

Abstract
Iron-sulfur (Fe-S) clusters are widely distributed protein cofactors that are vital to cellular biochemistry and the maintenance of bioenergetic homeostasis, but to our knowledge, they have never been identified in any phosphatase. Here, we describe an iron–sulfur cluster in Asp1, a dual-function kinase/phosphatase that regulates cell morphogenesis in Schizosaccharomyces pombe. Full-length Asp1, and its phosphatase domain (Asp1371–920), were each heterologously expressed in Escherichia coli. The phosphatase activity is exquisitely specific: it hydrolyzes the 1-diphosphate from just two members of the inositol pyrophosphate (PP-InsP) signaling family, namely, 1-InsP7 and 1,5-InsP8. We demonstrate that Asp1 does not hydrolyze either InsP6, 2-InsP7, 3-InsP7, 4-InsP7, 5-InsP7, 6-InsP7, or 3,5-InsP8. We also recorded 1-phosphatase activity in a human homologue of Asp1, hPPIP5K1, which was heterologously expressed in Drosophila S3 cells with a biotinylated N-terminal tag, and then isolated from cell lysates with avidin beads. Purified, recombinant Asp1371–920 contained iron and acid-labile sulfide, but the stoichiometry (0.8 atoms of each per protein molecule) indicates incomplete iron-sulfur cluster assembly. We reconstituted the Fe–S cluster in vitro under anaerobic conditions, which increased the stoichiometry to approximately 2 atoms of iron and acid-labile sulfide per Asp1 molecule. The presence of a [2Fe-2S]2+ cluster in Asp1371–920 was demonstrated by UV–visible absorption, resonance Raman spectroscopy, and electron paramagnetic resonance spectroscopy. We determined that this [2Fe-2S]2+ cluster is unlikely to participate in redox chemistry, since it rapidly degraded upon reduction by dithionite. Biochemical and mutagenic studies demonstrated that the [2Fe-2S]2+ cluster substantially inhibits the phosphatase activity of Asp1, thereby increasing its net kinase activity.
Graphical abstract
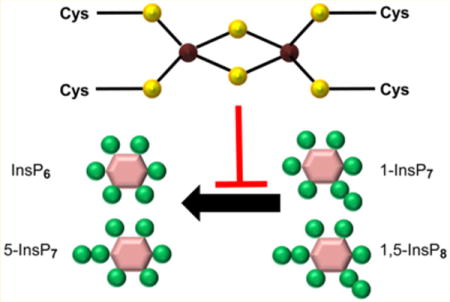
Iron–sulfur (Fe–S) clusters, which are found in proteins from all kingdoms of life, are vital to cellular biochemistry and the maintenance of bioenergetic homeostasis.1 The three most prevalent Fe–S centers in nature are rhombic [2Fe–2S] clusters composed of two irons bridged by two sulfides, a cubane [4Fe–4S] cluster comprising four irons and four triply bridging sulfides, and a cuboidal-type [3Fe-4S] cluster that is based on the cubane structure but has one less iron atom per cluster.2,3 Fe–S clusters are important for stabilizing protein structures, and they also play central roles in electron transport, enzyme catalysis, cellular regulation and iron homeostasis, as well as nucleic acid processing and repair.1–4 However, as far as we are aware, there has not been a previous description of a phosphatase that contains an Fe–S cluster. In the current study we characterize an Fe–S cluster in the phosphatase domain of Asp1, a regulator of cell morphogenesis in the yeast Schizosaccharomyces pombe.5,6 We also utilize chemical and biophysical techniques to establish that the cluster has a [2Fe–2S]2+ arrangement.
S. pombe is frequently used as a model system for studying gene function relevant to fundamental cellular processes that are conserved in human cells.7 Asp1 is of particular interest because it is a member of a conserved family of cell-signaling proteins that each host a kinase domain8–10 and a putative phosphatase domain.8,11 That is, Asp1 and its homologues are highly unusual, modular proteins which may synthesize and catabolize 1-InsP7 and 1,5-InsP8 (Figure 1A, and see below); such metabolites are among the most highly phosphorylated members of a family of signaling molecules known as inositol pyrophosphates, or occasionally, diphosphoinositol polyphosphates (PP-InsPs; Figure 1A).12,13 The PP-InsPs are considered to be highly “energetic”14 (i.e., release of a diphosphate group is associated with a relatively large free energy change15). There are several different molecular mechanisms of action of PP-InsPs. They can associate with specific “receptors”,16,17 they can antagonize PtdIns(3,4,5)P3-signaling,18–20 and they nonenzymatically phosphorylate certain proteins.21,22 Several studies have shown the participation of PP-InsPs in eukaryotic cellular and organismic homeostasis: 5-InsP7 regulates the balance between mitochondrial and glycolytic ATP production;23 there is reduced InsP8 synthesis during mild metabolic stress;24 5-InsP7 regulates both secretion of insulin25 and the efficacy of its actions upon cells.18 Additionally, Asp1, and the Vip1 homologue in Saccharomyces cerevisiae, both appear to mediate certain cellular responses to environmental challenges.6,11,16,17
Figure 1.
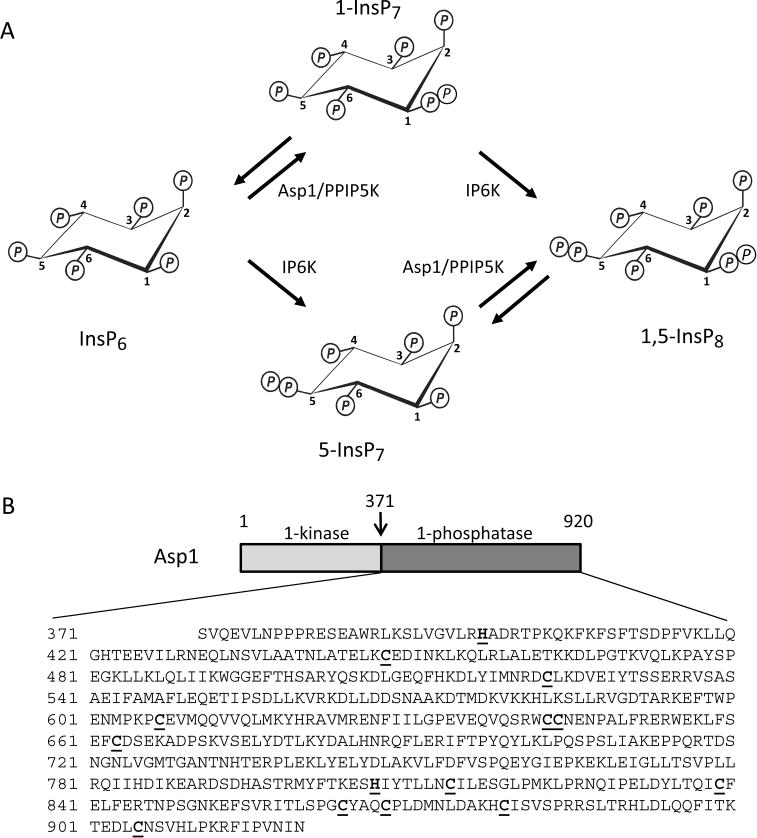
PP-InsP turnover by Asp1/PPIP5K. Panel A depicts the enzymatic reactions catalyzed by Asp1 (PIPP5K in mammals); see the Results and Discussion section for details. Panel B is a schematic of the domain structure of Asp1. A previously published domain alignment places Val375 at the N-terminus of the Asp1 phosphatase domain,9 but Ser371 is favored by our multiple sequence analysis (Supplementary Figure S1). Thus, we used Asp1371–920 as our phosphatase domain construct; its entire sequence is shown below the schematic. All Cys residues are underlined and in bold type. Similarly highlighted are two catalytically important His residues, His39711 and His807 (ref 9 and this work).
Although the existence of a putative 1-phosphatase activity against 1-InsP7 and 1,5-InsP8 (Figure 1A) is understood within the field,8 there has not yet been a published study that directly demonstrates this catalytic activity, in either Asp1 or any homologues. Nevertheless, it has been reported that, in vitro, the putative phosphatase domain in recombinant, full-length Asp1 is sufficient to significantly reduce the amount of InsP7 synthesized by the kinase domain.11 Also in vitro, the elimination of the apparent phosphatase activity in a recombinant Asp1H397A mutant increases the rate of InsP7 synthesis above that achieved by the wild-type protein.11 In vivo, a strain of S. pombe (asp1H397A) in which Asp1 was rendered phosphatase dead (and predicted to have higher PP-InsP levels than wild-type cells) exhibited heightened invasive growth, and a greater resistance to microtubule disruption by thiabendazole. Conversely, invasive growth and resistance to thiabendazole were impaired in an asp1D333A strain that expressed a putative kinase-dead version of Asp1 (predicted to have lower PP-InsP levels than wild-type cells).6,11 Such data indicate that a change in a dynamic balance between competing 1-kinase and 1-phosphatase activities does have predictable, biologically significant outcomes. However, a more complex situation emerged in further experiments which compared asp1Δ strains, in which either the kinase domain was expressed, or both the kinase and putative phosphatase domains were expressed as separate constructs; in each case, the same degree of thiabendazole tolerance was observed, even though PP-InsP levels should have been differentially affected.11 Furthermore, it was found that, compared to wild-type cells, mean microtubule length was longer in both asp1D333A and asp1H397A strains.11 These results hint at the possibility that, in vivo, intervention from other factors might modify either the 1-kinase and/or 1-phosphatase activities. In the current study, we show that such a role may be fulfilled by the new Fe–S cluster that we have characterized.
MATERIALS AND METHODS
Protein Expression and Purification
The plasmid pRSETB-Asp1 was kindly provided by Dr. Kathleen L. Gould (Vanderbilt University). The Gateway expression system (Invitrogen) was used to subclone either the full-length Asp1 or residues 371–920 (Asp1371–920; Figure 1B) into the pDest-566 vector. This vector encodes a 6X His tag, a maltose binding protein tag and TEV protease cleavage site at the N terminus. This vector was used to transform a strain of DE3 competent cells (Stratagene) expressing the chaperone plasmid pGro7 (Takara Clontech). Mutants of Asp1 were generated using a site-directed mutagenesis kit (Stratagene); the primers are described in Supplementary Table 1.
