

Abstract
Lysine acetylation on numerous mitochondrial proteins, targeted by the sirtuin deacylase SIRT3, has been proposed to play a major role in regulating diverse mitochondrial functions, particularly in the liver. A new study by Weinert, Choudhary, and colleagues, in this issue of The EMBO Journal, finds that the absolute levels of hepatic mitochondrial protein acetylation in wild‐type mice are extremely low and may be insufficient to exert regulatory effects.
Subject Categories: Metabolism; Post-translational Modifications, Proteolysis & Proteomics
Mitochondria play central roles in maintaining normal cellular and organismal physiology in many contexts: energy production, metabolism, intracellular signaling, apoptosis, and pathogen responses. Consequently, mitochondrial defects are associated with diverse pathologies in mammals, including metabolic dysfunction, cancer, aging, and others. How various mitochondrial functions are coordinated to meet cellular requirements represents a central question in mitochondrial biology. A potential role for lysine acetylation in this process was suggested by proteomic studies showing that a large fraction of mitochondrial proteins are acetylated (Kim et al, 2006; Choudhary et al, 2009; Zhao et al, 2010). The SIRT3 protein, a member of the sirtuin deacylase family, was initially shown to deacetylate and thereby activate a mitochondrial enzyme involved in acetyl‐CoA synthesis (see Kumar & Lombard, 2015 and references therein). A more general role for SIRT3 in deacetylating many protein targets within this organelle was subsequently revealed. These results provided a plausible mechanism by which mitochondrial acetylation could be regulated via SIRT3 activity. Indeed, a large literature has demonstrated roles for acetylation in modulating (typically inhibiting) activities of many mitochondrial proteins, work that has largely focused on the liver. Consistent with the physiologic importance of SIRT3 in this role, many groups have found that SIRT3‐deficient mice and cells show marked mitochondrial defects.
These and other findings have led to an intuitively appealing model, in which reversible acetylation represents a mechanism to regulate activities of diverse mitochondrial enzymes in a somewhat binary, “on–off” manner, thereby governing metabolic pathway flux (Fig 1A) (Zhao et al, 2010). More recent studies have identified a diverse array of other lysine post‐translational modifications (PTMs), present in the mitochondria and/or elsewhere in the cell: for example, succinylation, malonylation, and glutarylation—all of which are regulated by the sirtuin SIRT5—as well as many other PTMs (Hirschey & Zhao, 2015). These newly discovered modifications are likewise attractive candidates to play regulatory roles.
Figure 1. Roles of acetylation and SIRT3 in regulating mitochondrial functions.
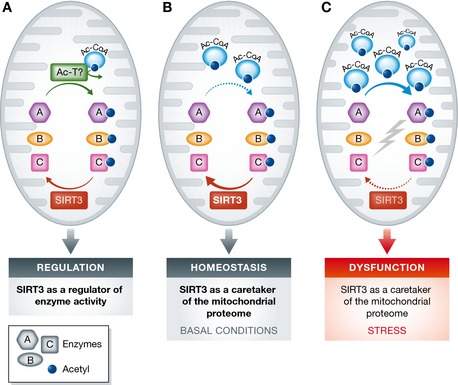
(A) A popular model posits that acetylation on mitochondrial proteins, regulated by SIRT3 and perhaps by putative mitochondrial acetyltransferases (Ac‐T), governs activities of various enzymes (designated A, B, and C) and flux through metabolic pathways, allowing mitochondria to adapt to varied cellular needs. This scheme would not predict a universally low stoichiometry of lysine acetylation in the mitochondria. (B) The results of Weinert and colleagues, and those of others, suggest that mitochondrial protein acetylation may occur largely as a passive consequence of non‐enzymatic reaction of proteins with acetyl‐CoA, and be efficiently removed by SIRT3 under most conditions in wild‐type animals. In this model, SIRT3 functions as a caretaker of the mitochondrial proteome, rather than as a regulator of enzyme activities in a strict sense. (C) Under stress conditions, mitochondrial acetyl‐CoA levels rise, and/or SIRT3 activity declines, allowing accumulation of acetylated proteins and consequently mitochondrial dysfunction. Such dysfunction might be amenable to correction by pharmacologic or genetic SIRT3 hyperactivity.
