

Abstract
Long non-coding RNAs (lncRNAs) are important players in diverse biological processes. Upon DNA damage, cells activate a complex signaling cascade referred to as the DNA damage response (DDR). Using a microarray screen, we identify here a novel lncRNA, DDSR1 (DNA damage-sensitive RNA1), which is induced upon DNA damage. DDSR1 induction is triggered in an ATM-NF-κB pathway-dependent manner by several DNA double-strand break (DSB) agents. Loss of DDSR1 impairs cell proliferation and DDR signaling and reduces DNA repair capacity by homologous recombination (HR). The HR defect in the absence of DDSR1 is marked by aberrant accumulation of BRCA1 and RAP80 at DSB sites. In line with a role in regulating HR, DDSR1 interacts with BRCA1 and hnRNPUL1, an RNA-binding protein involved in DNA end resection. Our results suggest a role for the lncRNA DDSR1 in modulating DNA repair by HR.
Keywords: BRCA1, hnRNPUL1, p53, RAP80, repair
Introduction
Genomes continuously experience a wide variety of damages. In order to maintain genome stability, cells have evolved complex mechanisms to sense and repair DNA damage caused by intrinsic and extrinsic insults. Among the various types of DNA damage, double-strand breaks (DSB) are the most toxic and difficult to repair. DSB repair, by either non-homologous end-joining (NHEJ) or homologous recombination (HR), is mediated by proteins of the phosphatidylinositol 3-kinase-like protein kinase (PIKKs) family, particularly ATM, ATR, and DNA-PKcs 1. The choice of repair pathway is influenced by the cell cycle phase 2. During G1, DSBs are primarily repaired by the error-prone NHEJ, which involves direct rejoining of DNA ends 3, whereas in S/G2, HR predominates, using a homologous DNA template sequence for error-free repair 4. For DNA repair by HR, DSBs are detected by the MRE11-RAD50-NBS1 (MRN) complex, which promotes ATM activity by autophosphorylation 5. ATM activation in turn results in phosphorylation of various DNA repair factors such as the core histone variant H2AX, CtIP, BRCA1, and exonuclease EXO1 4. The 53BP1 protein prevents BRCA1 accumulation at DSB sites in the G1 phase of the cell cycle and promotes NHEJ, whereas BRCA1 promotes end resection and HR 6–8. DSB repair also involves extensive reorganization and modification of chromatin near break sites to facilitate access for repair factors 9.
Genomewide transcriptome analysis has led to the identification of numerous long non-coding RNAs (lncRNAs), which are loosely defined as non-coding transcripts longer than 200 nucleotides 10,11. Numerous lncRNAs have been shown to play important regulatory roles in various biological processes ranging from cell cycle control, pluripotency, and differentiation, to disease 10–14. LncRNAs may affect the expression of genes in cis or trans via association with proteins such as chromatin modifiers or ribonucleoprotein complexes or with other RNAs, although the precise mode of action for individual lncRNAs is often unknown 10,11,14,15.
LncRNAs are also emerging as regulators of DNA damage-sensitive gene expression programs 16,17. ATM modulates the expression of lncRNAs in response to DNA damage 18,19. LncRNA expression altered in response to DNA damage appears to be modulated by transcription factor p53 20–23. LincRNA-p21 interacts with hnRNP-K and participates in p53-mediated gene repression 20, and the p53-induced lncRNA, PANDA, interacts with the transcription factor NF-YA to impede induction of apoptotic genes 21. p53 also induces lincRNA Pint which interacts with the polycomb repressive complex 2 (PRC2) to mediate gene silencing 22. LncRNAs have also been implicated in regulating DNA repair by HR. Loss of one such ATM-regulated lncRNA, ANRIL, has been shown to impair DNA repair by homologous recombination 19. Also, PCAT-1, a prostate-specific lncRNA, regulates BRCA2 and controls homologous recombination in cancer 24.
In the current study, we examined lncRNA expression profiles across the genome in response to DNA damage treatment and identified a novel lncRNA, DDSR1 (DNA damage-sensitive RNA1). We show that DDSR1 is induced in an ATM-NF-κB-dependent manner upon DNA damage and negatively regulates p53 target genes, analogous to other DNA damage-induced lncRNAs. However, in addition, DDSR1 contributes to HR by a transcription-independent mode of action by interacting with BRCA1 and modulating BRCA1 and RAP80 access to DSBs sites. Our results identify lncRNA DDSR1 as an integral player in the mammalian DNA damage response.
Results
Identification of DNA damage-induced lncRNAs
We sought to identify, in an unbiased fashion and at a genome-wide scale, lncRNAs whose expression is responsive to DNA damage. We performed gene expression analysis using the NCode™ microarray of total or nuclear RNA isolated from hTert-immortalized human skin fibroblasts treated for 3 h with DNA-damaging agents neocarzinostatin (NCS, 50 ng/ml), camptothecin (4 μM), or etoposide (10 μM). Cells treated with vehicle were used as controls. Using a 1.5-fold change and P-value < 0.05 as a threshold, we identified 127 and 54 non-coding transcripts showing differential expression in screens of total or nuclear RNA, respectively (Tables EV1 and EV2). Six transcripts were common between both screens, but only one was upregulated in both screens. The lncRNA clone AK0217744 (#NCBI GenBank) was found to be induced upon DNA damage both in total (2.5-fold) and in nuclear (2.6-fold) RNA extracts in response to all DNA-damaging agents, suggesting a global role in the DNA damage response. We refer to this lncRNA as DDSR1 (DNA damage-sensitive RNA1).
Using 5′ and 3′ RACE and sequencing, we identified DDSR1 as an intronless transcript of 1,616 nucleotides, located on human chromosome 12q23.3(+) (Fig 1A, Dataset EV1). DDSR1 contains no overlapping sequence with transcripts from its neighboring genes C12orf45 (5′) and ALDH1L2 (3′) and is an independent inter-genic transcript. Evaluation of the protein-coding capacity of DDSR1 using coding potential calculator software 25 confirmed a negligible protein coding potential for DDSR1. LncRNA-DDSR1 was detectable by qRT–PCR as a moderately expressed transcript in undamaged cells, but was upregulated upon DNA damage by ∼2-fold in response to the various DNA-damaging treatments (Fig 1B). DDSR1 induction was dose independent in response to treatment with NCS levels above 25 ng/ml for 4 h (Fig 1C) or camptothecin (4–50 μM) (Fig EV1A). Time-course analysis identified DDSR1 as a delayed transcript with an induction of ∼2-fold at 3 h of treatment, ∼2.5- at 6 h, ∼3-fold at 12 h, reaching a plateau thereafter (Fig 1D). Based on copy number measurement by PCR, we estimate its abundance in human fibroblast cells to be about twelve-fold lower than the common housekeeping gene mRNA TBP. DDSR1 induction upon DNA damage was not cell type specific as it was also expressed and induced upon DNA damage in PC3 (prostate), A549 (lung), U2OS (osteosarcoma), and HCT116 (colon) cells (Fig 1E). Moreover, DDSR1 induction was specific to DNA damage and was not indicative of a general stress response since no change in DDSR1 levels was observed upon heat shock (Fig EV1B). RNA fluorescence in situ hybridization (FISH) in U2OS cells confirmed DDSR1 expression at low levels in both the cytoplasm and nucleus and its induction upon DNA damage (Fig EV1C).
Figure 1. LncRNA DDSR1 is a DNA damage-inducible transcript.

- DDSR1 is an intronless transcript of 1,616 nt, located on human chromosome 12q23.3(+) in cis to ALDH1L2 gene.
- LncRNA-DDSR1 is induced upon DNA damage with different DNA-damaging agents. Human fibroblast cells were treated with the indicated DNA-damaging agent for 3 h and DDSR1 levels measured by qRT–PCR.
- DDSR1 induction is not dose dependent. Cells were treated with the indicated concentration of NCS for 3 h and analyzed for DDSR1 expression.
- DDSR1 induction upon DNA damage plateaus. Cells were treated with NCS (100 ng/ml) and DDSR1 levels measured at the indicated times.
- DDSR1 induction upon DNA damage is not cell type specific. The indicated cell lines were treated with 100 ng/ml NCS and DDSR1 transcript levels determined after 6 h.
Data information: RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from 4 independent experiments. *Significant change compared to control (P < 0.05). Statistical comparisons were made using Student’s t-test.
Figure EV1. Further characterization of DDSR1 levels and subcellular localization.
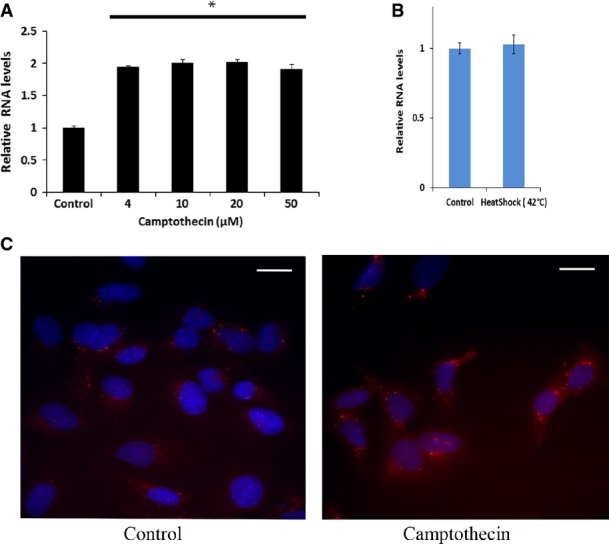
- DDSR1 induction is not dose dependent. Human fibroblast cells were treated with the indicated concentration of camptothecin for 3 h and analyzed for DDSR1 expression by quantitative RT–PCR.
