

Abstract
The SMC5/6 complex is the least understood of SMC complexes. In yeast, smc5/6 mutants phenocopy mutations in sgs1, the BLM ortholog that is deficient in Bloom's syndrome (BS). We here show that NSMCE2 (Mms21, in Saccharomyces cerevisiae), an essential SUMO ligase of the SMC5/6 complex, suppresses cancer and aging in mice. Surprisingly, a mutation that compromises NSMCE2‐dependent SUMOylation does not have a detectable impact on murine lifespan. In contrast, NSMCE2 deletion in adult mice leads to pathologies resembling those found in patients of BS. Moreover, and whereas NSMCE2 deletion does not have a detectable impact on DNA replication, NSMCE2‐deficient cells also present the cellular hallmarks of BS such as increased recombination rates and an accumulation of micronuclei. Despite the similarities, NSMCE2 and BLM foci do not colocalize and concomitant deletion of Blm and Nsmce2 in B lymphocytes further increases recombination rates and is synthetic lethal due to severe chromosome mis‐segregation. Our work reveals that SUMO‐ and BLM‐independent activities of NSMCE2 limit recombination and facilitate segregation; functions of the SMC5/6 complex that are necessary to prevent cancer and aging in mice.
Keywords: NSMCE2, SMC5/6, mouse models, SUMO, chromosome segregation
Subject Categories: DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The structural maintenance of chromosome (SMC) complexes play key roles in chromosome architecture and dynamics (Losada & Hirano, 2005; Nasmyth & Haering, 2005). SMC proteins contain globular N‐ and C‐terminal ATPase domains (Walker boxes), which are linked through two long α‐helixes folded against each other at a hinge domain. Bacteria present a single SMC protein, and six can be identified in eukaryotes. These six proteins are arranged in three independent heterodimeric complexes named SMC1/3 (cohesin), SMC2/4 (condensin), and SMC5/6. Whereas the functions of cohesin and condensin in chromosome structure are well established, what the SMC5/6 complex does is still a matter of debate (Murray & Carr, 2008; De Piccoli et al, 2009; Stephan et al, 2011b).
The SMC5/6 complex was initially identified in fission yeast as a heterodimer of Rad18 and Spr18 proteins that presented the architecture of SMC proteins (Lehmann et al, 1995; Fousteri & Lehmann, 2000). Human orthologs were subsequently identified and were shown to be particularly abundant during meiosis (Fousteri & Lehmann, 2000). The complex contains six additional polypeptides named non‐SMC elements (NSMCE) 1–6 (McDonald et al, 2003; Pebernard et al, 2004; Sergeant et al, 2005), although only four (NSMCE1‐4) have been identified in mammals (Taylor et al, 2008). Besides the ATPase activity of SMC5 and SMC6, 2 additional activities are found in the complex. NSMCE1 presents an E3 ubiquitin (UQ) ligase activity that is only detectable upon interaction with NSMCE3 (Pebernard et al, 2008a; Doyle et al, 2010). In addition, NSMCE2 has a C‐terminal Siz/PIAS‐RING (SP‐RING) domain with E3 SUMO ligase activity (Andrews et al, 2005; Potts & Yu, 2005; Zhao & Blobel, 2005). The last component found in mammals, NSMCE4, is the kleisin subunit that bridges SMC5 and SMC6 (Morikawa et al, 2004; Palecek et al, 2006). To date, and due to the viability of catalytically dead mutants, many of the studies on the SMC5/6 complex have focused on the SUMO ligase activity of NSMCE2.
The consensus is that the SMC5/6 complex is involved in genome maintenance, consistent with the original rad18 (Smc5) and mms21 (Nsmce2) mutants being isolated on the basis of sensitivity to genotoxic agents (Nasim & Smith, 1975; Prakash & Prakash, 1977). Mutations or RNAi studies for SMC5/6 have further confirmed sensitivity to genotoxic agents in plants, chicken, and human cells (reviewed in Stephan et al, 2011b). Still, the actual role of this complex in preventing DNA damage is not well defined. On one hand, the complex has been proposed to promote homologous recombination (HR). Accordingly, RNAi studies in human cells reported lower rates of sister chromatid exchanges (SCE) in NSMCE2‐depleted cells (Potts et al, 2006; Wu et al, 2012). The same group also reported reduced rates of telomere recombination (T‐SCE) upon SMC5/6 knockdown (Potts & Yu, 2007). On the other hand, these results are at odds with the fact that mutants of the SMC5/6 complex display increased recombination rates in every model organism tested. In fact, the mms21 mutant was first isolated in yeast for presenting a 23‐fold increase in mitotic recombination (Prakash & Prakash, 1977). Likewise, Nsmce2 or Smc5 knockouts in chicken (Stephan et al, 2011a; Kliszczak et al, 2012) and reduced levels of SMC6 in mice (Ju et al, 2013) all led to increased rates of SCE. To what extent these effects reflect species‐specific roles or technical differences remains to be solved.