This straightforward model may require significant reevaluation, based on a new study by Weinert, Choudhary, and coworkers (Weinert et al, 2015). These researchers set out to measure the absolute stoichiometry of protein acetylation, focusing on the liver. Several previous studies have characterized the mitochondrial acetylome and its regulation by SIRT3, measuring relative changes in acetylation occurring in SIRT3‐deficient cells and tissues relative to controls (e.g. Hebert et al, 2013; Rardin et al, 2013; Dittenhafer‐Reed et al, 2015). However, measurement of acetylation stoichiometry in an absolute sense has proven to be a technically challenging endeavor, particularly in vivo. Weinert and colleagues addressed this question using an elegant chemical labeling approach, coupled with quantitative mass spectrometry. For selected sites, they confirmed their results using labeled synthetic peptide controls as internal standards. Using these techniques, they found that most acetylation sites show remarkably low stoichiometry, with a median acetylation of only 0.05%. Acetylation on mitochondrial proteins is slightly more abundant, with a median acetyl occupancy of 0.11%.
Weinert and colleagues compare their datasets with those generated in prior surveys of the SIRT3‐dependent acetylome (Hebert et al, 2013; Rardin et al, 2013). They show that acetylation sites previously identified as SIRT3 targets possess low levels of acetylation in livers from wild‐type animals under basal conditions: the great majority (97%) of SIRT3 target sites are acetylated at an occupancy of < 1%. The very highest stoichiometry of any SIRT3‐regulated site in wild‐type liver is 5.5%, at a lysine residue in HMGCL (3‐hydroxymethyl‐3‐methylglutaryl‐CoA lyase), an enzyme involved in ketone body synthesis. Their data are at odds with the results of a previous study that estimated acetylation levels of two metabolic enzymes, EHHADH (enoyl‐CoA, hydratase/3‐hydroxyacyl CoA dehydrogenase) and MDH2 (mitochondrial malate dehydrogenase), at significantly higher levels (Zhao et al, 2010). However, as Weinert et al (2015) point out, the quantification by Zhao et al (2010) was performed using ectopically expressed proteins purified from cell lines and therefore may not accurately reflect levels of acetylation of these proteins in tissues. Unfortunately, due to technical limitations, Weinert and colleagues were unable to accurately assess acetylation stoichiometry on histone N‐terminal tails. Therefore, it is not possible to compare levels of acetylation measured by their approach to other estimates in the literature, in the context of histones, where several groups have previously measured absolute acetylation stoichiometry, and for which the functional significance of acetylation is already well established.
The findings of Weinert and colleagues potentially provide insight into mechanisms of mitochondrial protein acetylation. Outside mitochondria, acetyltransferases mediate acetylation on histones and other protein targets, using acetyl‐CoA as a cofactor. Whether or not acetyltransferases exist in the mitochondria represents an important open question in mitochondrial biology. Two groups have identified putative mitochondrial lysine acetyltransferases (Scott et al, 2012; Fan et al, 2014); however, the general roles of these factors in acetylating mitochondrial proteins remain to be established. An alternative model that has arisen in recent years proposes that most acetylation in mitochondria arises via a spontaneous, non‐enzymatic mechanism (Wagner & Hirschey, 2014). Under the chemical conditions present in the mitochondrial matrix—alkaline pH and high acetyl‐CoA concentrations—protein acetylation can occur efficiently without the need to invoke enzymatic acetylation (Wagner & Payne, 2013; Weinert et al, 2014). In yeast, acetyl‐CoA derived from glycolysis represents the primary acetyl‐CoA source for mitochondrial protein acetylation (Weinert et al, 2014). In mammals, acetyl‐CoA derived from fatty acid metabolism makes a major contribution to this process (Pougovkina et al, 2014), an observation corroborated by Weinert and colleagues. Weinert et al (2015) find that most high occupancy acetylation sites are present on enzymes directly involved in acetyl‐group metabolism: that is, proteins exposed to high local concentrations of acetyl metabolites. Altogether, these observations are most consistent with the notion that the majority of mitochondrial acetylation events occur as a consequence of spontaneous chemical reaction of acetyl‐CoA with lysine residues, rather than as a regulated process catalyzed by acetyltransferases.