An overnight culture of E. coli cells carrying the appropriate pDest-566-Asp1 construct was inoculated into nutrient-rich super optimal broth medium supplemented with 0.07% (w/v) L-arabinose at pH 7.5 and grown at 37 °C to A595 = 0.7. Isopropyl β-D-thiogalactopyranoside (0.1 mM) was then added, and cultures were continued at 15 °C for 20 h. The cells were disrupted using a constant cell disruption system (Constant Systems LTD) under 20 KPsi. All of the recombinant Asp1 proteins obtained in the current work were soluble; there was no significant delivery of insoluble and/or misfolded protein into bacterial inclusion bodies. Proteins were purified with a nickel-charged, nitrilotriacetic acid-agarose column (Qiagen) followed by a HiTrap Heparin HP column (GE Healthcare), cleavage by a tobacco etch virus protease, and another HiTrap Heparin HP column. As a final step, a Superdex 200 gel filtration column (GE Healthcare) was used with a running buffer of 150 mM NaCl and 40 mM HEPES, pH 7.2; purified proteins were concentrated under aerobic conditions using a centrifugal filter unit (EMD Millipore, molecular weight cutoff 10 kDa) and then stored at −80 °C. Recombinant human DIPP1 was prepared as previously described.26 Concentrations of purified proteins were measured by using a bicinchoninic acid protein assay kit (Thermo Scientific, Rockford IL).
The full-length cDNA for hPPIP5K1 was subcloned into the pDONOR entry vector (Genecoepia Inc., Rockville MD) before transfer to the pMT/BioEase-DEST destination vector using Gateway cloning, as directed by the manufacturer’s instructions (Life Technologies, Grand Island, NY). An empty vector was also constructed as a control. Vectors, along with the pCoBlast selection vector, were stably transfected into Drosophila S3 cells using Effectene transfection reagent (Qiagen). Blasticidin selection (25 μg/mL) was continued for 3 weeks. The S3 cells were maintained in Schneider’s complete medium, supplemented with 10% heat-inactivated fetal bovine serum (Gibco).
The expression of hPPIP5K1 was induced by addition of 0.7 mM CuSO4 for 72 h to approximately 7.5 × 108 S3 cells, propagated as adherent cultures in 11 T-75 flasks. Cells harboring empty vector were similarly processed for a control. Cells were harvested in PBS, transferred to an anaerobic chamber, and lysed on ice by suspension for 1 h in 14 mL of buffer containing 130 mM NaCl, 10 mM HEPES pH (7.3), 1% Triton X-100, 10 mM NaF, 10 mM Na2HPO4, 10 mM NaPPi,1 mM DTT plus protease inhibitors (Roche #11836170001) and phosphatase inhibitors (Roche #04906845001) at the vendor’s recommended concentrations. The lysate was cleared by centrifugation, prior to the addition of 285 μL of avidin-coated beads (SoftLink Soft Release, Promega) to bind hPPIP5K1 that was biotinylated intracellularly on the N-terminal BioEase tag; the vendor’s beads were prewashed twice before use (the “wash buffer” was 150 mM NaCl, 20 mM HEPES, pH 7.3, 1 mM DTT plus protease inhibitors (see above), and then beads were resuspended in the same buffer to the original volume). After 2 h of gentle agitation, beads were pelleted from the lysate by centrifugation and washed 5 times with more wash buffer. The final pellet of beads was resuspended in an equal volume of wash buffer and stored in aliquots at −80 °C. For Western analysis, hPPIP5K1 was released from the beads in 4× SDS loading buffer (Novagen) heated to 95 °C for 10 min. The supernatant was clarified by centrifugation and then resolved by SDS/PAGE. After transfer to a PVDF membrane, the hPPIP5K1 was visualized with Amersham ECL Western Blotting Detection Reagent (GE Healthcare). The primary and secondary antibodies were anti-hPPIP5K1 (Sigma SAB1401487; 1:3000 dilution in 5% BSA) and HRP-linked anti mouse IgG antibody (Cell Signaling; 1:3000 dilution in 5% milk), respectively.
In Vitro Reconstitution of the Fe–S Cluster in Recombinant Asp1
Reconstitution was performed on ice in an anaerobic chamber. Either full-length Asp1 or Asp1371–920 was combined with 4 equiv of both ferric citrate and (NH4)2Fe(SO4)2 and 8 equiv of Na2S under anaerobic conditions in buffer containing 40 mM HEPES (pH 7.2), 100 mM NaCl and 50 mM DTT. After 30 min, samples were removed from the anaerobic chamber and applied to a 10DG column (Bio-Rad) and Superdex200 to separate the protein from excess iron and sulfide. Protein was then concentrated and stored at −80 °C. Controls were performed which did not contain either Fe or S, as indicated in the figure legends.
Enzyme Assays and HPLC Analysis
Enzyme activity was recorded at either 25 °C (for DIPP1 or Asp1) or 37 °C (for hPPIP5K1). Phosphatase activity was measured after 30 min assays in 100 μL reaction mixtures containing 20 mM HEPES, pH 7.2, 100 mM KCl, 0.6 mM MgCl2. Inorganic phosphate release from individual chemically synthesized PP-InsPs26,27 was determined with a colorimetric assay.28 For HPLC analysis, the appropriate PP-InsP was spiked with approximately 2000 d.p.m. of the corresponding [3H]-labeled substrate.29 For the kinase assays, the buffer contained 100 mM KCl, 20 mM HEPES pH 7.2, 1.6 mM MgCl2 and 1 mM ATP.
Assays to be analyzed by HPLC were acid quenched (by addition of 0.2 vol of 2 M perchloric acid plus 1 mg/mL InsP6), neutralized, and typically chromatographed on a 4.6 × 125 mm Partisphere SAX HPLC using an ammonium phosphate gradient generated from Buffer A (1 mM Na2EDTA) and Buffer B (Buffer A plus 1.3 M (NH4)2HPO4, pH 3.85 with phosphoric acid). The gradient (1 mL/min) is as follows: 0–5 min, 0% B; 5–10 min, B increased linearly from 0 to 45%; 10–60 min, B increased linearly from 45 to 100%; 60–75 min, B was 100%. From each run 1 mL fractions were collected, vigorously mixed with 4 mL MonoFlow 4 scintillant (National Diagnostics, Manville NJ), and counted using a liquid scintillation counter.
In the later stages of this work, an alternative procedure was used for the HPLC analysis of PP-InsPs in the PCA-quenched assays. The PP-InsPs were extracted using TiO2 beads.30 The extract was augmented with “carriers” (0.5 nmol each of 1-InsP7, 5-InsP7 and 1,5-InsP8) and loaded onto a 3 × 250 mm CarboPac PA200 column plus 3 × 50 mm guard column. Samples were eluted with a gradient generated from Buffer C (1 mM Na2EDTA, 10 mM 1,4-piperazinedipropanesulfonic acid (Santa Cruz Biotechnology), pH 4.7 with NH4OH, 5% methanol) and Buffer D (Buffer C plus 0.5 M tetramethylammonium nitrate, pH 4.7). The gradient (0.5 mL/min) is as follows: 0–10 min, 0% D, 10 to 110 min, D increased linearly from 0 to 60%. From each run, 0.3 mL fractions were collected and mixed with 3 mL of MonoFlow 4 scintillant.
Resonance Raman and EPR Spectroscopy
Resonance Raman spectra were recorded at 20 K as previously described,31 using an Instruments SA Ramanor U1000 spectrometer coupled with a Coherent Sabre argon ion laser, with 20 μL frozen droplets of approximately 2 mM Asp1371–920 mounted on the coldfinger of an Air Products Displex Model CSA-202E closed cycle refrigerator. X-band (approximately 9.6 GHz) EPR spectra were recorded using a ESP-300D spectrometer (Bruker, Billerica, MA), equipped with an ESR 900 helium flow cryostat (Oxford Instruments, Concord, MA), and quantified under nonsaturating conditions by double integration against a 1.0 mM CuEDTA standard.
Determinations of Iron and Sulfide Content of Asp1371–920 by Colorimetric Methods
The acid-labile sulfide content was determined by incubating Asp1371–920 with N,N′-dimethyl-1,4-phenylenediamine under acidic and oxidizing conditions (FeCl3); methylene blue formation was recorded at 670 nm.32 The iron content was determined through complexation with 1,10-phenanthroline in the presence of hydroxylamine-HCl; absorbance was recorded at 500 nm.32
RESULTS AND DISCUSSION
The Asp1 Phosphatase Domain is Specific for the 1-Diphosphate Group
It is generally considered that Asp1/Vip1/PPIP5Ks harbor a putative 1-phosphatase domain.8 Nevertheless, there are no previously published data that directly demonstrate such a phosphatase activity. A recent study with Asp1 is consistent with the phosphatase domain being catalytically active, but as the authors themselves noted,11 phosphatase activity was not formally demonstrated.
We prepared a recombinant Asp1 construct that contains the putative phosphatase domain (Asp1371–920; Figure 1B), and we incubated it with a variety of chemically synthesized PP-InsPs.26,27 Using an inorganic phosphate assay, we found that 1-InsP7 and 1,5-InsP8 are dephosphorylated by Asp1371–920 (Figure 2A). Note that the rate of dephosphorylation of InsP8 was 10-fold higher than that for 1-InsP7 (Figure 2A). This is significant because the rates of synthesis of InsP8 by the kinase domains of this family of enzymes are also several-fold higher than are the rates of 1-InsP7 synthesis.29 That is, the potential rate of kinase/phosphatase metabolic cycling between 5-InsP7 and 1,5-InsP8 substantially exceeds that between InsP6 and 1-InsP7.
Figure 2.