Despite a role in HR, this cannot account for all SMC5/6 functions. All components of the SMC5/6 complex are essential in Saccharomyces cerevisiae (Zhao & Blobel, 2005), whereas HR is not. The essential role of SMC5/6 might relate to defects in chromosome segregation (Torres‐Rosell et al, 2005). These problems could derive from persistent DNA (Torres‐Rosell et al, 2005; Branzei et al, 2006; Bermudez‐Lopez et al, 2010) or protein (Outwin et al, 2009) links between sister chromatids during mitosis. Accordingly, yeast lacking SMC5/6 undergo catastrophic meiosis due to unresolved inter‐chromosome links (Farmer et al, 2011). Noteworthy, this role of SMC5/6 on the dissolution of joint DNA molecules (JM) is analogous to the one carried by Sgs1, the yeast ortholog of the BLM protein that is deficient in patients of the Bloom syndrome (OMIM 210900). BLM is one of the five helicases from the RecQ family, three of which (BLM, WRN, and RECQL4) are associated with cancer predisposition and premature aging in human hereditary disorders (Chu & Hickson, 2009). Similar to yeast smc5/6 mutants, BLM‐deficient human cells also present segregation problems due to anaphase bridges between unresolved chromatids (Chan et al, 2007, 2009; Naim & Rosselli, 2009). To what extent the similarities between the SMC5/6 complex and BLM are also present at the organism level remains unknown.
To explore the physiological roles of the SMC5/6 complex in mammals, we developed three independent murine alleles of NSMCE2, the essential SUMO ligase of the complex (constitutive knockout, conditional knockout, and SUMO‐dead knock‐in). Overall, our data support that the main role of the SMC5/6 complex is on facilitating segregation and suppressing recombination by an essential BLM‐ and SUMOylation‐independent pathway. Finally, this work reveals for the first time that the genome protection activity provided by the SMC5/6 complex is essential for the suppression of cancer and aging in mammals.
Results
NSMCE2 localizes with BRCA1 at sites distinct from DNA breaks
In order to explore the potential functions of NSMCE2 in mammals, we developed antibodies against the murine and human proteins. In humans, NSMCE2 expression was highest in proliferating tissues (Appendix Fig S1A and B). Likewise, NSMCE2 expression was most abundant on Ki67‐positive regions of mouse embryos (Appendix Fig S1C). NSMCE2 levels were distinctively high in human and mouse testis (Fig 1A; Appendix Fig S1B). To investigate the potential role that NSMCE2 could be playing during spermatogenesis, we analyzed the distribution of NSMCE2 on spermatocyte spreads. Preparations were co‐stained with antibodies detecting SCP3, a lateral element of the synaptonemal complex, which provided information about the degree of synapsis between homologous chromosomes (Fig 1B). In contrast to the punctuated pattern shown by proteins involved in the repair of meiotic DNA double‐strand breaks (DSB), we did not detect any focal NSMCE2 staining in leptotene. In contrast, a distinct NSMCE2 staining was detected in pachytene spermatocytes, which coincided with regions of weak SCP3 signal (Fig 1B). This localization is reminiscent of proteins that accumulate at regions of incomplete synapsis of meiotic chromosomes such as BRCA1 (Scully et al, 1997b). Consistently, NSMCE2 colocalized with BRCA1 along the non‐homologous regions of the XY chromosomes during pachytene (Fig 1C). The accumulation of NSMCE2 at sex chromosomes is also evident by immunohistochemistry (IHC) of testes, where XY chromosomes form a compact structure known as the “sex body” on spermatocytes (inset, Fig 1A). Earlier studies reported a similar accumulation of SMC5 and SMC6 at the sex chromosomes (Taylor et al, 2001; Gomez et al, 2013) and also failed to detect any DSB‐related localization of the complex during meiosis. Moreover, the recruitment of BRCA1 to the XY chromosomes during spermatogenesis is unrelated to DNA breaks, since the same localization is present on cells lacking the SPO11 nuclease (Mahadevaiah et al, 2008). In summary, the distribution of NSMCE2 during spermatogenesis suggests that NSMCE2 is recognizing structures that are also recognized by BRCA1, but which are different from meiotic DSB.
Figure 1. NSMCE2 colocalizes with BRCA1 at sites different to DSB .