- DDSR1 does not respond to heat shock. Human fibroblast were kept at 42 or 37°C (control) for 1 h and DDSR1 expression was determined.
- Visualization of DDSR1 by RNA fluorescence in situ hybridization (FISH) in U2OS cells untreated or treated with camptothecin (10 μM). Scale bar represents 20 μM.
Data information: For (A, B), RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from three independent experiments. *Significant change compared to control cells (P < 0.05). Statistical comparisons were made using Student’s t-test.
Regulation of DDSR1 expression by ATM, NF-κB, and p53
Since the ATM kinase is the primary responder to DSBs and a major regulator of the DDR signaling cascade, we tested whether induction of DDSR1 upon DNA damage is ATM dependent. Inhibition of ATM during DNA damage with its specific inhibitor KU55933 markedly reduced DDSR1 induction as compared to cells treated with NCS alone (Fig 2A). In order to identify the transcription factors involved in DDSR1 expression upon DNA damage, we analyzed the 2,000-bp putative promoter sequence upstream of the DDSR1 transcription start site for consensus transcription factor binding sites. In silico analysis using Alibaba2 revealed an NF-κB binding site in the DDSR1 promoter region ∼1.1 kb upstream of the TSS. Consistent with the known role of ATM as an activator of NF-κB in response to DNA damage 26, treatment of cells with the NF-κB inhibitor BAY11-7085 for 1 h before induction of DNA damage strongly suppressed DDSR1 induction upon DNA damage (Fig 2B). No effect of BAY11-7085 was observed in the absence of DNA damage. These results suggest that induction of DDSR1 upon DNA damage is regulated by the ATM-NF-κB signaling pathway.
Figure 2. Regulation of lncRNA DDSR1 expression.
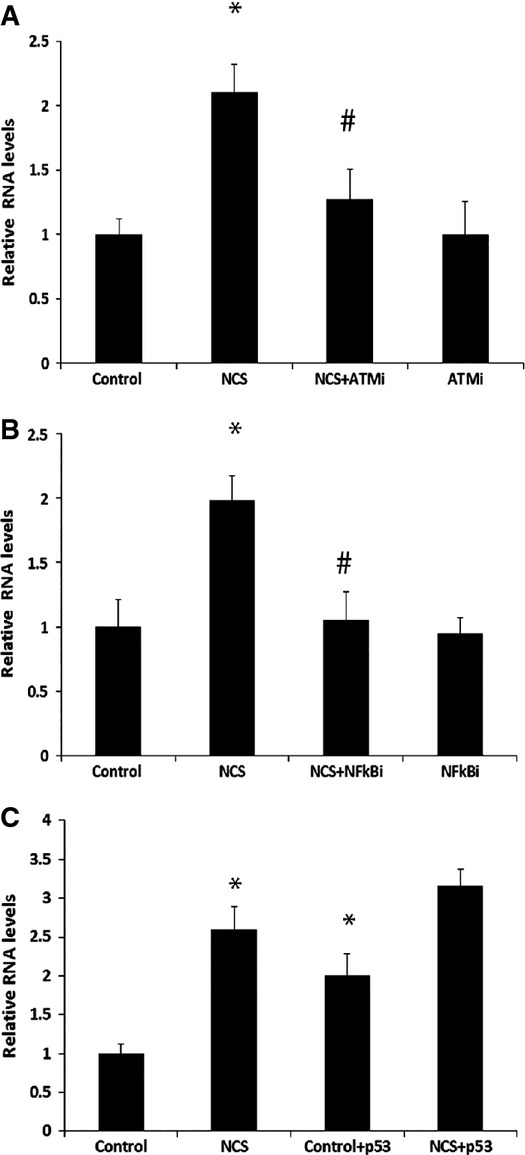
- DDSR1 induction upon DNA damage is ATM dependent. Human fibroblast cells were pretreated with 20 μM KU55933 (ATMi) for 1 h followed by co-treatment with NCS (100 ng/ml) for 3 h, and DDSR1 expression was determined by qRT–PCR.
- NF-κB inhibition abrogates DDSR1 induction upon DNA damage. Human fibroblast cells were pretreated with 10 μM BAY11-7085 (NF-κBi) for 1 h followed by co-treatment with NCS (100 ng/ml) for 3 h, and DDSR1 transcript levels were determined by qRT–PCR.
- p53 regulates DDSR1 expression but is not necessary for DDSR1 induction upon DNA damage. H1299 (p53-null cells) cells were transfected with p53 or control vector for 24 h, and DDSR1 levels were evaluated after 6-h treatment with NCS (100 ng/ml).
Data information: RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from three independent experiments. *Significant change compared to control (P < 0.05). #Significant change compared to NCS (P < 0.05). Statistical comparisons were made using Student’s t-test.
Another master regulator of the DNA damage response, and of numerous lncRNAs, is the tumor suppressor p53. DDSR1 induction was unaffected in the p53-null cell line H1299 upon DNA damage, suggesting p53 independence of DDSR1 induction (Fig 2C). On the other hand, expression of exogenous p53 was sufficient to induce DDSR1 in H1299 p53-null cells in the absence of DNA damage and DDSR1 transcript levels were higher in H1299 cells expressing p53 upon DNA damage as compared to p53-null H1299 cells (Fig 2C). These results suggest that p53 is not required for DDSR1 induction upon DNA damage, consistent with the absence of a p53 binding site in the promoter, but that p53 can promote DDSR1 expression, likely through other p53 targets associated with transcription or p53 binding to enhancer regions.
Regulation of gene expression by DDSR1
LncRNA transcripts often regulate gene expression, directly or indirectly, in cis and trans 10–12,14. Using microarray analysis, we identified 119 genes that show differential expression upon knockdown of DDSR1 by siRNA for 72 h as compared to control siRNAs in human fibroblasts (> 1.5-fold; P < 0.05) (Fig 3A, Table EV3). We verified microarray results by qRT–PCR validation of 15 select genes (Fig 3C), and similar results were obtained with another siRNA targeting a distinct sequence in DDSR1 as well as a locked nucleic acid antisense oligonucleotide (ASO) targeted to DDSR1 (Fig EV2A and B). Of the 119 mRNAs showing differential expression, 67 were upregulated and 52 were downregulated. IPA network analysis showed that they fall predominantly into the categories of cell death and survival (CDK6, E2F7, OLR1, JAG1, TNFSF18, MCM6, IFIT3) and DNA replication, recombination, and repair (CENPW, MCM6, ANP32E, HELLS). Interestingly, depletion of DDSR1 led to upregulation of a subset of p53 target genes (DRAM, DHRS3, HMOX1), suggesting that DDSR1 participates in transcriptional repression of p53 target genes. To further confirm that DDSR1 modulates p53 target gene expression, we performed qPCR analysis for additional p53-regulated mRNAs GADD45, TIGAR, p21, and PUMA, along with those identified in the microarray analysis upon DDSR1 knockdown in the presence and absence of DNA damage with camptothecin in U2OS cells. DDSR1 knockdown resulted in a significant upregulation of all p53 target mRNAs (Fig 3C). Moreover, the levels of DRAM1, GADD45, p21, and PUMA mRNAs were significantly higher in DDSR1 knockdown cells treated with camptothecin as compared to cells transfected with non-specific siRNA and treated with camptothecin (Fig 3C), suggesting that DDSR1 modulates p53 gene expression upon DNA damage.
Figure 3. DDSR1 modulates gene expression.
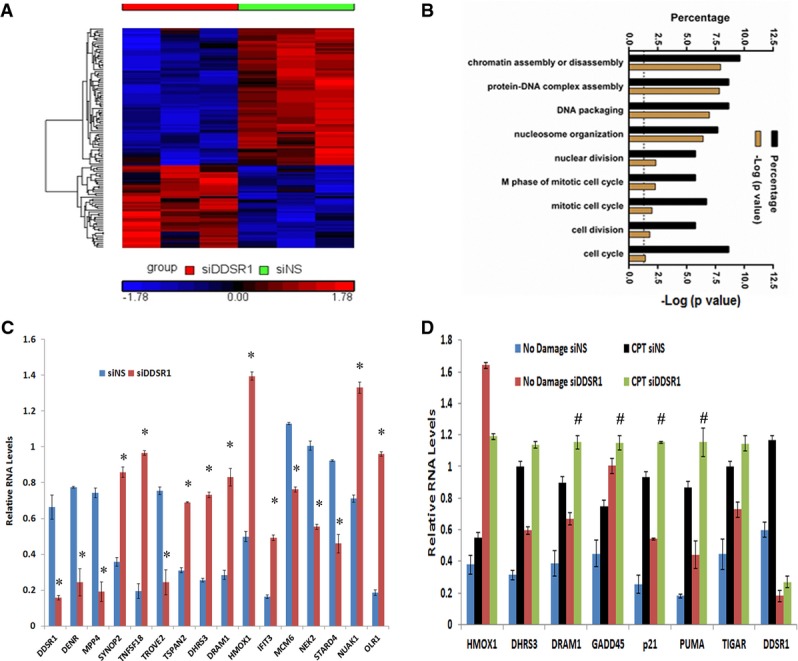
- Heatmap representing the relative abundance of differentially expressed genes (DEG, ≥ 1.5-fold, P < 0.05) in human fibroblasts upon treatment for 72 h with siDDSR1 or siNS.
- Gene ontology analysis of DDSR1-sensitive mRNAs.
- Validation of DDSR1-regulated mRNAs. Human fibroblasts were transfected with siNS or siDDSR1, and 72 h later, the transcript levels of the genes indicated were measured by qRT–PCR. RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from three independent experiments. *Significant change compared to cells transfected with siNS (P < 0.05).