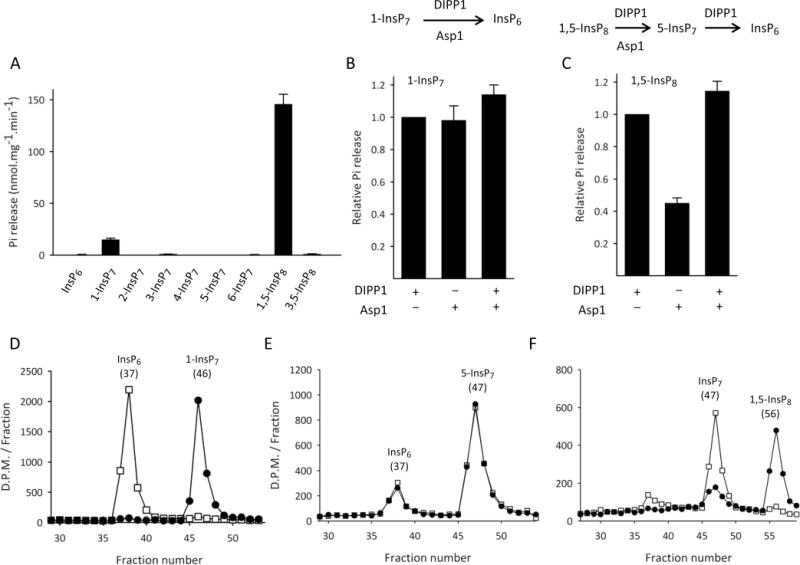
Asp1371–920 specifically hydrolyzes the 1-diester phosphate of 1-InsP7 and 1,5-InsP8. Panel A: Asp1371–920 (0.7 μg/mL) was incubated in phosphatase-assay buffer (see Materials and Methods) with 10 μM of the indicated test substrate (either InsP6 or the indicated PP-InsP). Reactions were assayed for phosphate release. Panel B: Phosphate release from 1-InsP7 (assayed as described for Panel A) was determined following incubations with either10 μg/mL Asp1371–920 for 30 min (left bar), or 10 μg/mL DIPP1 for 30 min (middle bar), or Asp1371–920 for 30 min followed by DIPP1 for 30 min (right bar). Panel C: Phosphate release was measured in the same conditions described for Panel B, except 1,5-InsP8 replaced 1-InsP7 as substrate. For both panels B and C, the reactions catalyzed by DIPP1 and Asp1371–920 are described above the bar graphs. Panel D, E, and F depict HPLC analysis of incubations (see Materials and Methods) in which trace amounts of 1-[3H]InsP7 or 5-[3H]InsP7 or 1,5-[3H]InsP8, respectively, were each incubated with Asp1371–920 (0.041 μg/mL) for either 0 min (circles) or 30 min (squares) in the phosphatase-assay buffer. The chromatographic peaks are labeled with the nature of the material and their elution peak (fraction number). HPLC data are representative of three experiments. Phosphate release data are means ± S.E., n ≥ 3.
The phosphatase domain of Asp1371–920 has a histidine acid phosphatase signature.9,33 Certain other inositol phosphate phosphatases, such as phytases and MINPPs, are also members of the histidine acid phosphatase family,33 but neither of these two groups of enzymes exhibit positional specificity for a particular phosphate group.34,35 Therefore, we scrutinized the specificity of 1-InsP7 and 1,5-InsP8 dephosphorylation. We found that Asp1371–920 does not dephosphorylate a number of other PP-InsPs that do not contain a 1-diphosphate: 2-InsP7, 3-InsP7, 4-InsP7, 5-InsP7, 6-InsP7, and 3,5-InsP8 (Figure 2A). Furthermore, there was no monophosphate hydrolysis from InsP6 (Figure 2A).
We found that the amount of phosphate that was hydrolyzed from 1-InsP7 by Asp1371–920 was not further increased by addition of the diphosphate-specific DIPP1 (Figure 2B). We therefore conclude that both enzymes hydrolyze only the diphosphate group. That is, Asp1371–920 does not show any significant monoester phosphate phosphatase activity toward 1-InsP7. Furthermore, the amount of phosphate that was hydrolyzed from 1,5-InsP8 by Asp1371–920 was approximately doubled by the further addition of DIPP1 (Figure 2C). Such data confirm that (i) Asp1371–920 only hydrolyzes one of the two diphosphate groups in 1,5-InsP8 (whereas DIPP1 attacks both diphosphates), and (ii) Asp1371–920 does not hydrolyze monoester phosphate from 1,5-InsP8.
We also used Partisphere SAX HPLC to analyze the phosphatase activity of Asp1371–920. The results confirm that 1-[3H]InsP7 is a substrate (Figure 2D), whereas 5-[3H]InsP7 is not (the 5-[3H]InsP7 substrate that we used was somewhat contaminated with [3H]InsP6, but there was no further substrate dephosphorylation during the assays; Figure 2E). Analysis by HPLC also confirms the nature of the product(s) of 1-InsP7 and 1,5-InsP8 dephosphorylation by Asp1371–920. For example, the 1-[3H]InsP7 was dephosphorylated to a product that coeluted with InsP6 (Figure 2D). That is, Asp1 shows 1-phosphatase activity toward 1-InsP7. In addition, the HPLC analysis of 1,5-[3H]InsP8 metabolism by Asp1371–920 indicated that an [3H]InsP7 was the major [3H]product; even in exhaustive reactions, only about 10% of substrate was further converted to InsP6 (Figure 2F). In these experiments, a diesterphosphate-specific hydrolysis of 1,5-InsP8 could in principle yield either 5-InsP7 and/or 1-InsP7. In this HPLC system, the peak of 5-[3H]InsP7 (Figure 2E) elutes one fraction after the peak of 1-[3H]InsP7 (Figure 2D); the [3H]InsP7 product of 1,5-[3H]InsP8 dephosphorylation coelutes with the HPLC peak of 5-InsP7 (Figure 2E). In conclusion, Asp1 cleaves the 1-diphosphate from 1,5-InsP8, thereby forming 5-InsP7.
The above data demonstrate that the phosphatase domain in Asp1 is an unusual, highly specialized member of the histidine acid phosphatase family. Much of this domain sequence is fairly well-conserved in hPPIP5K1 and hPPIP5K2, the human homologues of Asp1 (see Supplementary Figure S1 and refs 8 and 9). Nevertheless, in a previous study by the Shears group,36 no phosphatase activity toward either 1-InsP7 and 1,5-InsP8 was observed in assays containing recombinant constructs of the PtdIns(3,4,5)P3-binding domains of the human enzymes (hPPIP5K1382–917; hPPIP5K2371–901). However, the latter constructs excluded some of the more C-terminal residues that, in the native proteins, may be necessary for phosphatase activity (see Supplementary Figure S1 and refs 8 and 9). We therefore set out to assay for phosphatase activity in a full-length, recombinant version of hPPIP5K1; this was a technically challenging proposition, due in part to the large size of the protein (160 kDa) and its lability.10 Indeed, we were unable to obtain intact protein using either E. coli or Sf9 cells as an expression system, so we turned to Drosophila cells; stable transfection of these cells with genes for which expression is induced through the inducible metallothionein promoter has much less impact on cell viability than does viral expression in Sf9 cells.37 We expressed full-length hPPIP5K1 in Drosophila S3 cells (see under Materials and Methods). The protein construct contained an N-terminal BioEase tag that was biotinylated in the intact cells, allowing the hPPIP5K1 to be pulled-down with avidin-coated beads. Western analysis indicated the protein construct was 180 kDa in size (Figure 3A), close to the predicted size of 170 kDa, including the tag. To assay for 1-phosphatase activity we incubated recombinant enzyme with 1 μM 1,5-[3H]InsP8 and analyzed the products by HPLC. For these particular experiments we developed a new HPLC procedure (see under Materials and Methods) that slightly improved the peak-to-peak resolution of 5-InsP7 and 1-InsP7 (Figure 3B). Note that the 1,5-[3H]InsP8 used in these experiments was contaminated with approximately 12% InsP7 (Figure 3B). Under reaction conditions in which about 50% of the 1,5-[3H]InsP8 was dephosphorylated, the major product eluted at the position expected for 5-InsP7 (i.e., hPPIP5K1 removed the 1-phosphate from 1,5-InsP8; Figure 3C). These data indicate that the 1-phosphatase domain in Asp1 is conserved in hPPIP5K1. Next, we extended the incubation time 10-fold, well beyond that needed to completely dephosphorylate the 1,5-[3H]InsP8, and 90% of the products were still recovered as 5-InsP7 (Figure 3D). In control experiments, with avidin-bead pull-downs prepared from cells transformed with empty vector, there was no dephosphorylation of 1,5-[3H]InsP8 (Figure 3E), after taking into account the zero-time InsP7 contaminant.
Figure 3.
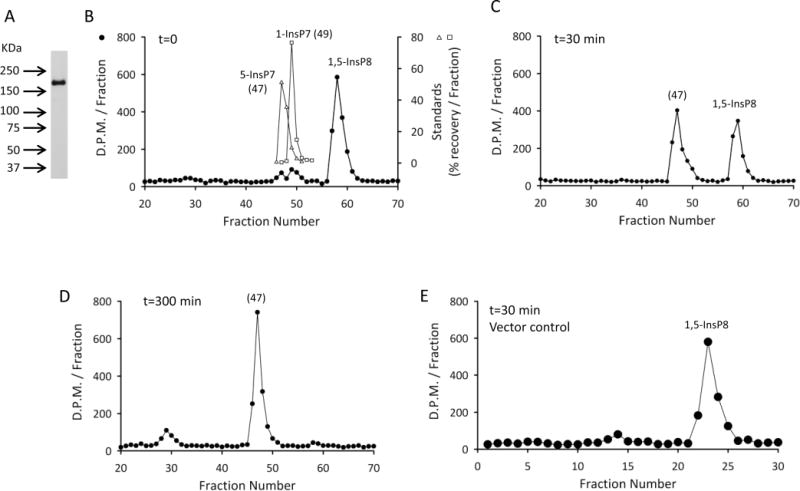
Recombinant, full-length hPPIP5K1 expresses 1-phosphatase activity. Panel A, Western blot of hPPIP5K1 expressed in Drosophila S3 cells and pulled down with avidin-beads (see Materials and Methods). Panel B, CarboPac HPLC analysis of zero-time incubations of 1 μM 1,5-[3H]InsP8 with 0.69 μg/mL recombinant hPPIP5K1 pulled down from S3 cells with avidin beads (filled symbols); open symbols show the elution of standards of 1-[3H]InsP7 and 5-[3H]InsP7 obtained in parallel runs. Panel C, CarboPac HPLC analysis of assays as described for panel B, except the incubation time was 30 min. Panel D, CarboPac HPLC analysis of assays as described for panel B, except the incubation time was 300 min. Panel E, HPLC analysis of assays as described for panel B, except the incubation time was 30 min, and “control” (no enzyme) avidin beads were added (see Materials and Methods). A Partisphere SAX column was used for these assays, and hence the fraction numbers are not comparable with those in panels B, C, and D.