Several interventions have been shown to provoke increases in mitochondrial acetylation, most markedly in the liver but also in other tissues: fasting, long‐term calorie restriction (CR), high‐fat diet, and chronic ethanol ingestion. All of these interventions are characterized by metabolic perturbations leading to elevated mitochondrial acetyl‐CoA levels. SIRT3‐deficient mice show physiologic and biochemical defects in the context of CR, prolonged high‐fat feeding, and extended fasting (Kumar & Lombard, 2015), suggesting that SIRT3 might play an active role in the organismal response to these challenges. However, Weinert and colleagues find that the absolute stoichiometry of hepatic mitochondrial protein acetylation remains low even in these states in wild‐type animals and is minimally affected at SIRT3 target sites, across multiple different tissues.
Pending validation of these results by independent methodologies, what are their implications for the study of SIRT3 and for mitochondrial biology more generally? They may cast doubt on a major role for reversible lysine acetylation in liver mitochondria as a general means by which mitochondrial enzyme activities are activated and inhibited, at least under most physiologic conditions. Conceptually similar studies of protein phosphorylation have revealed that the absolute stoichiometry of phosphorylation varies widely across different protein targets, from very low to nearly total. Phosphorylation levels can be modulated over a very wide dynamic range, making it an attractive means of target regulation. By contrast, mitochondrial acetylation, seemingly occurring at such a consistently low level in the liver, both basally and in response to various stimuli, might not appear to represent an intuitively appealing regulatory mechanism. The study of Weinert and colleagues also emphasizes the difficulty of extrapolating phenotypes observed in Sirt3 mutant animals, or in cultured cells, to physiologic roles for mitochondrial protein acetylation in wild‐type animals in vivo. The potential impact of acetylation on its targets is frequently assessed via generation of mutant proteins in which acetylated lysines are mutated to resemble a constitutively acetylated or deacetylated state—typically to glutamine or arginine, respectively. These mutants may or may not faithfully recapitulate the effects of reversible acetylation, since these amino acids are not ideal lysine mimics, and because they model a likely non‐physiologic scenario in which a given lysine residue is present in the fully acetylated or deacetylated state.