- DDSR1 modulates p53 target gene expression during DNA damage. U2OS cells transfected with siNS or siDDSR1#1 were treated with or without 2 μM with camptothecin for 1 h followed by 3-h release, and transcript levels of indicated genes were measured. RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from three independent experiments. #significant change compared to cells transfected with siNS and treated with camptothecin (P < 0.05).
Data information: Statistical comparisons were made using Student’s t-test.
Figure EV2. Validation of DDSR1-regulated mRNAs by additional siRNAs.
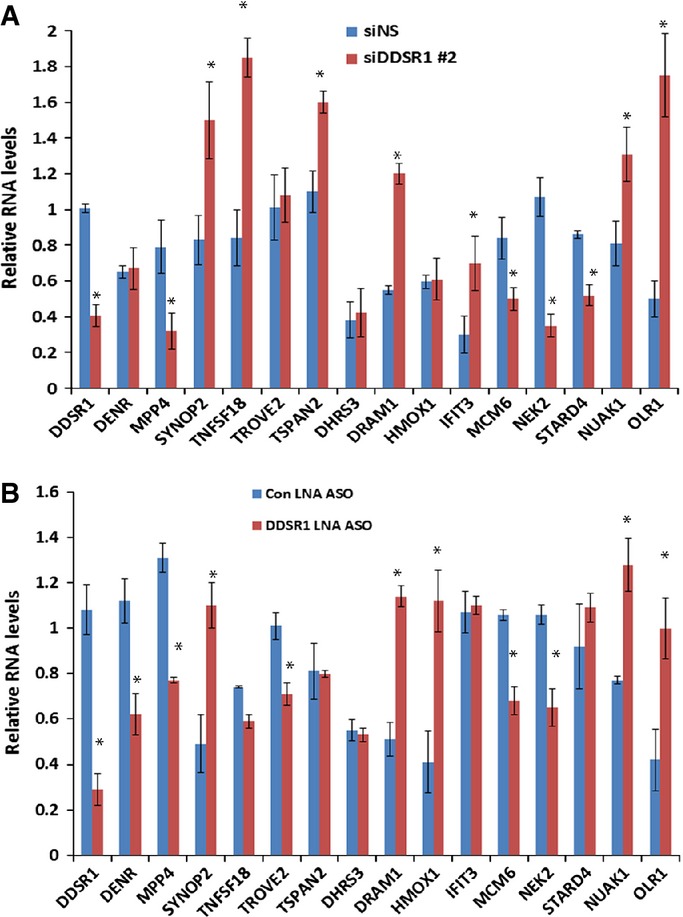
- Human fibroblasts cells were transfected with siNS or siDDSR1#2, and 72 h later, the transcript levels of the genes indicated were measured.
- Human fibroblasts cells were transfected with control locked nucleic acid antisense oligonucleotides or an antisense oligonucleotide against DDSR1, and 48 h later, the transcript levels of the genes indicated were measured.
Data information: RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from three independent experiments. *Significant change compared to control cells (P < 0.05). Statistical comparisons were made using Student’s t-test.
DDSR1 modulates cell proliferation and impairs DDR signaling
To assess the cellular effects of DDSR1 in the regulation of cell proliferation, we first examined cell proliferation in control and DDSR1-silenced human skin fibroblast cells. DDSR1 knockdown cells showed slower proliferation rates at days 3 and 4 than cells transfected with non-specific siRNA, consistent with DDSR1 modulating genes involved in cell proliferation and survival as determined by gene expression analysis (Fig 4A). Loss of DDSR1 also reduced proliferation of cancer cell lines such as U2OS, PC3, and A549 cells (Fig EV3A).
Figure 4. DDSR1 regulates cell proliferation and DDR signaling.
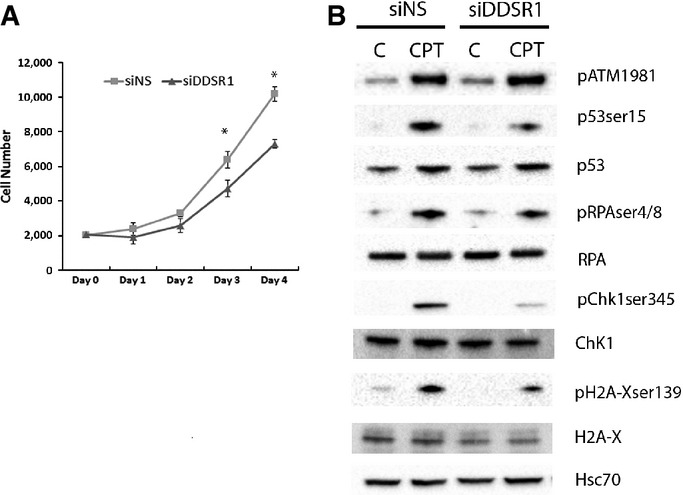
- Human fibroblasts were transfected with siNS or siDDSR1#1, and cell numbers were calculated at indicated times using WST1. Values represent mean ± SD from four independent experiments. *Significant change compared to siNS cells at corresponding time (P < 0.05). Statistical comparisons were made using Student’s t-test.
- DDSR1 impairs DDR signaling. Western blot analysis of DDR signaling molecules in U2OS cells treated with 2 μM camptothecin for 1 h followed by 3-h release after DDSR1 knockdown with siRNA#1. A representative blot is shown from three independent experiments with similar results. Blots were reprobed for Hsc-70 to establish equivalent loading.
Figure EV3. DDSR1 knockdown reduces cancer cell proliferation.
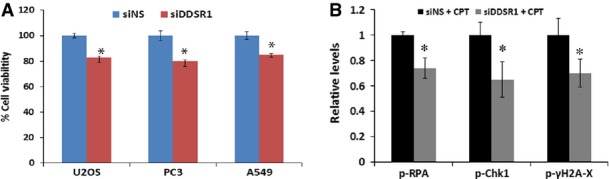
- Cell proliferation upon DDSR1 knockdown. Viability of A549, PC3, and U2OS cells was determined by WST1 assay 72 h after transfection with siNS or siDDSR1#1. The graph represents the percentage of viable cells as compared to control cells for each cell type. Viability of cells transfected with siNS for each cell line was taken as a control. Values represent mean ± SD of three independent experiments. *Significant change compared to control cells (P < 0.05).
- Densitometry measurements of Western blots shown in Fig 4B for the indicated antibodies. Values represent the means ± SD from three individual experiments. *Significant change compared to siNS cells treated with CPT (P < 0.05).
Data information: Statistical comparisons were made using Student’s t-test.
Since DDSR1 is induced upon DNA damage, we evaluated the effect of DDSR1 depletion on activation of DDR signaling molecules by Western blot analysis. Consistent with the fact the DDSR1 acts downstream of ATM, loss of DDSR1 did not alter phospho-ATM levels upon DNA damage. However, the levels of γ-H2AX, phospho-RPA, phospho-Chk1(Ser345), and phospho-p53(Ser15) were significantly reduced upon DNA damage by CPT in cells with silenced DDSR1 compared to control cells (Figs 4B and EV3B). These results suggest that DDSR1 acts downstream of ATM and modulates DDR signaling.
Loss of DDSR1 inhibits DNA repair by homologous recombination
Since DDSR1 knockdown impaired DDR signaling and its induction upon DNA damage is NF-κB dependent, combined with the fact that NF-κB is known to stimulate DNA repair by promoting homologous recombination (HR) 27, we hypothesized that DDSR1 might be involved in regulating DNA repair by HR. To evaluate this possibility, we determined HR repair efficiency upon DDSR1 knockdown using a previously characterized doxycycline-inducible I-SceI-mediated gene conversion assay in TRI-DR-U2OS cells 28, which was derived from DR-U2OS cells containing a mutant version of GFP with an internal I-SceI endonuclease restriction site, and another GFP mutant with 3′ and 5′ end truncations, neither of which expresses a functional protein 29. In this assay, a gene conversion event by recombination repair of an I-SceI-induced DSB restores wild-type GFP and indicates repair by HR 29. siRNA or antisense locked nucleic acid-mediated knockdown of DDSR1 in TRI-DR-U2OS or DR-U2OS cells reduced HR repair efficiency by ∼50% as compared to mock-transfected cells (Figs 5A and EV4A). This decrease in HR upon DDSR1 knockdown was not due to altered expression of common DNA repair genes as their transcript levels remained unaltered upon DDSR1 knockdown (Fig EV4B). DDSR1 knockdown also resulted in a modest, albeit statistically insignificant, increase in repair by NHEJ (Fig EV4C), but did not significantly change the cell cycle profile of cells (Fig 5B) or the proportion of cells in S/G2 phase as assessed by cyclin A staining (Fig EV4D), suggesting that HR repair defects due to loss of DDSR1 are not due to changes in the cell cycle profile of cells. Importantly, DDSR1 depletion did not markedly perturb DNA replication as revealed by BrDU incorporation analysis by FACS (Fig EV3E). To assess whether DDSR1 contributes to cell viability following genotoxic stress, we evaluated the ability of cells deficient in DDSR1 to sustain growth in clonogenic survival assays. Since cells deficient in HR are sensitive to PARP inhibitors due to synthetic lethality 30, we challenged DDSR1-depleted cells with the PARP inhibitor olaparib in clonogenic assays. Cells depleted for DDSR1 showed increased sensitivity to olaparib (Fig 5C). Similar results were obtained after challenge with neocarzinostatin (Fig EV3F). These results suggest a function for DDSR1 in supporting cell viability in response to genotoxic stress.
Figure 5. LncRNA DDSR1 is involved in regulating HR.

- TRI-DR-U20S cells were transfected with the indicated oligonucleotides and GFP expression was determined by flow cytometry following I-SceI induction by Dox treatment for 48 h. Efficiency of repair by HR is shown as % GFP+ cells. Values represent mean ± SEM from four independent experiments. *Significant change compared to corresponding control (P < 0.05).