The Asp1 C-Terminal Domain Contains a [2Fe-2S] Cluster
As the concentration of recombinant Asp1371–920 was increased above 1 mg/mL during its purification, we noticed that the preparations became brown (Figure 4A). Such a phenomenon is often indicative of the presence of an Fe–S cluster (for recent examples, see refs 38–40).
Figure 4.
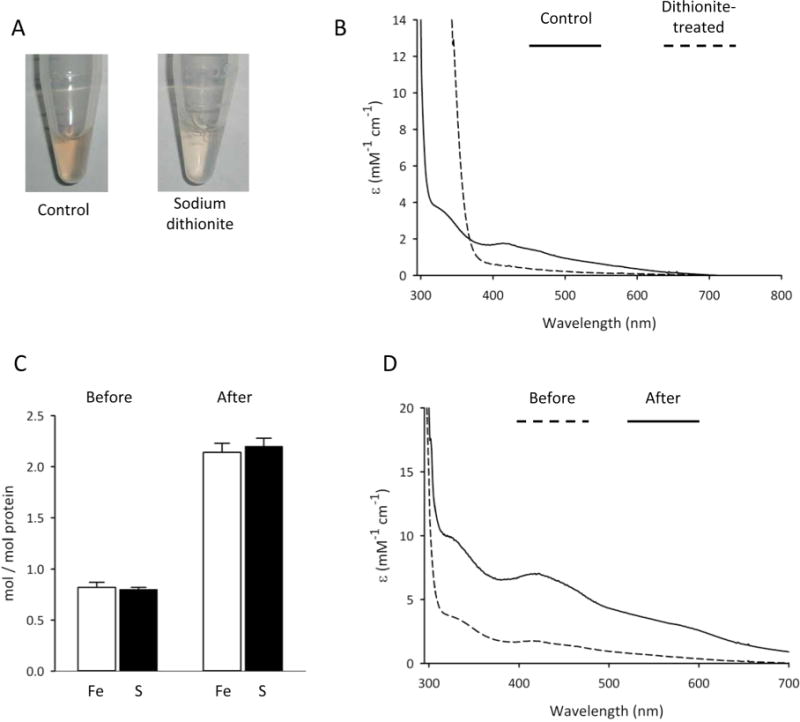
Iron and acid-labile sulfide content, and UV–visible absorption properties of Asp1371–920. Panel A shows Asp1371–920 (10 mg/mL), in buffer containing 40 mM HEPES (pH 7.2), 150 mM NaCl in the presence or absence of 1 mM sodium dithionite. Panel B shows the UV–visible absorption spectra of the same protein samples, either control (solid line) or dithionite-treated (broken line); spectra are corrected for buffer background. Panel C shows iron and acid-labile sulfide content (means ± S.E, n = 3) before and after reconstitution with Fe3+,2+ and S2− (see Materials and Methods). Panel D shows UV−visible absorption of Asp1371–920 both before (broken line) and after (solid line) reconstitution.
The presence and the type of Fe–S cluster can often be inferred from the UV–visible absorption spectrum. Asp1371–920 showed distinct bands at 325 and 420 nm, with a shoulder at 460 nm, and a broad tail extending out to at least 700 nm (Figure 4B). Such UV–visible absorption characteristics are generally indicative of biological [2Fe-2S]2+ clusters.41 The visible absorption was bleached on reduction of Asp1371–920 by addition of a 10-fold excess of dithionite (Figure 4A,B). Also informative was the lack of a pronounced band at approximately 550 nm that is characteristic of a one-electron-reduced, valence-localized [2Fe-2S]1+ cluster,42,43 and aerial oxidation did not significantly restore the UV–visible absorption spectrum of reduced Asp1371–920. These data suggest that dithionite induces irreversible reductive degradation of the cluster, a conclusion that is supported by EPR studies (see below).
Further evidence that Asp1371–920 hosts an Fe–S cluster came from chemical analyses which showed that approximately 0.8 irons and 0.8 acid-labile sulfides were bound to each molecule of protein (Figure 4C). Clearly this stoichiometry is less than maximal, since simple Fe–S centers found in nature contain [2Fe-2S], [3Fe-4S], or [4Fe-4S] clusters.2,3 Suboptimal formation of Fe–S clusters in our recombinant Asp1371–920 may reflect the only partial compatibility of the bacterial and eukaryotic Fe–S assembly machineries.1,44 Alternately, Fe–S clusters are often oxidatively labile, resulting in their partial or complete loss during purification under aerobic conditions. Nevertheless, Fe–S clusters can often be reconstituted in proteins upon treatment with inorganic Fe2+/3+ and S2−, under anaerobic conditions and in the presence of a reducing agent.45–47 We used this methodology to reconstitute an Fe–S cluster into Asp1371–920, after which approximately two irons and two acid-labile sulfides per molecule of protein were detected (Figure 4C). The latter stoichiometry is consistent with the reconstitution of one [2Fe-2S] cluster per Asp1 monomer. Moreover, after the reconstitution process, the only significant change to the UV–visible absorption spectrum was a substantial increase in the molar extinction coefficients of the [2Fe-2S] cluster absorption bands (Figure 4D). The extinction coefficients of the reconstituted Asp1371–920 at 325 and 420 nm (10 000 and 7500 M−1 cm−1 respectively) are close to the lower end of the range of values that are considered typical for [2Fe-2S] clusters (11 000 and 8000 M−1 cm−1, respectively41).
Additional evidence for the presence of a [2Fe-2S] cluster, as well as an assessment of cluster ligation and redox properties, came from resonance Raman and EPR studies of Asp1371–920 (Figures 5 and 6), before and after the protein was reconstituted with iron and sulfide. Both as-purified and reconstituted samples exhibited analogous resonance Raman and EPR properties, although the signal-to-noise properties were enhanced after reconstitution due to the higher cluster content. Figure 5 shows low temperature (20 K) resonance Raman spectra in the Fe–S stretching region for reconstituted Asp1371–920 recorded using 458 and 488 nm laser excitation. The spectra are uniquely indicative of a [2Fe-2S]2+ cluster.42,46,48,49 Indeed, the Fe-S stretching frequencies and vibrational mode relative intensities are very similar to those of the all-cysteinyl ligated [2Fe-2S] centers in thioredoxin-like ferredoxins, such as Clostridium pasteurianum ferredoxin,46,50 other than a lower resolution in the 300–375 nm region, which suggests some heterogeneity in the [2Fe-2S] cluster binding environment in Asp1371–920. The lack of bands involving Fe-His stretching in the 250–280 cm−1 region (Figure 5) excludes the possibility of one or two histidyl ligands.51,52 Mutagenesis and 34/32Sb isotope labeling studies have indicated that the frequencies of the Agt and B3ut predominantly Fe–S(Cys) stretching modes for all-cysteinyl ligated [2Fe-2S]2+ clusters generally occur between 326 and 340 cm−1 and 281–291 cm−1, respectively, compared to 332–356 cm−1 and 289–302 cm−1 and for [2Fe-2S]2+ clusters with one oxygenic ligand.53 Thus, the participation of four ligating Cys residues is consistent with the observed Fe–S stretching frequencies (Figure 5), but three Cys and a single oxygenic ligand (e.g., Ser or Asp)54–56 cannot be completely ruled out.
Figure 5.
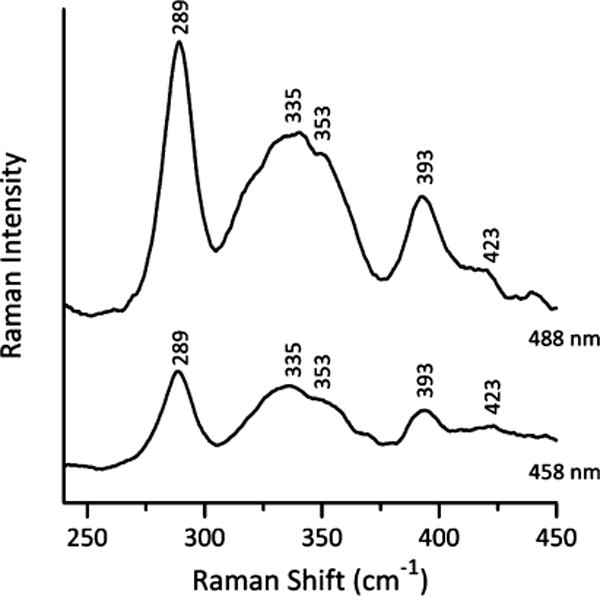
Resonance Raman spectra of reconstituted Asp1371–920 recorded with 457.9 and 487.9 nm laser excitation. Sample of reconstituted Asp1371–920 (approximately 2 mM) was in the form of a frozen droplet at 20 K. Each spectrum is the sum of 100 scans, with each scan involving photon counting for 1 s at 0.5 cm−1 increments with 7 cm−1 spectral resolution. Bands corresponding to the frozen buffer solution have been subtracted from both spectra.
Figure 6.