It should be noted that low stoichiometry on its own does not conclusively rule out a regulatory role in a more general sense. For example, if acetylation on certain mitochondrial proteins marked them for rapid degradation, then this modification would be observed at low stoichiometry, due to the short half‐life of modified proteins. Similarly, for specific proteins, acetylation might be targeted to a small, functionally important pool—that present in a complex with key cofactors, for example. Although as a general matter most investigators have found that acetylation inhibits mitochondrial enzymes, one report showed that acetylation actually enhanced the enzymatic activity of MDH2 (Zhao et al, 2010). Even low‐level acetylation could exert important biological effects, if associated with activation rather than inhibition. It is also possible that low stoichiometry acetylation, particularly when present on multiple lysines per protein, and/or several proteins acting in the same biochemical pathway, could exert a functional impact, subject to regulation by SIRT3. However, overall the results of Weinert and colleagues are most consistent with the idea that the great majority of acetylation events in hepatic mitochondria do not play a regulatory role per se. Instead, in this model, acetylation occurs passively, as a consequence of spontaneous chemical reaction of proteins with acetyl‐CoA, and is rapidly and efficiently removed by SIRT3. In this regard, lysine acetylation in mitochondria has recently been conceptualized as a form of “carbon stress,” for which SIRT3 represents a protein repair factor (Wagner & Hirschey, 2014), a model fully consistent with the results of Weinert et al (2015). In this scheme, SIRT3 is demoted from a full‐blown metabolic regulator, to the perhaps somewhat less glamorous (but still vital) role of a protein caretaker (Fig 1B). Another observation that is consistent with such a caretaker role for SIRT3 is that the deleterious phenotypes of SIRT3 deficiency tend to manifest most strongly in response to prolonged stress conditions, such as extended fasting, high‐fat diet, or advanced age (Kumar & Lombard, 2015). These conditions might allow for gradual accumulation of acetylated, dysfunctional mitochondrial proteins and thereby represent situations where the caretaker function of SIRT3 becomes most crucial. In this regard, SIRT3 has previously been proposed to possess three distinct functions: (i) regulation of common metabolic pathways, such as the TCA cycle, in all tissues; (ii) coordination of specific metabolic pathways, such as ketone body metabolism, between tissues; and (iii) allowing certain organs, such as kidney and liver, to cope with their high acetate burden, which would otherwise lead to “bulk” mitochondrial protein hyperacetylation in the absence of SIRT3 (Dittenhafer‐Reed et al, 2015).
In contrast to the low‐level acetylation stoichiometry present in wild‐type liver, by extrapolating from previous studies (Hebert et al, 2013; Rardin et al, 2013), Weinert et al (2015) estimate that acetyl occupancy at individual lysines can reach relatively high levels in liver mitochondria of Sirt3 mutants, particularly under CR conditions, and could therefore easily rationalize the varied phenotypic defects observed in SIRT3‐deficient animals (Kumar & Lombard, 2015). Clearly, the mitochondrial hyperacetylation provoked the absence of SIRT3 entails significant long‐term deleterious consequences for the organism. Moreover, SIRT3 deficiency is also associated with perturbations in key regulators such as PGC1α and AMPK, alterations that likely also contribute to the negative consequences of SIRT3 deficiency.
The results of Weinert et al (2015) highlight the key question of whether there are conditions in which SIRT3 expression and/or activity is suppressed in wild‐type organisms, mimicking the effects of SIRT3 depletion and its downstream sequelae. The answer is an unambiguous yes (see Kumar & Lombard, 2015 for references). During prolonged high‐fat feeding, hepatic SIRT3 levels decline, inducing hyperacetylation and reduced function of LCAD (long chain acyl‐CoA dehydrogenase), a key β‐oxidation enzyme. In skeletal muscle, SIRT3 levels are reduced in response to fasting, contributing to a metabolic shift from glucose oxidation to fatty acid utilization. During aging, SIRT3 levels decline markedly in mouse hematopoietic stem cells (HSCs), impairing ROS management. Reconstitution of aged HSCs cells with ectopically expressed SIRT3 enhances their regenerative capacity, formally demonstrating that SIRT3 activity becomes a limiting factor for proper function of this cell type during aging. Increased SIRT3 activity protects against hearing loss and degeneration of spiral ganglion neurites induced by loud noise.
As with other sirtuins, SIRT3 requires the metabolic cofactor NAD+ for its enzymatic activity. An emerging literature suggests that NAD+ levels decline with age in diverse organisms, potentially leading to reduced function of SIRT3 and other sirtuins. Indeed, in mouse heart, cyclophilin D, a SIRT3 target and a component of the mitochondrial permeability transition pore (mPTP), becomes hyperacetylated in older animals, an effect that may contribute to age‐associated cardiac hypertrophy and fibrosis (Hafner et al, 2010). One recent report showed that during normal cardiac aging, overall mitochondrial acetylation increases to levels qualitatively resembling those present in Sirt3 heterozygous animals (Porter et al, 2014). Weinert et al (2015) quantitate acetylation in the livers of young and middle aged mice; it would be quite revelatory to repeat their analysis in older animals, particularly in cell types such as HSCs and cardiomyocytes that seem especially sensitive to reductions in SIRT3 function. Taken together, these results suggest that under stress conditions, including natural aging, SIRT3 activity becomes limiting for mitochondrial function in critical tissues and cell types (Fig 1C). Enhancement of SIRT3 activity, genetically or pharmacologically, may exert beneficial effects specifically in these contexts.