- Cell cycle analysis of U2OS cells transfected with siNS or siDDSR1#1 by flow cytometry. A representative from two independent experiments is shown.
- DDSR1 regulates cell survival upon genotoxic stress. Clonogenic survival assay in U2OS cells transfected with siNS and siDDSR1#1 in response to treatment with the indicated doses of PARP inhibitor olaparib. Samples were analyzed in triplicates. Values represent mean ± SD from three independent experiments. *Significant change compared to siNS cells treated with olaparib (P < 0.05).
- Native RNA IP with hnRNPUL1. RNA levels in the IP materials were measured by qRT–PCR analysis, normalized to GAPDH mRNA levels in each IP reaction, and represented as “fold enrichment” relative to control IgG IP. Values represent mean ± SD from three experiments. *Significant change compared to control IgG or NCS IgG (P < 0.05).
- GFP-RPA recruitment in S phase cells (2–3 h post-double-thymidine block) upon DDSR1 knockdown. A box plot for GFP-RPA levels 15 min post-DSB is shown. The black line between boxes indicates the median. The box shows the 25th–75th percentile. Whiskers show the range between minimum and maximum values. The data were analyzed 15 min post-DSB upon DDSR1 knockdown with siRNA#1 (n = 39) and control cells (n =45) from two independent experiments. Representative images are shown on the right. *Significant change compared to cells transfected with siNS (P < 0.05).
- Relative quantification of ssDNA (native BrDU nuclear intensity) in S/G2 (cyclin A-positive) cells upon DDSR1 knockdown. A box plot for relative ssDNA (native BrDU nuclear intensity) levels 90 min post-12-Gy radiation upon DDSR1 knockdown is shown. The black line between the boxes indicates the median. The box shows the 25th–75th percentile. Whiskers show the range between minimum and maximum values. Cells from three independent experiments siNS (n = 926) and siDDSR1#1 (n = 1018) were analyzed. *Significant change compared to cells transfected with siNS (P < 0.05).
Data information: Statistical comparisons were made using Student’s t-test.
Figure EV4. HR inhibition upon DDSR1 knockdown is not due to cell proliferation changes.

- HR efficiency in DRGFP-U2OS cells. Cells were transfected with siNS or siDDSR1#1 followed by I-SceI expression by transient transfection. HR efficiencies were analyzed by fluorescence-activated cell sorting. Efficiency of repair by HR is shown as % GFP+ cells. Values represent mean ± SEM from three independent experiments. *Significant change compared to control cells (P < 0.05).
- DDSR1 knockdown does not affect DNA repair gene expression. U2OS cells were transfected with siNS or siDDSR1#1, and expression levels of DNA repair genes were determined by qRT–PCR. RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from three independent experiments. *Significant change compared to cells transfected with siNS (P < 0.05).
- DNA repair by NHEJ is not affected by DDSR1 knockdown. NHEJ efficiencies were analyzed by fluorescence-activated cell sorting after transient transfection of I-SceI in NHEJ-U2OS reporter cells transfected with siNS or siDDSR1#1. Efficiency of repair by NHEJ is shown as % GFP+ cells. Values represent mean ± SEM from three independent experiments.
- DDSR1 knockdown does not cause cell cycle defects. U2OS cells were transfected with the indicated oligonucleotides for 72 h and probed for cyclin A expression by immunofluorescence. Samples were analyzed in quadruplicates with ≥ 300 cells per sample and values expressed as mean ± SD.
- DDSR1 knockdown does not perturb DNA replication. Cells transfected with the indicated oligonucleotides were pulsed with BrDU for 2 h and analyzed by fluorescence-activated cell sorting. Values are expressed as the % BrDU-positive cells per 10,000 7-AAD events from triplicates. Bars represent mean values ± SD.
- DDSR1 regulates cell survival upon genotoxic stress. Clonogenic survival assay in U2OS cells transfected with siNS and siDDSR1#1 in response to treatment with the indicated doses of NCS. Samples from three independent experiments were analyzed in triplicates. Values represent mean ± SD. *Significant change compared to siNS cells treated with NCS (P < 0.05).
Data information: Statistical comparisons were made using Student’s t-test.
DDSR1 interacts with mediators of HR
LncRNAs often exert their functions via interaction with protein complexes. To identify proteins that associate with DDSR1, we used MS2-TRAP-tagged RNA affinity purification 31. DDSR1 tagged with a multimer of viral MS2 stem loops was co-expressed with a MS2 coat protein (MCP)-GST fusion protein recognizing the MS2 RNA hairpins, and MS2-DDSR1 RNA/protein complexes were affinity purified using GST beads (see Materials and Methods for details). Mass spectrometry analysis identified several DNA damage-related proteins including factors involved in HR, such as heterogeneous nuclear ribonucleoprotein U-like 1 (hnRNPUL1, also known as E1B-AP5), previously implicated in DNA repair, UBR2, the BRCA1-interacting partner BRIP/FANCJ, and RAD54B, which all specifically associated with DDSR1 upon DNA damage, but not with MS2 control RNA (Table EV4). Association of lncRNA DDSR1 with RNA-binding protein hnRNPUL1 in the presence and absence of DNA damage was confirmed by immunoprecipitation (IP) of ribonucleoprotein (RNP) complexes (RIP analysis) with anti-hnRNPUL1 in parallel with control immunoglobulin G (IgG) IP. DDSR1 was enriched ∼1.8-fold in hnRNPUL1 IP samples (P < 0.05) compared with IgG IP samples, and this association was found to be further increased upon DNA damage (Fig 5D). Since hnRNPUL1 depletion has previously been shown to inhibit DNA repair by regulating DNA end resection 32, we evaluated resection upon DDSR1 depletion by GFP-RPA recruitment to laser-induced DSBs and by detection of ssDNA formation by native BrDU staining upon irradiation in S/G2 cells. DDSR1 depletion led to significantly reduced GFP-RPA recruitment to DSBs (Fig 5E; P < 0.02) and also reduced ssDNA formation in S/G2 cells (Fig 5F; P < 0.001), demonstrating reduced resection efficiency in the absence of DDSR1. These observations are consistent with reduced pRPA and pChk1 levels observed upon DNA damage in DDSR1-depleted cells (Fig 4B) and confirm a role for DDSR1 in regulating HR by modulating DNA end resection.
DDSR1 regulates BRCA1 recruitment to DSBs
Having established a role for DDSR1 in modulating DNA repair by HR, we next asked how the depletion of DDSR1 accounts for the observed HR defects. Since BRCA1 is a key regulator of DNA repair by HR 33, we probed the effect of DDSR1 depletion on BRCA1 recruitment to laser-induced DSBs by live cell imaging in a previously described BRCA1-GFP stable cell line 34. siRNA-mediated depletion of DDSR1 significantly increased BRCA1 accumulation at laser-induced DSBs (Fig 6A). At 4 min after damage, BRCA1 levels were 1.25-fold higher in the absence of DDSR1 compared to control cells (P < 0.05) and the difference was maintained over the duration of the measurement with about 1.4-fold more BRCA1 at in DDSR1 knockdown cells as compared to control cells at 10 min post-damage (Fig 6B; P < 0.05, see heatmap in figure).
Figure 6. DDSR1 modulates recruitment of repair factor to DSBs sites.

- Recruitment kinetics of GFP-BRCA1 to laser-induced DSBs in siNS (n = 47) and siDDSR1#1 (n = 50) cells.
- Recruitment kinetics of GFP-RAP80 in synchronized cells to laser-induced DSBs in siNS (n = 32) and siDDSR1 (n = 42) cells.
- DDSR1 knockdown alters 53BP1 recruitment. U2OS cells transfected with siNS or siDDSR1#1 were treated with or without 10 μM camptothecin for the indicated times and probed for 53BP1 expression by immunofluorescence. y-axis represents average 53BP1 spot number per cell. Samples were analyzed in quadruplicates with ≥ 225 cells per sample and values expressed as mean ± SD.
- DDSR1 knockdown alters γ-H2AX recruitment. U2OS cells transfected with siNS or siDDSR1#1 were treated with or without 10 μM camptothecin for the indicated times and probed for γ-H2AX expression by immunofluorescence. y-axis represents average intensity of nuclei. Samples were analyzed in quadruplicates with ≥ 225 cells per sample and values expressed as mean ± SD.
Data information: For (A, B), two independent experiments were combined. Datasets were subjected to Student’s two-tailed t-test at each imaging time point. The heatmap at top of the graph depicts the P-value distribution over time. Gray indicates non-significance. Representative images are shown on the top. (C, D) *Significant change compared to cells transfected with siNS at corresponding time point (P < 0.05). Statistical comparisons were made using Student’s t-test.