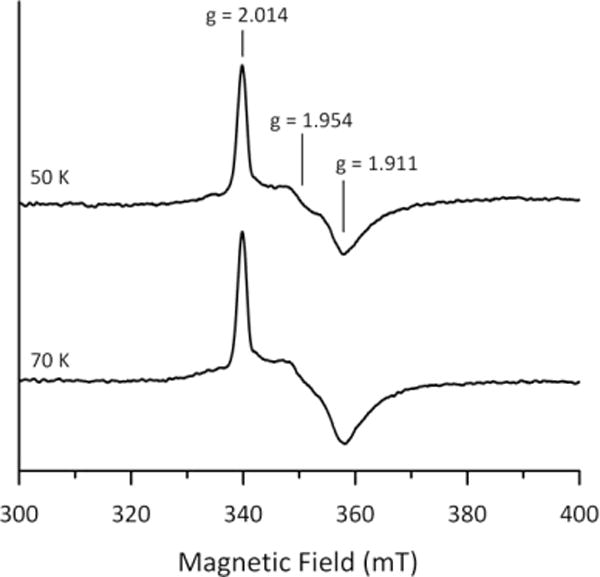
X-band EPR spectra of dithionite-reduced reconstituted Asp1371–920 recorded at 50 and 70 K. Asp1371–920 (0.74 mM) was reduced in an EPR tube under anaerobic conditions by addition a 2-fold stoichiometric excess of sodium dithionite (i.e., a 4-fold excess of reducing equivalents) and frozen immediately in liquid nitrogen. EPR conditions: microwave frequency, 9.60 GHz; modulation frequency, 100 kHz; modulation amplitude, 0.65 mT; microwave power, 5 mW; temperatures as indicated.
We did not observe any EPR signals characteristic of a S = 1/2 [2Fe-2S]1+ upon anaerobic reduction of reconstituted Asp1371–920, following its incubation with a 10-fold excess of sodium dithionite at room temperature for 10 min, prior to freezing in liquid nitrogen. However, weak rhombic EPR signals, g = 2.014, 1.954, 1.911, accounting for 0.06 spins/Asp1371–920, were observed when samples were reduced anaerobically with a 2-fold excess of dithionite in the EPR tube and frozen in liquid nitrogen within 10 s of mixing (Figure 6). The relaxation properties, i.e., observable without broadening at 50 and 70 K, and readily broadened due to power saturation at 10 K, are indicative of slow-relaxing S = 1/2 [2Fe-2S]1+ clusters rather than fast relaxing S = 1/2 [4Fe-4S]1+ clusters.2,3 In addition, the gav value (~1.96) is typical of a [2Fe-2S]1+ cluster ligated by four Cys residues, but does not rule out the possibility of an oxygenic ligand bound at the ferric site of the valence-localized cluster.54–56 These EPR results, taken together with the observation that prolonged reduction with a 10-fold excess of dithionite results in irreversible bleaching of the visible absorption, demonstrate that the reduced S = 1/2 [2Fe-2S]1+ cluster is unstable. This implies that the cluster is very unlikely to be reduced or involved in redox chemistry.
The Fe-S Cluster Inhibits the Phosphatase Activity of Asp1371–920
To investigate if the Fe–S cluster modifies the catalytic activity of Asp1371–920, we exploited our observation that there is incomplete cluster assembly in “as-purified” recombinant enzyme expressed in E. coli (Figure 4C); full [2Fe-2S]2+ cluster occupancy was accomplished through anaerobic reconstitution in vitro (Figure 4C). This enabled us to compare phosphatase activities in two Asp1 preparations exhibiting different degrees of [2Fe-2S]2+ cluster assembly. We found that phosphatase activity in reconstituted Asp1371–920 was almost 95% lower than that observed with as-purified enzyme (Figure 7A). Control experiments, in which the Asp1371–920 protein was incubated with iron or sulfide individually, had no significant effect upon phosphatase activity (Figure 7A). The inhibitory effect of the reconstituted [2Fe-2S]2+ cluster upon phosphatase activity was also observed in full-length, wild-type Asp1 (Figure 7B). The quantitative significance of this inhibition is underscored by the further observation that a similar loss of phosphatase activity was observed upon mutagenesis of a catalytically important residue, His807 (Figure 7B).
Figure 7.
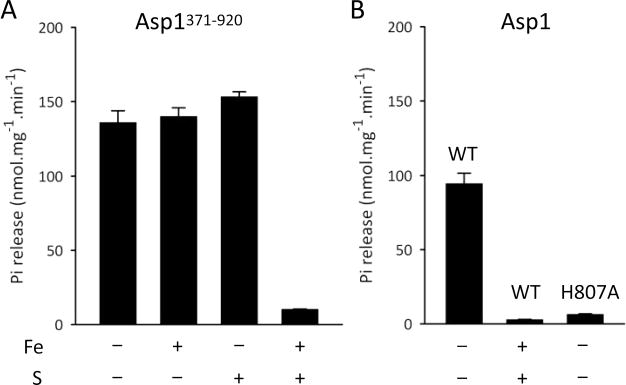
The Fe–S cluster of Asp inhibits its phosphatase activity and unmasks its kinase activity. Panel A shows the phosphatase activity of Asp1371–920 (0.5 μg/mL) against 10 μM 1,5-InsP8 as substrate, following “reconstitution” of the protein in buffer with or without Fe3+,2+ and/or S2− as indicated. Panel B shows similar experiments except the added protein was either full length wild-type (“WT”) or the Asp1H807A mutant (0.7 μg/mL).
We next investigated if removal of the Fe–S cluster would activate phosphatase activity. Since our “as-purified” preparations of Asp1371–920 appear to have about 40% [2Fe-2S]2+ cluster occupancy (Figure 4C,D), we examined the consequence of removing virtually all of the Fe by treating the Asp1371–920 with dithionite/EDTA (Figure 8). This procedure elicited only an approximately 15% activation of the phosphatase activity of our preparations (Figure 8). Next, the reconstituted Asp1371–920 that was fully occupied with the [2Fe-2S]2+ cluster was treated with dithionite/EDTA; phosphatase activity was again only marginally increased (Figure 8). We therefore propose that the [2Fe-2S]2+ cluster may inhibit phosphatase activity indirectly, perhaps through a noncatalytic, structural modification that cannot readily be reversed when the cluster is subsequently removed. Indeed, as far as we are aware, there is no precedent for an Fe–S cluster to have a direct catalytic influence upon any phosphatase, whereas there are a number of examples of Fe–S clusters enforcing and/or stabilizing the conformation of certain proteins.40,57–61 However, we must also consider that, in vivo, there might be a mechanism for disassembling the [2Fe-2S]2+ cluster that involves an oxidative process and/or the involvement of an accessory protein; such a process may not be well-replicated by treatment of Asp1 with dithionite/EDTA. Thus, our experiments do not exclude the possibility that the phosphatase domain may be activated upon removal of the [2Fe-2S]2+ cluster in vivo.
Figure 8.
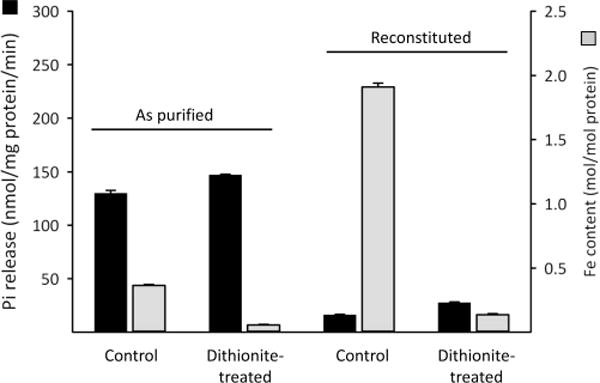
The effect of dithionite in the presence of EDTA on the Fe content and phosphatase activity of Asp1371–920. Either “as-purified” or reconstituted Asp1371–920 was incubated for 30 min with 10 mM sodium dithionite plus 2 mM EDTA. The enzyme preparations were then repurified by gel filtration and assayed for Fe content (gray bars) or Pi release (black bars) from 10 μM 1,5-InsP8 substrate.
Asp1 hosts a kinase as well as a phosphatase domain (Figure 1B). In previous experiments with full-length Asp1, the degree of kinase activity that could be detected was higher in a putative phosphatase-dead mutant as compared to wild-type enzyme, which suggested that phosphatase activity reduced the amount of PP-InsP product that can accumulate.11 To investigate this hypothesis, full-length “as-purified” Asp1 was incubated with ATP and 5-InsP7, and very little 1,5-InsP8 product accumulated (Figure 9A). Following reconstitution of the Fe–S cluster into full-length Asp1, which attenuates the phosphatase activity (Figure 7B), the kinase activity was able to produce a considerable quantity of 1,5-InsP8 product (Figure 9A). A similar degree of phosphorylation of 5-InsP7 to 1,5-InsP8 was observed in incubations containing the full-length Asp1H807A phosphatase-dead mutant (Figure 9B). These data (Figure 9A,B) reveal the extent to which full occupation of the [2Fe-2S]2+ cluster can unmask the kinase activity in full-length Asp1. That is, under iron-replete conditions in vivo, the dominant catalytic function for Asp1 may be its kinase activity. Since similarly high kinase activities were recorded in both reconstituted Asp1 and the non-reconstituted Asp1H807A phosphatase-dead mutant, we conclude that the reconstitution process does not inherently damage the protein.
Figure 9.
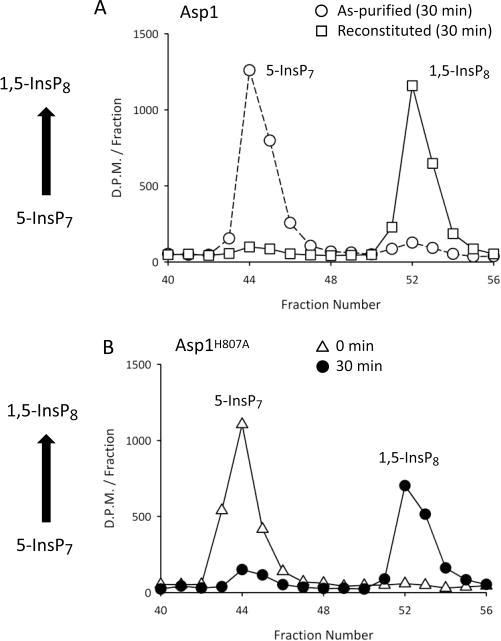
HPLC analysis of 5-InsP7 1-kinase activity of full-length Asp1 and the Asp1H807A mutant. Panel A shows HPLC analysis of of 5-[3H]InsP7 phosphorylation to 1,5-[3H]InsP8 after full-length Asp1 (8.6 μg/mL) was incubated with 10 μM 5-[3H]InsP7 in kinase-assay buffer (see Materials and Methods) at 25 °C for 30 min, either in “as-purified” enzyme (circles) or after reconstitution with Fe3+,2+ and S2− (squares). Panel B shows HPLC analysis of similar kinase assays for the as-purified, full-length Asp1H807A mutant (8.6 μg/mL) incubated with 10 μM 5-[3H]InsP7 for either 0 min (triangles) or 30 min (circles).