The results reported by Weinert and colleagues will no doubt spark much vigorous discussion in the study of mitochondrial acetylation. With respect to other lysine PTMs in mitochondria, notably the stoichiometry of lysine succinylation appears to be significantly higher than that of acetylation (Park et al, 2013). Moreover, SIRT5‐deficient cells and liver mitochondria show elevated mitochondrial respiration, suggesting that SIRT5 and its target lysine modifications may represent better candidates than acetylation to regulate mitochondrial enzyme activities in vivo. Overall, the regulatory role of acetylation and other PTMs in mitochondrial biology is far from a settled question.
Acknowledgements
The authors thank Drs. John Denu, Matthew Hirschey, and Yingming Zhao for critical comments on the manuscript, and sincerely apologize to colleagues whose work was not cited due to strict space limitations. Supported by NIH (R01GM101171), the Glenn Foundation for Medical Research, the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000433, and the John S. and Suzanne C. Munn Cancer Fund of the University of Michigan Comprehensive Cancer Center.
See also: BT Weinert et al (November 2015)
References
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science 325: 834–840 [DOI] [PubMed] [Google Scholar]
- Dittenhafer‐Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, Denu JM (2015) SIRT3 mediates multi‐tissue coupling for metabolic fuel switching. Cell Metab 21: 637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, Gu TL, Aguiar M, Lonning S, Chen H, Mohammadi M, Britton LM, Garcia BA, Aleckovic M, Kang Y, Kaluz S, Devi N, Van Meir EG, Hitosugi T, Seo JH et al (2014) Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell 53: 534–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA (2010) Regulation of the mPTP by SIRT3‐mediated deacetylation of CypD at lysine 166 suppresses age‐related cardiac hypertrophy. Aging 2: 914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer‐Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, Westphall MS, Pagliarini DJ, Prolla TA, Assadi‐Porter F, Roy S, Denu JM, Coon JJ (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell 49: 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Zhao Y (2015) Metabolic regulation by lysine malonylation, succinylation and glutarylation. Mol Cell Proteomics 14: 2308–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell 23: 607–618 [DOI] [PubMed] [Google Scholar]
- Kumar S, Lombard DB (2015) Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxid Redox Signal 22: 1060–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, Lombard DB, Zhao Y (2013) SIRT5‐mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50: 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM (2014) SIRT3 deficiency exacerbates ischemia‐reperfusion injury: implication for aged hearts. Am J Physiol Heart Circ Physiol 306: H1602–H1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pougovkina O, te Brinke H, Ofman R, van Cruchten AG, Kulik W, Wanders RJ, Houten SM, de Boer VC (2014) Mitochondrial protein acetylation is driven by acetyl‐CoA from fatty acid oxidation. Hum Mol Genet 23: 3513–3522 [DOI] [PubMed] [Google Scholar]
- Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW (2013) Label‐free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci USA 110: 6601–6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott I, Webster BR, Li JH, Sack MN (2012) Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J 443: 655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Payne RM (2013) Widespread and enzyme‐independent Nepsilon‐acetylation and Nepsilon‐succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem 288: 29036–29045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Hirschey MD (2014) Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell 54: 5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Iesmantavicius V, Moustafa T, Scholz C, Wagner SA, Magnes C, Zechner R, Choudhary C (2014) Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol Syst Biol 10: 716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Moustafa T, Iesmantavicius V, Zechner R, Choudhary C (2015) Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J 34: 2620–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q et al (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327: 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]