Although BRCA1 recruitment at DSBs primarily promotes DNA repair by HR 35,36, BRCA1 interacts with the ubiquitin-binding protein RAP80, which promotes its recruitment to DSBs 37 and the aberrant activity of this BRCA1-RAP80 complex limits HR by inhibiting DSB end resection 38–40. Hence, we probed the effect of DDSR1 depletion on RAP80 recruitment to laser-induced DSBs. As expected, DDSR1 knockdown resulted in an increase in RAP80 recruitment to sites of damage as compared to cells transfected with non-specific siRNA (Fig 6B). While initial recruitment of RAP80 during the first 90 s was unaffected by DDSR1 loss, the subsequent rate of accumulation of RAP80 was significantly increased in the absence of DDSR1 (Fig 6B; P < 0.05, see heatmap in figure). Knockdown of DDSR1 by siRNA in U2OS cells did not result in any significant changes in accumulation of γ-H2AX and 53BP1 upon DNA damage by camptothecin compared to cells transfected with non-specific siRNA at early stages of DNA damage (Fig 6C). However, 1 h post-DNA damage, 53BP1 recruitment was modestly increased and γ-H2AX levels concomitantly decreased in DDSR1 knockdown cells as compared to cells transfected with non-specific siRNA (Fig 6D). These results suggest that loss of DDSR1 leads to increased recruitment of BRCA1 and RAP80 and in this way limits HR.
hnRNPUL1 modulates BRCA1 and RAP80 recruitment to DSBs
Since DDSR1 interacts with hnRNPUL1 and alters BRCA1 recruitment to laser-induced DSBs, we next examined whether hnRNPUL1 plays any role in BRCA1 recruitment to DSBs. Depletion of hnRNPUL1 led to a significant increase in BRCA1 recruitment to laser-induced DSBs similar to the effect of DDSR1 depletion on BRCA1 recruitment (Fig 7A). We then evaluated the effect of hnRNPUL1 depletion on RAP80 recruitment to laser-induced DSBs. Consistent with an increase in BRCA1 recruitment upon hnRNPUL1 depletion, RAP80 recruitment to laser-induced DSBs was also elevated upon hnRNPUL1 depletion as compared to cells transfected with non-specific siRNA (Fig 7B). These changes in BRCA1 and RAP80 recruitment upon hnRNPUL1 knockdown were not due to altered DDSR1 expression, since DDSR1 transcript levels remain unaltered upon hnRNPUL1 depletion (Fig 7C). These results point to a functional interaction between DDSR1 and hnRNPUL1 which likely modulates HR by regulating access of BRCA1 and RAP80 to DSBs.
Figure 7. hnRNPUL1 modulates recruitment kinetics of BRCA1.
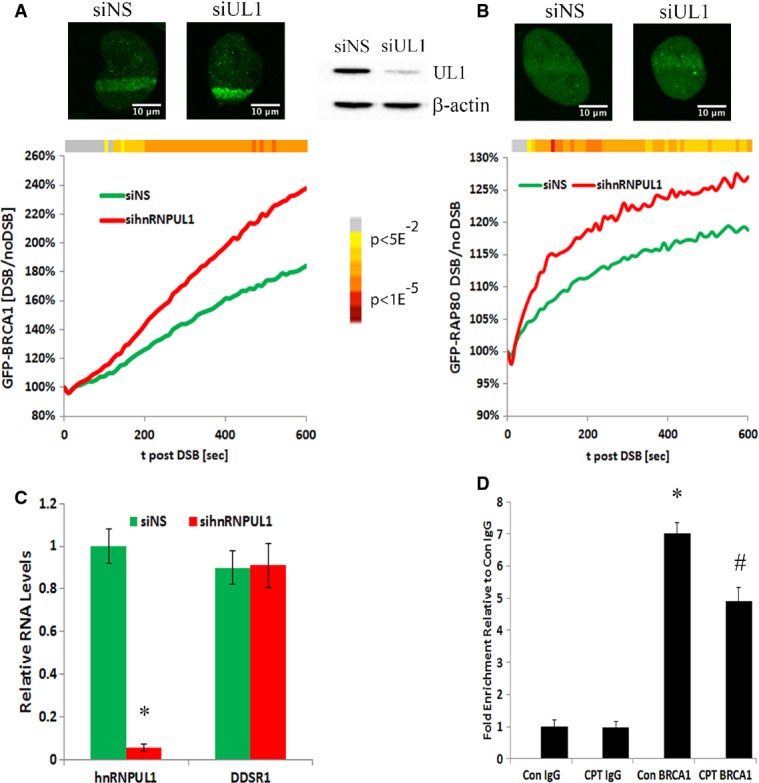
- Recruitment kinetics of GFP-BRCA1 to laser-induced DSBs in siNS (n = 54) and sihnRNPUL1 (n = 45) cells.
- Recruitment kinetics of GFP-RAP80 in synchronized cells to laser-induced DSBs in siNS (n = 36) and sihnRNPUL1 (n = 41) cells.
- DDSR1 levels upon hnRNPUL1 depletion. RNA samples were analyzed by quantitative RT–PCR, and error bars represent the mean ± SEM from three independent experiments. *Significant change compared to control (P < 0.05).
- Formaldehyde-cross-linked RNA IP with BRCA1. DDSR1 RNA levels in immunoprecipitated samples were measured by qRT–PCR analysis, normalized to input, and represented as “fold enrichment” relative to control IgG IP. Values represent mean ± SD from four independent experiments. *Significant change compared to control IgG or CPT IgG (P < 0.05). #Significant change compared to Con BRCA1 (P < 0.05).
Data information: For (A, B), two independent experiments were combined. Datasets were subjected to Student’s two-tailed t-test at each imaging time point. The heatmap at top of the graph depicts the P-value distribution over time. Gray indicates non-significance. Representative images are shown on the top. (C, D) Statistical comparisons were made using Student’s t-test.
BRCA1 interacts with DDSR1
We did not pull down BRCA1 in our RNA affinity mass spectrometry screen, since DDSR1 knockdown altered recruitment of BRCA1 to DSBs sites and DDSR1 interacts with BRCA1-interacting proteins such as FANCJ and MSH6 (Table EV4). We hypothesized that DDSR1 binds to BRCA1 to modulate its access to DNA repair sites. To test this hypothesis, we performed formaldehyde-cross-linked RIP with BRCA1. We detect significant BRCA1 binding to DDSR1 (7-fold enrichment compared to IgG IP; P < 0.05) and this association was decreased by ∼30% upon DNA damage with camptothecin (Fig 7D). These results are in line with our observation in imaging experiments, suggesting a role of DDSR1 in controlling the access of BRCA1 to sites of DNA damage.
Discussion
We describe here a lncRNA involved in the DNA damage response and characterize its role in homologous recombination. DDSR1 is a 1.6-kb transcript induced with intermediate kinetics by the ATM-NF-kB pathway in response to DNA damage by several agents. DDSR1 interacts with several proteins involved in promoting HR including BRCA1 and hnRNPUL1. DDSR1 loss impairs DDR signaling, alters recruitment of HR repair factors BRCA1 and RAP80 to DSBs, and affects DNA repair by HR. Based on these observations, we suggest that DDSR1 is a regulatory factor in DDR and plays an important role in maintaining genome stability.
The activation of the cellular DDR results in a complex signaling cascade leading to cell cycle arrest so as to allow cells to repair damage or, if the damage is irreparable, to eliminate them via apoptosis or entry into senescence. Both miRNAs and lncRNAs are emerging as important contributors to regulation of the cell cycle and of apoptosis and are as such likely key players in the DDR 16. DROSHA- and DICER-dependent small RNAs called DDRNAs, generated from regions near the DSB site, contribute to DNA repair foci formation 41. Moreover, two RNA-binding proteins, RBMX and hnRNPUL 1, are recruited to the site of DSBs and promote HR 32,42. It is likely that additional RNA species interacting with these and other repair proteins may be involved in DNA repair by HR. Two lncRNAs are known to be involved in promoting HR. Depletion of lncRNA ANRIL inhibits DNA repair by HR 19, although the molecular mechanisms of ANRIL-mediated HR are unknown. Interestingly, just like DDSR1, ANRIL modulates both gene expression and DNA repair by HR 19,43. A second lncRNA known to be involved in regulating HR is prostate-specific lincRNA PCAT1, whose expression is not responsive to DNA damage, but it inhibits HR in prostate tissue by causing posttranscriptional repression of BRCA2 24.
ATM-mediated DNA damage signaling regulates the expression of lncRNAs and micro-RNA biogenesis 18,44. Similar to the DNA damage-responsive lncRNA Jade1 18, DDSR1 induction upon DNA damage is regulated by the ATM-NF-κB pathway. DDSR1 induction upon DNA damage is p53 independent. On the other hand, we find p53 is sufficient to induce DDSR1 expression, although we did not find any p53-responsive elements on the putative promoter region of DDSR1, suggesting that induction of DDSR1 by p53 is not by direct binding at its promoter, but likely through other p53 targets associated with its transcription or p53 binding to enhancer regions.
Expression of several lncRNAs has been linked to p53, and they participate in the p53 network 20–22. Interestingly, loss of DDSR1 leads to upregulation of p53 target genes, suggesting that DDSR1 negatively regulates p53-mediated gene expression. This is not surprising as several p53-induced lncRNAs, such as TUG1, lincRNA-p21, and PINT1, are involved in negatively regulating p53 targets by various mechanisms 20,22,45. In addition, knockdown of DDSR1 leads to mis-regulation of numerous genes involved in DNA damage and repair such as CENPW, MCM6, ANP32E, HELLS, HIST1H2A, and HIST1H2B. Considering that many lncRNAs appear to act in trans on target genes, it is possible that some of these effects are direct gene regulatory events rather than secondary events. Based on these observations, it is tempting to speculate that DDSR1 interacts with protein complexes to transcriptionally or epigenetically repress DNA damage response genes, including some in the p53 pathway.