Impact upon Phosphatase Activity of Mutating Individual Cys residues in Asp1371–920
We took a mutagenic approach to further pursue our discovery that the [2Fe-2S]2+ cluster in Asp1 inhibits 1-phosphatase activity. Fe–S clusters are predominantly or exclusively ligated by Cys residues.2,3 As Cys-based consensus motifs for [2Fe-2S] centers are so diverse, it is difficult to predict which might be the coordinating residues.4 There are 12 Cys residues in Asp1371–920 (Figure 1B). Each of these residues was individually mutated (except the Cys663/Cys664 pair, which were mutated together), and then we recorded the impact upon phosphatase activities of the “as-purified” enzymes. Ser was chosen as the substitute residue, to preserve as much as possible the hydrogen-bonding character and the side chain geometry of the original Cys residues.
Six individual Cys to Ser mutations—at residues 607, 663, 864, 868, 879, and 905—each led to a reduction in the Fe content of Asp1371–920 (Figure 10A). Since no more than four Cys residues are required to ligate a [2Fe-2S]2+ cluster,2,3 these mutagenic data raise the possibility that two of these six mutations alter protein structure and thereby indirectly impact Fe–S cluster assembly. Another possibility to consider is that Asp1371–920 has alternate cluster ligation states, with varying affinities for the alternate Fe–S clusters, each of which is associated with a unique protein conformation.62 Indeed, three of the mutants that we isolated, Cys643/644Ser, Cys814Ser, and Cys839Ser, each contained more iron than wild-type enzyme (Figure 10A), perhaps reflecting the formation of a higher-affinity Fe–S cluster. In any case, we observed a striking, inverse correlation between the degree of iron content and the catalytic activity of the phosphatase domain (Figure 10B).
Figure 10.
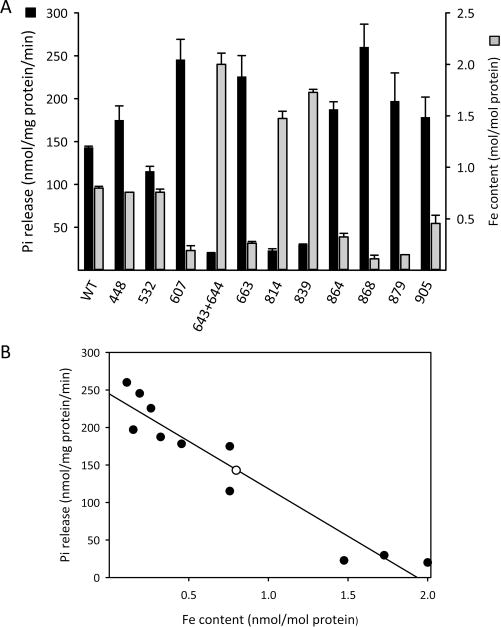
Effect of Cys to Ser mutations upon Fe content and phosphatase activity of Asp1371–920. Panel A: “As-purified” wild-type (“WT”) Asp1371–920, or the indicated Cys to Ser mutants (residue number noted on the X-axis), were assayed for Fe content (gray bars) or Pi release (black bars) from 10 μM 1,5-InsP8 substrate. Panel B: Regression analysis of Fe content versus Pi release for each of the constructs described in panel A (wild-type is the open circle).
CONCLUSIONS
The impact of our study lies in the new information we provide concerning the characteristics of inositol pyrophosphate turnover by Asp1, a dual-domain cell-signaling protein that catalyzes both 1-kinase and 1-phosphatase activities. We have described the exquisite specificity of a conserved phosphatase domain in Asp1 that selectively hydrolyzes the β-phosphate from a 1-diphosphate group of 1-InsP7 and 1,5-InsP8. We have also demonstrated that Asp1 hosts an [2Fe-2S]2+ cluster which inhibits the 1-phosphatase activity. It is of further significance that we determine Asp1 expressed in E. coli has an iron and sulfide stoichiometry that is less than that associated with a fully occupied [2Fe-2S]2+ cluster. Thus, the degree of phosphatase activity associated with these “as-purified” preparations may not be representative of the degree of phosphatase activity that may prevail in intact cells.
There are rare examples of the non-physiological association of iron and sulfide with recombinant proteins expressed in E. coli. However, to our knowledge such an artifact has only been observed when the expressed proteins misfold and are delivered into inclusion bodies.63–67 In contrast, the Asp1 constructs that we have prepared are all fully soluble (see Materials and Methods). Moreover, we found that the initially substoichiometric [2Fe-2S]2+ cluster of “as-purified” Asp1 could be fully reconstituted in vitro (Figure 4C); such an outcome is also considered a hallmark of biological relevance.68 Thus, we conclude that the Fe–S cluster in Asp1 is physiologically relevant.
Genetic studies to investigate the relationship between Fe–S occupancy and 1-phosphatase activity in vivo are beyond the scope of the current study; we first need a more advanced characterization of the cluster’s coordinating residues that, for example, may arise out of a detailed structural characterization of the protein. Nevertheless, our mutagenic studies identified six Cys residues that are candidates for coordinating the Asp1 [2Fe-2S]2+ cluster (Figure 10). Using PROMALS3D,69 we assembled multiple sequence alignments to determine if any of these six Cys residues might be conserved in homologues of Asp1 from certain other fungal species (Figure 11); three such Cys residues from Asp1 align with Cys residues in some, but not all, Asp1 homologues: Cys663, Cys864, and Cys879 (Figure 11). A fourth residue, Cys868 in Asp1, aligns with a highly conserved aspartate residue (Figure 11), a potential oxygenic ligand for an Fe–S cluster.54–56 Among these four candidate Fe–S cluster coordinating residues that are conserved in certain fungal species, there are three—Cys864, Cys868 and Cys879—that may also be conserved in hPPIP5K1, although not in hPPIP5K2 (Supplemental Figure S1). Cys905 in Asp1, which is not conserved in other yeasts, can also be aligned with hPPIP5K1 (Supplemental Figure S1). Thus, if Fe–S cluster coordination is relevant to both yeast and mammalian forms of PPIP5K1, different binding domains are likely involved. To examine if mammalian PPIP5K1 or PPIP5K2 host an Fe–S cluster, milligram quantities of protein would be required for spectroscopic and structural studies. Since Fe–S clusters are inherently vulnerable to aerial oxidation and degradation, purification of recombinant PPIP5Ks may best be undertaken under anaerobic conditions.
Figure 11.

Multiple sequence alignment of Cys-containing regions of Asp1 and selected fungal homologues. Figure shows a multiple sequence alignment of Cys-containing regions of Asp1 and selected fungal homologues, created with PROMALS3D. The full species names are as follows (accession numbers in parentheses): Schizosaccharomyces pombe (O74429); Taphrina deformans (CCG84632.1); Pneumocystis jirovecii (CCJ29735.1); Tuber melanosporum Mel28 (XP_002842355.1); Drechslerella stenobrocha 248 (EWC44731.1); Togninia minima UCRPA7 (XP_007916255.1); Saccharomyces cerevisiae (NP_013514.1); Saitoella complicata NRRL Y-17804 (GAO50791.1); Aspergillus terreus NIH2624 (XP_001213430.1). All Cys residues are highlighted.
We have shown a dramatic increase in the net amount of PP-InsP product formed by the kinase domain of Asp1, when the activity of the phosphatase domain is inhibited by full occupancy of the [2Fe-2S]2+ cluster (Figures 7,9). Thus, it is possible that under iron-replete conditions in vivo, the dominant catalytic function for Asp1 is its kinase activity. It remains to be determined under what circumstances changes in [2Fe-2S]2+ cluster assembly in vivo may represent a cell-signaling mechanism that modulates net PP-InsP turnover. Our EPR data (Figure 6) indicate the [2Fe-2S]2+ cluster in Asp1 is not redox active. Furthermore, we found that inhibition of phosphatase activity was not relieved upon removal of the [2Fe-2S]2+ cluster by dithionite/EDTA treatment (Figure 8). Such data argue against a rapid, freely reversible mechanism of regulation of phosphatase activity by the [2Fe-2S]2+ cluster. However, we must not exclude the possibility that, in vivo, there is a biological mechanism by which the phosphatase can be activated through disassembly of the [2Fe-2S]2+ cluster. Nevertheless, there may also be circumstances in which the regulatory impact of altered [2Fe-2S]2+ cluster assembly upon PP-InsP metabolism reflects relatively long-term adaptive processes. For example, when S. pombe is experiencing suboptimal levels of iron, the yeast may synthesize apo-Asp1 with active phosphatase activity. In this event, net PP-InsP synthesis by the kinase will be constrained. Nonetheless, apo-Asp1 could remain competent to accept an Fe–S cluster. In which case, the dynamic balance of the kinase/phosphatase substrate cycle could be poised to respond as soon as iron repletion restores Fe–S cluster homeostasis. We should also remain cognizant of the possibility that other regulatory factors might modulate the balance between competing kinase and phosphatase activities of Asp1 in vivo.
Supplementary Material
Supplementary Table 1. A list of mutagenic primers;
Supplementary Figure S1. Multiple sequence alignment of hPPIP5K2, hPPIP5K1, and Asp1 (PDF)
Acknowledgments
The authors thank Dr. John York for sharing unpublished data in 2009 that describe the 1-phosphatase activity.
Funding: This research was supported by the Intramural Research Program of the NIH/National Institute of Environmental Health Sciences, and by a grant from the National Institutes of Health (GM62524 to M.K.J.).
