.1093/brain/awq250. [DOI] [PubMed] [Google Scholar]
- Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, Bennett CL. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Clifford DB, De Luca A, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol. 2010;9:438–446. doi: 10.1016/S1474-4422(10)70028-4. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Ances B, Costello C, Rosen-Schmidt S, Andersson M, Parks D, Perry A, Yerra R, Schmidt R, Alvarez E, Tyler KL. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol. 2011;68:1156–1164. doi: 10.1001/archneurol.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbinyan A, White MK, Akan S, Radhakrishnan S, Del Valle L, Amini S, Khalili K. Alterations of DNA damage repair pathways resulting from JCV infection. Virology. 2007;364:73–86. doi: 10.1016/j.virol.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Valle L, Piña-Oviedo S. HIV disorders of the brain: pathology and pathogenesis. Front Biosci. 2006;11:718–732. doi: 10.2741/1830. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Mason JL, Marcus JR, Stull M, Levinson R, Matsushima GK, Popko B. Oligodendrocytes assist in the maintenance of sodium channel clusters independent of the myelin sheath. Neuron Glia Biol. 2004;1:179–192. doi: 10.1017/S1740925X04000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Jeffrey A, Wakeman DR, Kim SU, Snyder EY, de Vellis J. Culture system for rodent and human oligodendrocyte specification, lineage progression, and maturation. Curr Protoc Stem Cell Biol. 2009 doi: 10.1002/9780470151808.sc02d04s10. Chapter 2, Unit 2D.4. [DOI] [PubMed] [Google Scholar]
- Garcia ADR, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principle source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Gheuens S, Bord E, Kesari S, Simpson DM, Gandhi RT, Clifford DB, Berger JR, Ngo L, Koralnik IJ. Role of CD4+ and CD8+ T-cell responses against JC virus in the outcome of patients with progressive multifocal leukoencephalopathy (PML) and PML with immune reconstitution inflammatory syndrome. J Virol. 2011;85:7256–7263. doi: 10.1128/JVI.02506-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura T, Kornblum HI, Sofroniew MV. The predominant neural stem cell isolated from postnatal and adult forebrain but not from early embryonic forebrain expresses GFAP. J Neurosci. 2003;23:2824–2832. doi: 10.1523/JNEUROSCI.23-07-02824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadi L, Selvaraju R, de Lys P, Proudfoot AE, Wells TN, Boschert U. Differential effects of chemokines on oligodendrocyte precursor proliferation and myelin formation in vitro. J Neuroimmunol. 2006;174:133–146. doi: 10.1016/j.jneuroim.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp Neurol. 2009;220:44–56. doi: 10.1016/j.expneurol.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili K, Gordon J, White MK. The polyomavirus, JCV and its involvement in human disease. Adv Exp Med Biol. 2006;577:274–287. doi: 10.1007/0-387-32957-9_20. [DOI] [PubMed] [Google Scholar]
- Lassman H. Comparative neuropathology of chronic experimental allergic encephalomyelitis and multiple sclerosis. Berlin: Springer-Verlag; 1983. [PubMed] [Google Scholar]
- Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA. Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci U S A. 2000;97:13883–13888. doi: 10.1073/pnas.250471697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, Miller RH, Ransohoff RM. CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci. 2010a;13:319–326. doi: 10.1038/nn.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Darnall L, Hu T, Choi K, Lane TE, Ransohoff RM. Myelin repair is accelerated by inactivating CXCR2 on nonhematopoietic cells. J Neurosci. 2010b;30:9074–9083. doi: 10.1523/JNEUROSCI.1238-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- Manley K, Gee GV, Simkevich CP, Sedivy JM, Atwood WJ. Microarray analysis of glial cells resistant to JCV infection suggests a correlation between viral infection and inflammatory cytokine gene expression. Virology. 2007;366:394–404. doi: 10.1016/j.virol.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzocchetti A, Cingolani A, Giambenedetto SD, Ammassari A, Giancola ML, Cauda R, Antinori A, Luca AD. Macrophage chemoattractant protein-1 levels in cerebrospinal fluid correlate with containment of JC virus and prognosis of acquired immunodeficiency syndrome-associated progressive multifocal leukoencephalopathy. J Neurovirol. 2005;11:219–224. doi: 10.1080/13550280590924539. [DOI] [PubMed] [Google Scholar]
- Mateen FJ, Muralidharan R, Carone M, van de Beek D, Harrison DM, Aksamit AJ, Gould MS, Clifford DB, Nath A. Progressive multifocal leukoencephalopathy in transplant recipients. Ann Neurol. 2011;70:305–322. doi: 10.1002/ana.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mázló M, Ressetar HG, Stoner GL. The neuropathology and pathogenesis of progressive multifocal leukoencephalopathy. In: Khalili K, Stoner GL, editors. Human polyomaviruses: molecular and clinical perspectives. New York: Wiley-Liss; 2001. pp. 257–335. [Google Scholar]
- Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabova N, Kaniowska D, Kaminski R, Deshmane SL, White MK, Amini S, Darbinyan A, Khalili K. JC virus agnoprotein inhibits in vitro differentiation of oligodendrocytes and promotes apoptosis. J Virol. 2008;82:1558–1569. doi: 10.1128/JVI.01680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabova N, Kaminski R, Krynska B, Amini S, Khalili K, Darbinyan A. JCV agnoprotein-induced reduction in CXCL5/LIX secretion by oligodendrocytes is associated with activation of apoptotic signaling in neurons. J Cell Physiol. 2012;227:3119–3127. doi: 10.1002/jcp.23065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messam CA, Hou J, Gronostajski RM, Major EO. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann Neurol. 2003;53:636–646. doi: 10.1002/ana.10523. [DOI] [PubMed] [Google Scholar]
- Miller RJ, Rostene W, Apartis E, Banisadr G, Biber K, Milligan ED, White FA, Zhang J. Chemokine action in the nervous system. J Neurosci. 2008;28:11792–11795. doi: 10.1523/JNEUROSCI.3588-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll NM, Rietsch AM, Ransohoff AJ, Cossoy MB, Huang D, Eichler FS, Trapp BD, Ransohoff RM. Cortical demyelination in PML and MS: similarities and differences. Neurology. 2008;70:336–343. doi: 10.1212/01.WNL.0000284601.54436.e4. [DOI] [PubMed] [Google Scholar]
- Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon–glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Omari KM, John G, Lango R, Raine CS. Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia. 2005a;53:24–31. doi: 10.1002/glia.20246. [DOI] [PubMed] [Google Scholar]
- Omari KM, John GR, Sealfon SC, Raine CS. CXC chemokine receptors on human oligodendrocytes: implications for multiple sclerosis. Brain. 2005b;128:1003–1015. doi: 10.1093/brain/awh479. [DOI] [PubMed] [Google Scholar]
- Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Brück W, Lucchinetti C, Lassmann H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129:3165–3172. doi: 10.1093/brain/awl217. [DOI] [PubMed] [Google Scholar]
- Piaton G, Gould RM, Lubetzki C. Axon–oligodendrocyte interactions during developmental myelination, demyelination and repair. J Neurochem. 2010;114:1243–1260. doi: 10.1111/j.1471-4159.2010.06831.x. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S, Otte J, Enam S, Del Valle L, Khalili K, Gordon J. JC virus-induced changes in cellular gene expression in primary human astrocytes. J Virol. 2003;77:10638–10644. doi: 10.1128/JVI.77.19.10638-10644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan S, Gordon J, Del Valle L, Cui J, Khalili K. Intracellular approach for blocking JC virus gene expression by using RNA interference during viral infection. J Virol. 2004;78:7264–7269. doi: 10.1128/JVI.78.13.7264-7269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine CS, Wu E. Multiple sclerosis: remyelination in acute lesions. J Neuropathol Exp Neurol. 1993;52:199–204. [PubMed] [Google Scholar]
- Ransohoff RM, Trebst C. Chemokines and chemokine receptors in multiple sclerosis: a few answers and many more questions. In: Ransohoff RM, Suzuki K, Proudfoot A, Hickey WF, Harrison JK, editors. Universes in delicate balance chemokines and the nervous system. New York: Elsevier; 2002. pp. 317–332. [Google Scholar]
- Rasband MN, Tayler J, Kaga Y, Yang Y, Lappe-Siefke C, Nave KA, Bansal R. CNP is required for maintenance of axon–glia interactions at nodes of Ranvier in the CNS. Glia. 2005;50:86–90. doi: 10.1002/glia.20165. [DOI] [PubMed] [Google Scholar]
- Robinson S, Franic LA. Chemokine GRO1 and the spatial and temporal regulation of oligodendrocyte precursor proliferation. Dev Neurosci. 2001;23:338–345. doi: 10.1159/000048717. [DOI] [PubMed] [Google Scholar]
- Robinson S, Tani M, Strieter RM, Ransohoff RM, Miller RH. The chemokine growth-regulated oncogene-alpha promotes spinal cord oligodendrocyte precursor proliferation. J Neurosci. 1998;18:10457–10463. doi: 10.1523/JNEUROSCI.18-24-10457.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 2010;9:425–437. doi: 10.1016/S1474-4422(10)70040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazzi E, White MK, Khalili K. Molecular insights into polyomavirus JC and clinical updates on progressive multifocal leukoencephalopathy. Rev Med Virol. 2012;22:18–32. doi: 10.1002/rmv.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Frost E, To V, Robinson S, Ffrench-Constant C, Geertman R, Ransohoff RM, Miller RH. The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell. 2002;110:373–383. doi: 10.1016/s0092-8674(02)00838-3. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Pathophysiology of demyelinated and remyelinated axons In: Cook SD, editor Handbook of multiple sclerosis. New York: Dekker; 1996. pp. 257–294. [Google Scholar]
- White MK, Khalili K. Pathogenesis of progressive multifocal leukoencephalopathy revisited. J Infect Dis. 2011;203:578–586. doi: 10.1093/infdis/jiq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollebo HS, Safak M, Del Valle L, Khalili K, White MK. Role for tumor necrosis factor-α in JC virus reactivation and progressive multifocal leukoencephalopathy. J Neuroimmunol. 2011;233:46–53. doi: 10.1016/j.jneuroim.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Miller RH, Ransohoff RM, Robinson S, Bu J, Nishiyama A. Elevated levels of the chemokine GRO-1 correlate with elevated oligodendrocyte progenitor proliferation in the jimpy mutant. J Neurosci. 2000;20:2609–2617. doi: 10.1523/JNEUROSCI.20-07-02609.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC, Ge B, Duncan ID. Adult brain retains the potential to generate oligodendroglial progenitors with extensive myelination capacity. Proc Natl Acad Sci U S A. 1999;96:4089–4094. doi: 10.1073/pnas.96.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]