DDSR1 is induced relatively late upon DNA damage (∼3 h), at a time when recruitment of repair factors to DSBs and initial phases of DNA repair have already occurred. Yet, we find that loss of DDSR1 acutely reduces DNA repair efficiency by HR, indicating that the basal levels of DDSR1 are important for DNA repair. In line with this observation, while DDSR1 is induced about 2- to 3-fold over several hours in response to DNA damage, it is also expressed in undamaged cells at moderate basal levels. Intriguingly, the decrease in repair efficiency by HR upon DDSR1 knockdown is accompanied by increased recruitment of BRCA1 and its recruitment factor, the ubiquitin-binding protein RAP80 37, to the sites of DNA damage. These observations, coupled with the fact that DDSR1 interacts with BRCA1 and this interaction is reduced upon DNA damage, suggest that DDSR1 sequesters the BRCA1-RAP80 complex and prevents it from promiscuous DNA binding. This model is consistent with our finding that we cannot detect DDSR1 at sites of laser-induced DSBs, although we cannot strictly exclude the possibility that low levels of endogenous DDSR1 are present but undetectable at DSB sites. One possibility the cells require a mechanism to carefully control the amount of BRCA1-RAP80 cells binding to DSBs is because of the ability of this complex to restrict DNA end resection, thereby limiting DNA repair by HR 38,39. This interpretation agrees well with our finding that DDSR1 knockdown results in reduced DNA end resection. It is possible that that low basal levels of DDSR1 regulate BRCA1 recruitment and access to DSB sites. However, upon persistent DNA damage, when DDSR1 levels become limited, its induction is triggered, via the ATM and NF-κB signaling pathways, to maintain the correct levels of BRCA1 at sites of DNA damage so as to ensure efficient repair or to remove BRCA1 from the DSB sites once repair is complete. Our findings suggest that DDSR1 functions in DDR repair progression and BRCA1 turnover at DNA repair sites.
In line with a role of DDSR1 in HR, we also observed that DDSR1 interacts with hnRNPUL1, a protein which is transiently recruited to DSBs sites and is required for effective DNA end resection and DSB repair by HR 32. Recruitment of hnRNPUL1 to DSBs is dependent on the MRN complex 32 and poly(ADP-ribosyl)ation mediated by PARP1 46, but is RNA independent 32. Hence, DDSR1 may not be involved in recruitment of hnRNPUL1 to DSBs. Depletion of hnRNPUL1 is thought to limit DNA repair by inhibiting recruitment of the BLM helicase to DSB, but the resection defect upon hnRNPUL1 depletion cells is not overcome by BLM overexpression 32. This suggests the existence of additional defects, which lead to DNA repair inhibition upon hnRNPUL1 depletion. In contrast to the earlier finding that hnRNPUL1 knockdown has no effect on BRCA1 recruitment at DSBs 32, we observe that knockdown of hnRNPUL1 leads to increased recruitment of BRCA1 at laser-induced DSBs similar to the effect observed upon DDSR1 depletion. This discrepancy is likely due to different technical and experimental conditions used for measurement of BRCA1 recruitment (BRCA1 foci formation in fixed cells after IR and camptothecin versus BRCA1-GFP recruitment in living cells to laser-induced DSBs). Similar to what is seen in DDSR1-depleted cells, BRCA1 recruitment at DSBs in hnRNPUL1-depleted cells was accompanied by enhanced recruitment of RAP80. These results allow for the possibility that the DNA end resection and HR defects observed upon hnRNPUL1 depletion are, at least in part, due to increased BRCA1 and RAP 80 recruitment. Taken together, these results suggest that the interaction of hnRNPUL1 with DDSR1 plays a role in regulating BRCA1 and RAP80 access to DSBs in modulating HR. Intriguingly, hnRNPUL1 is also implicated in regulation of transcription and p53-mediated gene expression 47,48 and it is plausible that the interaction of DDSR1 with hnRNPUL1 may also contribute to its role in transcription regulation.
In sum, in this study we have identified and characterized the lncRNA DDSR1 as a regulator of DNA repair by HR. The involvement of lncRNAs in DNA repair pathways expands the number and diversity of molecular components in the DDR and suggests that the DDR is even more complex than currently appreciated.
Materials and Methods
Cell culture and treatments
Human hTert-immortalized fibroblast cells 49 were grown in MEM containing high glucose supplemented with glutamine and 15% fetal bovine serum (FBS) (Atlanta Biologicals). U2OS, A549, PC3, H1299 (p53-null) were obtained from ATCC and were grown in DMEM containing high glucose, supplemented with glutamine and 10% FBS. To induce DNA damage, cells were treated with DNA-damaging agents neocarzinostatin (NCS) (#N9162), camptothecin (CPT) (#C9911), etoposide (#1383), or bleomycin (#2434) all from Sigma-Aldrich for indicated doses and times. To examine the role of ATM and NF-κB in lncRNA DDSR1 induction, cells were pretreated with 20 μM KU55933 (ATMi, Tocris) or 10 μM NF-κB inhibitor BAY11-7085 (Tocris) 1 h prior to DNA damage treatment and treatment was continued throughout the course of the experiment along with DNA damage-inducing agent. For Western blotting, cells were treated with 2 μM camptothecin (CPT) for 1 h, washed, and kept for 3 h before protein isolation. For I-SceI induction, cells were treated with 5 μg/ml doxycycline for the indicated time periods. For laser micro irradiation-induced DSB generation, cells were presensitized with Hoechst 33342 (0.1 μg/ml) for 60 min. For double-thymidine block, cells were treated with 2.5 mM thymidine (Sigma) for 18 h, followed by a 9-h release and a second 17-h thymidine treatment. For clonogenic survival assays, cells were transfected with siRNAs and were treated overnight with the indicated doses of olaparib (AZD-2281, Selleckchem).
Cell proliferation assays
For cell proliferation assay, cells were seeded at a concentration of 2,500–5,000 cells/well in 96-well plates and transfected with siRNAs at 100 nM. Cell numbers were quantified using reagent WST-1 (Cat#. 05015944001, Roche) at the indicated time according to the manufacturer’s instructions.
Colony formation assay
For clonogenic assays, U2OS cells were seeded into 60-mm dishes and treated with or without olaparib or neocarzinostatin at indicated doses 72 h after transfection with 100 nm siRNA against lncRNA-DDSR1 or non-specific siRNA. Colonies, which formed between 14 and 16 days after plating, were stained with a crystal violet solution and counted. Assays were done in triplicate.
Flow cytometric analysis of DNA content and BrDU incorporation in dsDNA
U2OS cells 72 h post-transfection with siNS or siDDSR1#1 were fixed in 70% ethanol and stored at −20°C. The fixed cells were washed in PBS, resuspended in propidium iodide solution (BD Biosciences) for 20 min at room temperature, and analyzed on FACS Calibur (Becton Dickinson). BrDU incorporation in dsDNA (replicating cells) of U2OS cells upon DDSR1 knockdown for 72 h was determined using FITC BrdU Flow Kit (BD Biosciences San Jose, CA) as per manufacturer’s instructions. Analysis was carried out using FlowJo software package.
RNA FISH
RNA FISH for DDSR1 detection was performed using a pool of 20 fluorescent probes purchased from Stellaris Biosearch Technologies, following the manufacturer’s protocol.
qRT–PCR
Nuclear RNA was isolated using the Ambion® PARIS™ system (Life Technologies). Total RNA was isolated using the RNeasy plus mini kit (Qiagen). cDNA synthesis was carried out with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was performed in triplicates with the iQ™ SYBR® Green supermix (Bio-Rad) on a Bio-Rad CFX-96 real-time PCR system. The comparative Ct method was employed to quantify transcripts. Normalization was performed to TBP expression. Primers used in this study are provided in Table EV5.
5′–3′ RACE
Total RNA extracted from human fibroblast cells was used to perform RACE to identify the full-length sequence of lncRNA DDSR1. 3′ RACE was performed using the RLM RACE kit (Life Technologies) following the manufacturer’s protocol. cDNA ends were amplified with universal primer mix and gene-specific primers. The first PCR product was used to perform a “nested” PCR with the nested universal primer and the nested gene-specific primers. 5′ RACE-PCR experiments were performed using 5′/3′ RACE kit (Roche Molecular Biochemicals) according to the manufacturer’s instructions. Primer sequences were as follows (5′ to 3′):
Reverse transcription for 5′ RACE: TCTCTGATAAATGGAGTTGACCC
GSP1 (3′ RACE): AAGAGTGGAAAATTCATCCCCA
GSP1 (5′ RACE): TGTCATGTTTCAGATATTTTCCCCA
Nested GSP2 (3′ RACE): TGCAACAATAGCTGGTACTTACT
Nested GSP2 (5′ RACE): CCCCAGTATTTCCCTTATCTTTTGA
The resulting DNA fragments were cloned by TA cloning and sequenced (Genewiz). Sequence data of the full-length human lncRNA-DDSR1 can be found in Dataset EV1. Using the sequence information from the two ends, we cloned the full-length DDSR1 sequence and tagged it with MS2 haipins.
siRNA and plasmid transfections
Transfections of siRNAs or antisense locked nucleic acid gapmers were done using Dharmafect 1 as per manufacturer’s instructions. Fibroblasts were transfected once with 100 nM duplex siRNA. U2OS or its derivative cell lines were transfected twice with 75 nM duplex siRNA on successive days or once with 50 nM antisense locked nucleic acid oligonucleotides. The siRNA duplexes (Dharmacon) used in this study are as follows siRNA #1 for DDSR1: sense 5′CUAGUGUGUUGCAAAUAAAUU-3′, antisense 5′P-UUUAUUUGCAACACACUAGUU-3′; siRNA #2 for DDSR1: sense 5′-GCUACUAGUCUAAGAGUUAUU-3′, antisense 5′P-UAACUCUUAGACUAGUAGCUU-3′; siRNA against hnRNPUL1: sense 5′-GCAGUGGAACCAGUACUAUUU-3′, antisense 5′-AUAGUACUGGUUCCACUGCUU-3′.
ON-TARGET plus non-targeting pool (# D-001810-10) was used as a control. The sequence of antisense oligonucleotide against DDSR1 and negative control purchased from Exiqon is as follows: 5′-TTCGATGCGCAATTAA-3′ and 5′-AACACGTCTATACGC-3′, respectively. The p53 overexpression construct was a gift from Dr. Bert Vogelstein and is described in 50. MS2 coat protein (MCP)-GST construct is described in 51. GFP-RAP80 was a gift from Roger Greenberg 52. GFP-RPA plasmid was a gift from Marc Wold 53. Plasmids were transfected using X-tremeGENE HP DNA Transfection Reagent (Roche) following the manufacturer’s instructions.