ABBREVIATIONS
- PP-InsPs
diphosphoinositol polyphosphates, or “inositol pyrophosphates” (individual PP-InsPs are distinguished by using the following nomenclature: x-InsP7 and x,y-InsP8, for diphosphoinositol pentakisphosphate and bis-diphospho-inositol tetrakisphosphate respectively (in the text, values for “x” and “y”, ranging from 1 to 6, denote the positions of the diphosphate groups around the inositol ring and the subscripts denote total number of phosphate groups))
- InsP6
inositol hexakisphosphate
- DIPP1
diphosphoinositol polyphosphate phosphohydrolase, type 1
- DDT
dithiothreitol
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IP6K
inositol hexakisphosphate kinase
- MINPP
multiple inositol polyphosphate phosphatase
- PBS
phosphate-buffered saline
- hPPIP5K1 and hPPIP5K2
human diphosphoinositol pentakisphosphate kinase types 1 and 2
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00532.
Notes
The authors declare no competing financial interest.
References
- 1.Rouault TA. Mammalian iron-sulphur proteins: novel insights into biogenesis and function. Nat Rev Mol Cell Biol. 2015;16:45–55. doi: 10.1038/nrm3909. [DOI] [PubMed] [Google Scholar]
- 2.Beinert H, Holm RH, Munck E. Iron-sulfur clusters: nature’s modular, multipurpose structures. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 3.Johnson MK, Smith AD. Iron-sulfur proteins. In: King RB, editor. Encyclopedia of Inorganic Chemistry. 2nd. John Wiley & Sons; Chichester: 2005. pp. 2589–2619. [Google Scholar]
- 4.White MF, Dillingham MS. Iron-sulphur clusters in nucleic acid processing enzymes. Curr Opin Struct Biol. 2012;22:94–100. doi: 10.1016/j.sbi.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Feoktistova A, McCollum D, Ohi R, Gould KL. Identification and characterization of Schizosaccharomyces pombe Asp1(+), a gene that interacts with mutations in the Arp2/3 complex and actin. Genetics. 1999;152:895–908. doi: 10.1093/genetics/152.3.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pohlmann J, Fleig U. Asp1, a conserved 1/3 inositol polyphosphate kinase, regulates the dimorphic switch in S. pombe. Mol Cell Biol. 2010;30:4535–4547. doi: 10.1128/MCB.00472-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botstein D, Fink GR. Yeast: an experimental organism for 21st century biology. Genetics. 2011;189:695–704. doi: 10.1534/genetics.111.130765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulugu S, Bai W, Fridy PC, Bastidas RJ, Otto JC, Dollins DE, Haystead TA, Rbeiro AA, York JD. A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science. 2007;316:106–109. doi: 10.1126/science.1139099. [DOI] [PubMed] [Google Scholar]
- 9.Fridy PC, Otto JC, Dollins DE, York JD. Cloning and characterization of two human vip1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J Biol Chem. 2007;282:30754–30762. doi: 10.1074/jbc.M704656200. [DOI] [PubMed] [Google Scholar]
- 10.Choi JH, Williams J, Cho J, Falck JR, Shears SB. Purification, sequencing, and molecular identification of a mammalian PP-InsP5 kinase that is activated when cells are exposed to hyperosmotic stress. J Biol Chem. 2007;282:30763–30775. doi: 10.1074/jbc.M704655200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pohlmann J, Risse C, Seidel C, Pohlmann T, Jakopec V, Walla E, Ramrath P, Takeshita N, Baumann S, Feldbrugge M, Fischer R, Fleig U. The Vip1 inositol polyphosphate kinase family regulates polarized growth and modulates the microtubule cytoskeleton in fungi. PLoS Genet. 2014;10:e1004586. doi: 10.1371/journal.pgen.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson MS, Livermore TM, Saiardi A. Inositol pyrophosphates: between signalling and metabolism. Biochem J. 2013;452:369–379. doi: 10.1042/BJ20130118. [DOI] [PubMed] [Google Scholar]
- 13.Shears SB. Diphosphoinositol polyphosphates: metabolic messengers? Mol Pharmacol. 2009;76:236–252. doi: 10.1124/mol.109.055897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakraborty A, Kim S, Snyder SH. Inositol pyrophosphates as mammalian cell signals. Sci Signaling. 2011;4:re1. doi: 10.1126/scisignal.2001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hand CE, Honek JF. Phosphate transfer from inositol pyrophosphates InsP5PP and InsP4(PP)2: a semi-empirical investigation. Bioorg Med Chem Lett. 2007;17:183–188. doi: 10.1016/j.bmcl.2006.09.066. [DOI] [PubMed] [Google Scholar]
- 16.Lee YS, Huang K, Quiocho FA, O’Shea EK. Molecular basis of cyclin-cdk-cki regulation by reversible binding of an inositol pyrophosphate. Nat Chem Biol. 2008;4:25–32. doi: 10.1038/nchembio.2007.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee YS, Mulugu S, York JD, O’Shea EK. Regulation of a cyclin-cdk-cdk inhibitor complex by inositol pyrophosphates. Science. 2007;316:109–112. doi: 10.1126/science.1139080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, Saleh M, Snowman AM, Moran TH, Mezey E, Snyder SH. Inositol pyrophosphates inhibit AKT signaling, thereby regulating insulin sensitivity and weight gain. Cell. 2010;143:897–910. doi: 10.1016/j.cell.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, Huang Y, Nagata E, Devreotes P, Snyder SH. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-PtdIns(3,4,5)P3 interactions. Cell. 2003;114:559–572. doi: 10.1016/s0092-8674(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 20.Gokhale NA, Zaremba A, Janoshazi AK, Weaver JD, Shears SB. PPIP5K1 modulates ligand competition between diphosphoinositol polyphosphates and PtdIns(3,4,5)P3 for polyphosphoinositide-binding domains. Biochem J. 2013;453:413–426. doi: 10.1042/BJ20121528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhandari R, Saiardi A, Ahmadibeni Y, Snowman AM, Resnick AC, Kristiansen TZ, Molina H, Pandey A, Werner JK, Jr, Juluri KR, Xu Y, Prestwich GD, Parang K, Snyder SH. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci U S A. 2007;104:15305–15310. doi: 10.1073/pnas.0707338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saiardi A, Bhandari A, Resnick R, Cain A, Snowman AM, Snyder SH. Inositol pyrophosphate: physiologic phosphorylation of proteins. Science. 2004;306:2101–2105. doi: 10.1126/science.1103344. [DOI] [PubMed] [Google Scholar]
- 23.Szijgyarto Z, Garedew A, Azevedo C, Saiardi A. Influence of inositol pyrophosphates on cellular energy dynamics. Science. 2011;334:802–805. doi: 10.1126/science.1211908. [DOI] [PubMed] [Google Scholar]
- 24.Choi K, Mollapour E, Choi JH, Shears SB. Cellular energetic status supervises the synthesis of bis-diphosphoinositol tetrakisphosphate independently of amp-activated protein kinase. Mol Pharmacol. 2008;74:527–536. doi: 10.1124/mol.107.044628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Illies C, Gromada J, Fiume R, Leibiger B, Yu J, Juhl K, Yang SN, Barma DK, Falck JR, Saiardi A, Barker CJ, Berggren PO. Inositol pyrophosphates determine exocytic capacity. Science. 2007;318:1299–1302. doi: 10.1126/science.1146824. [DOI] [PubMed] [Google Scholar]
- 26.Capolicchio S, Wang H, Thakor DT, Shears SB, Jessen HJ. Synthesis of densely phosphorylated bis-1,5-diphospho-myo-inositol tetrakisphosphate and its enantiomer by bidirectional p-anhydride formation. Angew Chem, Int Ed. 2014;53:9508–9511. doi: 10.1002/anie.201404398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capolicchio S, Thakor DT, Linden A, Jessen HJ. Synthesis of unsymmetric diphospho-inositol polyphosphates. Angew Chem, Int Ed. 2013;52:6912–5916. doi: 10.1002/anie.201301092. [DOI] [PubMed] [Google Scholar]
- 28.Hoenig M, Lee RJ, Ferguson DC. A microtiter plate assay for inorganic phosphate. J Biochem Biophys Methods. 1989;19:249–252. doi: 10.1016/0165-022x(89)90031-6. [DOI] [PubMed] [Google Scholar]
- 29.Weaver JD, Wang H, Shears SB. The kinetic properties of a human PPIP5K reveal that its kinase activities are protected against the consequences of a deteriorating cellular bioenergetic environment. Biosci Rep. 2013;33:228–241. doi: 10.1042/BSR20120115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson MS, Bulley SJ, Pisani F, Irvine RF, Saiardi A. A novel method for the purification of inositol phosphates from biological samples reveals that no phytate is present in human plasma or urine. Open Biol. 2015;5:150014. doi: 10.1098/rsob.150014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cosper MM, Jameson GN, Hernandez HL, Krebs C, Huynh BH, Johnson MK. Characterization of the cofactor composition of Escherichia coli biotin synthase. Biochemistry. 2004;43:2007–2021. doi: 10.1021/bi0356653. [DOI] [PubMed] [Google Scholar]
- 32.Lovenberg W, Buchanan B, Rabinowitz JC. Studies on the chemical nature of clostridial ferredoxin. J Biol Chem. 1963;238:3899–3913. [PubMed] [Google Scholar]
- 33.Rigden DJ. The histidine phosphatase superfamily: structure and function. Biochem J. 2008;409:333–348. doi: 10.1042/BJ20071097. [DOI] [PubMed] [Google Scholar]
- 34.Nogimori K, Hughes PJ, Glennon MC, Hodgson ME, Putney JW, Jr, Shears SB. Purification of an inositol (1,3,4,5)-tetrakisphosphate 3-phosphatase activity from rat liver and its substrate specificity. J Biol Chem. 1991;266:16499–16506. [PubMed] [Google Scholar]
- 35.Konietzny U, Greiner R. Molecular and catalytic properties of phytate-degrading enzymes (phytases) Int J Food Sci Technol. 2002;37:791–812. [Google Scholar]
- 36.Gokhale NA, Zaremba A, Shears SB. Receptor-dependent compartmentalization of PPIP5K1, a kinase with a cryptic polyphosphoinositide binding domain. Biochem J. 2011;434:415–426. doi: 10.1042/BJ20101437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernard AR, Kost TA, Overton L, Cavegn C, Young J, Bertrand M, Yahia-Cherif Z, Chabert C, Mills A. Recombinant protein expression in a drosophila cell line: comparison with the baculovirus system. Cytotechnology. 1994;15:139–144. doi: 10.1007/BF00762388. [DOI] [PubMed] [Google Scholar]
- 38.Spiller MP, Ang SK, Ceh-Pavia E, Fisher K, Wang Q, Rigby SE, Lu H. Identification and characterization of mitochondrial MIA40 as an iron-sulfur protein. Biochem J. 2013;455:27–35. doi: 10.1042/BJ20130442. [DOI] [PubMed] [Google Scholar]
- 39.Jain R, Vanamee ES, Dzikovski BG, Buku A, Johnson RE, Prakash L, Prakash S, Aggarwal AK. An iron-sulfur cluster in the polymerase domain of yeast DNA polymerase epsilon. J Mol Biol. 2014;426:301–308. doi: 10.1016/j.jmb.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stiban J, Farnum GA, Hovde SL, Kaguni LS. The N-terminal domain of the Drosophila mitochondrial replicative DNA helicase contains an iron-sulfur cluster and binds DNA. J Biol Chem. 2014;289:24032–24042. doi: 10.1074/jbc.M114.587774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dailey HA, Finnegan MG, Johnson MK. Human ferrochelatase is an iron-sulfur protein. Biochemistry. 1994;33:403–407. doi: 10.1021/bi00168a003. [DOI] [PubMed] [Google Scholar]
- 42.Fu W, Drozdzewski PM, Davies MD, Sligar SG, Johnson MK. Resonance Raman and magnetic circular dichroism studies of reduced [2Fe-2S] proteins. J Biol Chem. 1992;267:15502–15510. [PubMed] [Google Scholar]
- 43.Subramanian S, Duin EC, Fawcett SE, Armstrong FA, Meyer J, Johnson MK. Spectroscopic and redox studies of valence-delocalized [Fe2S2]+ centers in thioredoxin-like ferredoxins. J Am Chem Soc. 2015;137:4567–4580. doi: 10.1021/jacs.5b01869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 45.Malkin R, Rabinowitz JC. The reconstitution of clostridial ferredoxin. Biochem Biophys Res Commun. 1966;23:822–827. doi: 10.1016/0006-291x(66)90561-4. [DOI] [PubMed] [Google Scholar]
- 46.Han S, Czernuszewicz RS, Kimura T, Adams MWW, Spiro TG. Fe2S2 protein resonance Raman spectra revisited: structural variations among adrenodoxin, ferredoxin, and red paramagnetic protein. J Am Chem Soc. 1989;111:3505–3511. [Google Scholar]
- 47.Poor CB, Wegner SV, Li H, Dlouhy AC, Schuermann JP, Sanishvili R, Hinshaw JR, Riggs-Gelasco PJ, Outten CE, He C. Molecular mechanism and structure of the Saccharomyces cerevisiae iron regulator AFT2. Proc Natl Acad Sci U S A. 2014;111:4043–4048. doi: 10.1073/pnas.1318869111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spiro TG, Czernuszewicz RS, Han S. Iron-sulfur proteins and analog complexes. In: Spiro TG, editor. Resonance Raman Spectra of Heme and Mettaloproteins. Wiley; New York: 1988. pp. 523–554. [Google Scholar]
- 49.Spiro TG, Czernuszewicz RS. Resonance Raman spectroscopy of metalloproteins. Methods Enzymol. 1995;246:416–460. doi: 10.1016/0076-6879(95)46020-9. [DOI] [PubMed] [Google Scholar]
- 50.Golinelli MP, Chatelet C, Duin EC, Johnson MK, Meyer J. Extensive ligand rearrangements around the [2Fe-2S] cluster of Clostridium pasteurianum ferredoxin. Biochemistry. 1998;37:10429–10437. doi: 10.1021/bi9806394. [DOI] [PubMed] [Google Scholar]
- 51.Li H, Mapolelo DT, Dingra NN, Naik SG, Lees NS, Hoffman BM, Riggs-Gelasco PJ, Huynh BH, Johnson MK, Outten CE. The yeast iron regulatory proteins GRX3/4 and FRA2 form heterodimeric complexes containing a [2Fe-2S] cluster with cysteinyl and histidyl ligation. Biochemistry. 2009;48:9569–9581. doi: 10.1021/bi901182w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuila D, Schoonover JR, Dyer RB, Batie CJ, Ballou DP, Fee JA, Woodruff WH. Resonance Raman studies of Rieske-type proteins. Biochim Biophys Acta, Bioenerg. 1992;1140:175–183. doi: 10.1016/0005-2728(92)90007-o. [DOI] [PubMed] [Google Scholar]
- 53.Agar JN, Krebs C, Frazzon J, Huynh BH, Dean DR, Johnson MK. IscU as a scaffold for iron-sulfur cluster biosynthesis: sequential assembly of [2Fe-2S] and [4Fe-4S] clusters in IscU. Biochemistry. 2000;39:7856–7862. doi: 10.1021/bi000931n. [DOI] [PubMed] [Google Scholar]
- 54.Bertrand P, Gayda JP. A theoretical interpretation of the variations of some physical parameters within the [2Fe-2S] ferredoxin group. Biochim Biophys Acta, Protein Struct. 1979;579:107–121. doi: 10.1016/0005-2795(79)90091-6. [DOI] [PubMed] [Google Scholar]
- 55.Orio M, Mouesca JM. Variation of average g-values and effective exchange coupling constants among [2Fe-2S] clusters: a density functional theory study of the impact of localization (trapping forces) versus delocalization (double-exchange) as competing factors. Inorg Chem. 2008;47:5394–5416. doi: 10.1021/ic701730h. [DOI] [PubMed] [Google Scholar]
- 56.Werth MT, Cecchini G, Manodori A, Ackrell BA, Schroder I, Gunsalus RP, Johnson MK. Site-directed mutagenesis of conserved cysteine residues in Escherichia coli fumarate reductase: modification of the spectroscopic and electrochemical properties of the [2Fe-2S] cluster. Proc Natl Acad Sci U S A. 1990;87:8965–8969. doi: 10.1073/pnas.87.22.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rietzschel N, Pierik AJ, Bill E, Lill R, Muhlenhoff U. The basic leucine zipper stress response regulator Yap5 senses high-iron conditions by coordination of [2Fe-2S] clusters. Mol Cell Biol. 2015;35:370–378. doi: 10.1128/MCB.01033-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin AE, Burgess BK, Stout CD, Cash VL, Dean DR, Jensen GM, Stephens PJ. Site-directed mutagenesis of Azotobacter vinelandii ferredoxin I: [Fe-S] cluster-driven protein rearrangement. Proc Natl Acad Sci U S A. 1990;87:598–602. doi: 10.1073/pnas.87.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shand O, Volz K. The solution structure of apo-iron regulatory protein 1. Gene. 2013;524:341–346. doi: 10.1016/j.gene.2013.03.112. [DOI] [PubMed] [Google Scholar]
- 60.Crooks DR, Ghosh MC, Haller RG, Tong WH, Rouault TA. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood. 2010;115:860–869. doi: 10.1182/blood-2009-09-243105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu Y, Brosh RM., Jr DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res. 2012;40:4247–4260. doi: 10.1093/nar/gks039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonomi F, Iametti S, Morleo A, Ta D, Vickery LE. Facilitated transfer of IscU-[2Fe2S] clusters by chaperone-mediated ligand exchange. Biochemistry. 2011;50:9641–9650. doi: 10.1021/bi201123z. [DOI] [PubMed] [Google Scholar]
- 63.Archer VE, Breton J, Sanchez-Garcia I, Osada H, Forster A, Thomson AJ, Rabbitts TH. Cysteine-rich Lim domains of Lim-homeodomain and Lim-only proteins contain zinc but not iron. Proc Natl Acad Sci U S A. 1994;91:316–320. doi: 10.1073/pnas.91.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li PM, Reichert J, Freyd G, Horvitz HR, Walsh CT. The Lim region of a presumptive Caenorhabditis elegans transcription factor is an iron-sulfur-and zinc-containing metallodomain. Proc Natl Acad Sci U S A. 1991;88:9210–9213. doi: 10.1073/pnas.88.20.9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leartsakulpanich U, Antonkine ML, Ferry JG. Site-specific mutational analysis of a novel cysteine motif proposed to ligate the 4Fe-4S cluster in the iron-sulfur flavoprotein of the thermophilic Methanoarchaeon Methanosarcina thermophila. J Bacter-iol. 2000;182:5309–5316. doi: 10.1128/jb.182.19.5309-5316.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerardi G, Biasiotto G, Santambrogio P, Zanella I, Ingrassia R, Corrado M, Cavadini P, Derosas M, Levi S, Arosio P. Recombinant human hepcidin expressed in Escherichia coli isolates as an iron containing protein. Blood Cells, Mol, Dis. 2005;35:177–181. doi: 10.1016/j.bcmd.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 67.Stepanyuk GA, Xu H, Wu CK, Markova SV, Lee J, Vysotski ES, Wang BC. Expression, purification and characterization of the secreted luciferase of the copepod Metridia longa from Sf9 insect cells. Protein Expression Purif. 2008;61:142–148. doi: 10.1016/j.pep.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Urzica E, Pierik AJ, Muhlenhoff U, Lill R. Crucial role of conserved cysteine residues in the assembly of two iron-sulfur clusters on the CIA protein Nar1. Biochemistry. 2009;48:4946–4958. doi: 10.1021/bi900312x. [DOI] [PubMed] [Google Scholar]
- 69.Tong J, Pei J, Otwinowski Z, Grishin NV. Refinement by shifting secondary structure elements improves sequence alignments. Proteins: Struct, Funct, Genet. 2015;83:411–427. doi: 10.1002/prot.24746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. A list of mutagenic primers;
Supplementary Figure S1. Multiple sequence alignment of hPPIP5K2, hPPIP5K1, and Asp1 (PDF)