DNA repair assays
HR assays upon DDSR1 knockdown were done in TRI-DR-U2OS cells expressing inducible I-SceI cells or after transient I-SceI transfection in DR-GFP U2OS cells as described previously 28. NHEJ assay after transient I-SceI transfection was done in stable, NHEJ-U2OS reporter cells generated as described 28 using the pEJ5 construct 54. HR or NHEJ efficiencies were analyzed by fluorescence-activated cell sorting and are shown as % GFP+ cells.
Immunofluorescence microscopy
After siRNA knockdowns, cells were treated with camptothecin for indicated time and fixed with 4% buffered paraformaldehyde in PBS for 15 min at room temperature followed by permeabilization with 0.5% Triton X-100 in PBS. Following permeabilization cells were blocked in 5% BSA in 0.05% PBS Tween-20 for 1 h. Blocked cells were stained with antibodies for cyclin A [1:100, Neomarkers # Ab6 (6E6)] or co-stained [γH2AX (1:1,000, Millipore #05636) and 53BP1 (1:2,000, Novus# 100304)]. Alexa Fluor donkey anti-mouse 488 (1:500) or Alexa Fluor donkey anti-mouse 568 (1:500) and Alexa Fluor donkey anti-rabbit 647 (1:500) were used for secondary labeling. Nuclei were labeled using 5 ng/μl DAPI and imaged on opera microscope using a 20× objective. For native BrDU staining to visualize ssDNA in S/G2 cells, cells transfected with siNS or siDDSR1#1 were pulsed with 10 μM BrdU for 36 h and exposed to 12 Gy radiation. Ninety minutes after irradiation, cells were extracted and fixed in methanol at −20°C for 30 min, followed by rinsing in 100% cold acetone. Cells were then rehydrated in PBS solution and blocked in 10% FCS in PBS solution for 1 h, followed by incubation with primary antibodies for BrDU (1:500; BD Biosciences #347580) and cyclin A (1:50; Santacruz # sc-751) diluted in 2.5% BSA in PBS solution for 2 h at room temperature. Cells were then washed 3× with PBS (0.05% Tween-20) solution and incubated with goat anti-mouse Alexa 568(1:500) and goat anti-rabbit Alexa 488 (1:500) for 1 h. The cells were then washed 4× with PBS (0.05% Tween-20) solution, and mounted in vectashield mounting medium containing DAPI from Vector Laboratories. Images of the cells were captured with a CCD camera on a Zeiss LSM 780 Meta confocal microscope using a 40× objective. The BrDU intensity of cyclin A-positive cells (S/G2) was analyzed using a custom-built macro plugin for Image J.
Immunoblotting
Cells lysates were prepared using RIPA buffer supplemented with protease and phosphate inhibitors. Protein samples were run on a 4–12% bis-tris gel and transferred to a PVDF membrane. Primary antibodies against pATM (1:5,000; Abcam# ab81292), pp53 (1:1,000, Cell Signalling #9284), p53 (Santa Cruz# sc126), pChk1 (1:1,000; Cell Signalling #2341), pRPA (1:2,000, Bethyl #A300-245), RPA (1:5,000; Abcam# ab2175), Chk1 (1:1,000; Santa Cruz #sc-8408), H2AX (1:2,000; Novus #NB100-638), hnRNPUL1 (Novus#NB110-40586), and γ-H2AX (1:2,000; Millipore) were incubated overnight at 4°C. HRP-anti-rabbit or mouse immunoglobulins (1:20,000; Santa Cruz) were incubated for 1 h at room temperature and ECL-plus kit (Amersham) was used for blot development. Anti-Hsc70 antibody (1:20,000; Abcam #ab19136) was used for normalization. All membrane images were acquired using a ChemiDocTM MP imaging system (Bio-Rad).
Microarray
RNA labeling and hybridization were carried out by the Laboratory of Molecular Technology, SAIC-Frederick. To identify ncRNAs induced upon DNA damage, total RNA or nuclear RNA was reverse-transcribed, labeled, and hybridized to the NCode™ Human array following the Agilent’s Quick Amp Labeling Kit protocol. Fold changes relative to control were calculated by averaging fold change observed with each drug. To identify genes regulated by DDSR1, total RNA was reverse-transcribed, labeled, and hybridized using GeneChiP® Human Gene 2.0 ST array following the Ambion ®WT Expression Kit Protocol. Data were analyzed using Partek Genomics Suite (Partek Incorporated), and differentially expressed genes (DEG) were defined as genes having ≥ 1.5-fold change of expression in pairwise comparisons, with a P-value < 0.05.
Native RNA immunoprecipitation
Native RIP was performed as described 55, with minor modifications. Briefly, control and DNA-damaged cells were suspended in ice-cold cell disruption buffer (20 mM Tris pH 7.5, 10 mM KCl, and 1.5 mM MgCl2, 0.1% Triton X-100) supplemented with protease, phosphatase, and RNAse inhibitors for 10 min on ice and centrifuged at 1,500 g for 5 min at 4°C to isolate the nuclei. To isolate RNA-containing nuclear extracts, nuclei were resuspended in polysome lysis buffer (10 mM HEPES pH 7.0, 100 mM KCl, 5 mM MgCl2, 0.5% NP-40, 10 μM DTT) supplemented with protease, phosphatase, and RNAse inhibitors for 10 min on ice and centrifuged at 10,000 g for 15 min at 4°C. The supernatants were incubated with antibodies against hnRNPUL1 (Novus #NB110-40586) and control IgG (Cell Signalling #2729) over night at 4°C. Antibody-bound protein and RNA was collected using 40 μl protein G magnetic beads (Pierce). The beads were then washed 4× for 5 min with NT2 buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1 mM MgCl2, and 0.05% NP-40) supplemented with protease, phosphatase, and RNAse inhibitors. To isolate RNA, beads were resuspended in TRIzol followed by isolation of RNA from the aqueous fraction using the RNeasy Micro Kit (Qiagen). RNPs isolated from the IP materials were further analyzed by qRT–PCR. Normalization of RIP results was carried out by quantifying in parallel the relative levels of GAPDH mRNA in each IP sample.
Formaldehyde-cross-linked RIP
BRCA1 RIP was performed as described 56, with minor modifications. Briefly, 30 million cells treated with or without 10 μM camptothecin for 30 min were fixed with 1% formaldehyde in PBS to cross-link protein-bound RNA/DNA complexes, and formaldehyde was quenched by the addition of glycine to a concentration of 0.125 M. Cells were then washed with PBS and incubated in 1 ml collection buffer [100 mM Tris–HCl (pH 9.4) and 100 mM DTT, protease inhibitor, RNaseOUT] on ice for 15 min. Nuclei were isolated by resuspension in 1 ml buffer A (10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, and 0.25% Triton X-100, protease inhibitor, RNaseOUT) and centrifugation at 3,000 g at 4°C. Nuclear pellets were then sonicated using bioruptor (Diagenode) in 900 μl buffer B (1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, and 200 mM NaCl) to produce DNA/RNA fragments of about 500 nucleotides. The sonicated extract was split into equal aliquots, made up to a total of 1.3 ml with IP buffer (1% Triton X-100, 0.1%DOC, 1× TE, RNaseOUT), 50 μl of the Dynabeads and 4 μg BRCA1 (Ab-1; Calbiochem) or IgG (12-371; Millipore) antibodies, and incubated overnight at 4°C. The beads-bound RNA/DNA complexes were washed six times with RIPA buffer (50 mM Hepes pH 8, 1 mM EDTA, 1%NP-40, 0.7% DOC, 0.5 M LiCl, and protease inhibitor mix, RNaseOUT), and once with TE buffer. The DNA/RNA was extracted from the beads by the addition of 100 μl of elution buffer (10 mM Tris pH 8, 1 mM EDTA, 1% SDS, RNaseOUT) and incubated at 65°C with gentle shaking for 15 min. For reversing the cross-linking, elution buffer containing RNA/DNA was supplemented with 2 μl of 5 M NaCl and 2 μl proteinase K (NEB) and made up to a total volume of 170 μl and incubated for 5 h at 70°C. After reverse cross-linking, 750 μl of TRIzol was added per sample and RNA extracted as per manufacturer’s instructions. DNase I (Roche) treatment was performed as per manufacturer’s protocol, and cDNA synthesis was carried out using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was performed in triplicates with 2 μl cDNA using the iQ™ SYBR® Green supermix (Bio-Rad) on a Bio-Rad CFX-96 real-time PCR system. The primers used for DDSR1 are forward 5′-AAGAGTGGAAAATTCATCCCCA-3′ and reverse 5′-TGTCATGTTTCAGATATTTTCCCCA-3′. The results are expressed as fold enrichment over input and normalized to IgG.
Laser microirradiation and imaging
Laser microirradiation was performed using a Zeiss LSM780 confocal microscope. Before imaging, cells were incubated with 0.1 μg/ml Hoechst 33324 for 1 h and then switched to FluoroBrite phenol red-free media containing 10% fetal bovine serum and glutamine, antibiotics without Hoechst and 5 mM HEPES. The cells were damaged with a 355-nm (10%) laser beam, with total UV laser output set to 20%, ten iterations, and laser scan speed set to 7 (pixel dwell time 3.15 μs) as described previously 57. The cells were laser-microirradiated in a rectangle and images taken every 10 s for 10 min, maintaining cells at 37°C and 5% CO2. To measure the recruitment of GFP-BRCA1 or GFP-RAP80 in laser-microirradiated cells, MIPAV software (v.5.1) was used. For measurements, the regions of interest were drawn within the laser-microirradiated regions, within a non-laser-microirradiated regions in the nucleus and a region outside the cells 28. The mean fluorescence intensity within the region of interest was measured for each time point (10 s) within the time series. The fluorescence intensity values were background subtracted, and the ratio of intensity within the microirradiated nuclear area to non-micro-irradiated area was calculated. The ratios were normalized to prelased time point.
MS2-TRAP for tagged RNA affinity purification
Human fibroblasts in 10-cm dishes were co-transfected with 6 μg of plasmid containing DDSR1 tagged with MS2 stem loop RNA or MS2 stem loop RNA alone as a control together with 3 μg MS2 coat protein (MCP)-GST construct using X-tremeGENE (Roche) according to the manufacturer’s instructions. Twelve hours post-transfection, cells were treated with NCS (50 ng/ml), or not treated as a control for 24 h. Cells were collected and lysed in lysis buffer (20 mM Tris–HCl at pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.3% IGEPAL CA-630) supplemented with protease (Sigma), phosphatase (Calbiochem), and RNAse inhibitors (Promega), and 10 mM DTT for 30 min on ice. This was followed by two freeze–thaw cycles on dry ice to help nuclear lysis. Cell lysates were cleared of debris by centrifugation at 10,000 g for 15 min at 4 °C. For isolation of MCP-GST-bound RNPs, cleared lysates were incubated at 4°C for 3 h with glutathione magnetic beads (Thermo Scientific # 88821) and washed as per manufacturer’s instructions. After washing, glutathione beads were suspended in 25 mM ammonium bicarbonate pH 8.4 and processed for mass spectrometry.
Statistical analysis
Results are presented as mean ± SEM, unless otherwise stated. We used paired Student’s t-test for comparisons between two experimental groups. Additional statistical tests information is described in the figure legends. P < 0.05 was considered statistically significant.
Accession numbers
The lncRNA DDSR1 sequence has been deposited to GenBank with accession number KT318134. The NCBI Gene Expression Omnibus accession number for the microarray data is GSE72358.
Acknowledgments
We thank David Sun, Xiaolin Wu, Li Jia, and Anand Merchant for Microarray experiments and analysis; Katherine McKinnon for help with FACS experiments; Tatiana Karpova (NCI Fluorescence Imaging Microscopy Facility) for help with microscopy; Murali Palangat for FISH imaging; Andy Tran for ssBrDU/cyclin A staining analysis; and Sudipto Das and Thorkell Anderson for help with mass spectrometry experiments. The authors would like to thank Misteli Lab members for helpful feedback and discussions. This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), NIA, NCI Center for Cancer Research, and by a Khorana Nirenberg Fellowship to VS.
Author contributions
VS and TM designed the study; VS performed majority of the experiments and data analysis; NK aided in the execution and analysis of high-throughput imaging; SK aided in execution and analysis of HR experiments and PO advised on the HR experiments; KA and MG did DDSR1-MS2 cloning and advised on MS2 pull-down and native RIP experiments. VS and TM wrote the manuscript. All the authors read and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Expanded View Figures PDF
Dataset EV1
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152:1344–1354. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14:699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KW, Ponting CP. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014;30:348–355. doi: 10.1016/j.tig.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass PG, Luft FC, Bahring S. Long non-coding RNA in health and disease. J Mol Med. 2014;92:337–346. doi: 10.1007/s00109-014-1131-8. [DOI] [PubMed] [Google Scholar]
- Bonasio R, Shiekhattar R. Regulation of transcription by long noncoding RNAs. Annu Rev Genet. 2014;48:433–455. doi: 10.1146/annurev-genet-120213-092323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David R. RNA: a new layer of regulation. Nat Rev Mol Cell Biol. 2011;12:766. doi: 10.1038/nrm3225. [DOI] [PubMed] [Google Scholar]
- Sharma V, Misteli T. Non-coding RNAs in DNA damage and repair. FEBS Lett. 2013;587:1832–1839. doi: 10.1016/j.febslet.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan G, Liu Y, Han C, Zhang X, Lu X. Noncoding RNAs in DNA repair and genome integrity. Antioxid Redox Signal. 2014;20:655–677. doi: 10.1089/ars.2013.5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan G, Hu X, Liu Y, Han C, Sood AK, Calin GA, Zhang X, Lu X. A novel non-coding RNA lncRNA-JADE connects DNA damage signalling to histone H4 acetylation. EMBO J. 2013;32:2833–2847. doi: 10.1038/emboj.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan G, Mathur R, Hu X, Liu Y, Zhang X, Peng G, Lu X. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal. 2013;25:1086–1095. doi: 10.1016/j.cellsig.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–629. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Bejar O, Marchese FP, Athie A, Sanchez Y, Gonzalez J, Segura V, Huang L, Moreno I, Navarro A, Monzo M, et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013;14:R104. doi: 10.1186/gb-2013-14-9-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Segura V, Marin-Bejar O, Athie A, Marchese FP, Gonzalez J, Bujanda L, Guo S, Matheu A, Huarte M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat Commun. 2014;5:5812. doi: 10.1038/ncomms6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner JR, Chen W, Iyer MK, Cao Q, Ma T, Han S, Sahu A, Malik R, Wilder-Romans K, Navone N, et al. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014;74:1651–1660. doi: 10.1158/0008-5472.CAN-13-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(Web Server issue):W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center stage. Cell Res. 2011;21:116–130. doi: 10.1038/cr.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volcic M, Karl S, Baumann B, Salles D, Daniel P, Fulda S, Wiesmuller L. NF-kappaB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res. 2012;40:181–195. doi: 10.1093/nar/gkr687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Kruhlak MJ, Kim J, Tran AD, Liu J, Nyswaner K, Shi L, Jailwala P, Sung MH, Hakim O, et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014;8:1049–1062. doi: 10.1016/j.celrep.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The elephant and the blind men: making sense of PARP inhibitors in homologous recombination deficient tumor cells. Front Oncol. 2013;3:228. doi: 10.3389/fonc.2013.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Srikantan S, Gorospe M. MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods. 2012;58:81–87. doi: 10.1016/j.ymeth.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Blackford AN, Chapman JR, Baskcomb L, Gravel S, Rusch A, Thomas A, Blundred R, Smith P, Kzhyshkowska J, et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol Cell. 2012;45:505–516. doi: 10.1016/j.molcel.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Sato K, Wu W. The BRCA1 ubiquitin ligase and homologous recombination repair. FEBS Lett. 2011;585:2836–2844. doi: 10.1016/j.febslet.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25:685–700. doi: 10.1101/gad.2011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KA, Greenberg RA. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Petit SA, Ficarro SB, Toomire KJ, Xie A, Lim E, Cao SA, Park E, Eck MJ, Scully R, et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014;4:1430–1447. doi: 10.1158/2159-8290.CD-13-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d’Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–235. doi: 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol. 2012;14:318–328. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congrains A, Kamide K, Ohishi M, Rakugi H. ANRIL: molecular mechanisms and implications in human health. Int J Mol Sci. 2013;14:1278–1292. doi: 10.3390/ijms14011278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41:371–383. doi: 10.1016/j.molcel.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Z, Jiang J, Ma J, Dai S, Xu T, Li H, Yasui A. The role of hnRPUL1 involved in DNA damage response is related to PARP1. PLoS ONE. 2013;8:e60208. doi: 10.1371/journal.pone.0060208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral PM, Rusch A, Turnell AS, Gallimore PH, Byrd PJ, Dobner T, Grand RJ. The interaction of the hnRNP family member E1B-AP5 with p53. FEBS Lett. 2005;579:2752–2758. doi: 10.1016/j.febslet.2005.03.095. [DOI] [PubMed] [Google Scholar]
- Kzhyshkowska J, Rusch A, Wolf H, Dobner T. Regulation of transcription by the heterogeneous nuclear ribonucleoprotein E1B-AP5 is mediated by complex formation with the novel bromodomain-containing protein BRD7. Biochem J. 2003;371(Pt 2):385–393. doi: 10.1042/BJ20021281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. In vitro generation of human cells with cancer stem cell properties. Nat Cell Biol. 2011;13:1051–1061. doi: 10.1038/ncb2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256:827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- Dutko JA, Schafer A, Kenny AE, Cullen BR, Curcio MJ. Inhibition of a yeast LTR retrotransposon by human APOBEC3 cytidine deaminases. Curr Biol. 2005;15:661–666. doi: 10.1016/j.cub.2005.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkila J, Coleman KA, Morrissey D, Pylkas K, Erkko H, Messick TE, Karppinen SM, Amelina A, Winqvist R, Greenberg RA. Familial breast cancer screening reveals an alteration in the RAP80 UIM domain that impairs DNA damage response function. Oncogene. 2009;28:1843–1852. doi: 10.1038/onc.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring SJ, Mason AC, Binz SK, Wold MS. Cellular functions of human RPA1. Multiple roles of domains in replication, repair, and checkpoints. J Biol Chem. 2008;283:19095–19111. doi: 10.1074/jbc.M800881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Abdelmohsen K, Hutchison ER, Mitchell SJ, Grammatikakis I, Guo R, Noh JH, Martindale JL, Yang X, Lee EK, et al. HuD regulates coding and noncoding RNA to induce APP-A beta processing. Cell Rep. 2014;7:1401–1409. doi: 10.1016/j.celrep.2014.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage KI, Gorski JJ, Barros EM, Irwin GW, Manti L, Powell AJ, Pellagatti A, Lukashchuk N, McCance DJ, McCluggage WG, et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol Cell. 2014;54:445–459. doi: 10.1016/j.molcel.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Burman B, Kruhlak MJ, Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014;9:1703–1717. doi: 10.1016/j.celrep.2014.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File