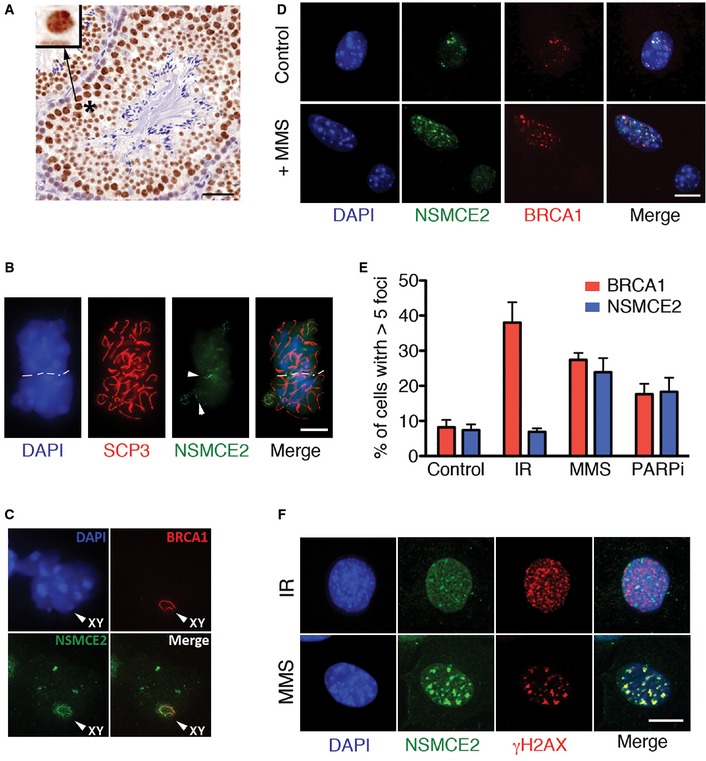
- Immunohistochemistry of NSMCE2 on mouse testis. The inset illustrates the accumulation of NSMCE2 on the XY body. Scale bar, 50 μm.
- Immunofluorescence (IF) of NSMCE2 and SCP3 on isolated mouse spermatocytes. Threads of NSMCE2 can be detected on regions of weak SCP3 signal, indicative of asynapsis (arrowheads). Scale bar, 2.5 μm.
- Colocalization of NSMCE2 and BRCA1 at the XY body of mouse spermatocytes.
- Representative image of NSMCE2 and BRCA1 foci in control and MMS‐treated MEFs. Scale bar, 2.5 μm.
- Quantification of NSMCE2 and BRCA1 foci in wild‐type MEFs exposed to IR (10 Gy; 1‐h recovery), MMS (1 mM for 1 h; 24‐h recovery), and PARP inhibitor (1 μM for 1 h; 24‐h recovery). Data indicate mean values and are representative of three independent experiments. Error bars indicate SD.
- Representative image of IR‐ (10 Gy; 1‐h recovery) and MMS‐ (1 mM for 1‐h; 24‐h recovery) induced NSMCE2 and γH2AX foci in MEFs. Scale bar, 2.5 μm.
To determine whether this colocalization between NSMCE2 and BRCA1 was also present in somatic cells, we performed immuno‐stainings on mouse embryonic fibroblasts (MEFs). BRCA1 accumulates at endogenous foci during replication, but relocalizes to DSB upon DNA breakage (Scully et al, 1997a; Tashiro et al, 2000). NSMCE2 colocalized with endogenous BRCA1 foci in unchallenged wild‐type MEFs (Fig 1D). However, and in contrast to BRCA1, NSMCE2 did not form ionizing radiation (IR)‐induced foci (Fig 1E and F). Previous studies in yeast suggested that NSMCE2 might be involved in the handling of recombinogenic structures induced by alkylating agents (Branzei et al, 2006). To investigate whether a similar response operates in mammals, MEFs were exposed for 1 h to methyl methanesulfonate (MMS) and let to recover for 24 h in the absence of the drug. These conditions promoted the accumulation of NSMCE2 foci that colocalized with BRCA1 (Fig 1D and E). The induction of NSMCE2 foci was not linked to the alkylating nature of the drug, since it could also be seen in response to other drugs that perturb DNA replication, such as poly‐ADP ribosyl transferase (PARP) inhibitors (Fig 1E). Moreover, catalytic inhibition of TOPO2A generated NSMCE2 foci, further suggesting that the complex can be recruited to specific sites on DNA in the absence of DNA breaks (Appendix Fig S2). In support of this, whereas NSMCE2 and H2AX phosphorylation (γH2AX) colocalized in response to MMS, NSMCE2 failed to colocalize with IR‐induced γH2AX foci, a bona fide marker of DSB (Rogakou et al, 1999) (Fig 1F). In summary, NSMCE2 accumulates at endogenous structures, which are distinct from DSB and which can be induced with reagents that challenge DNA replication and/or promote the accumulation of topological defects.
NSMCE2, but not its SUMO ligase activity, is essential for mouse development
In order to evaluate the role of NSMCE2 in vivo, we generated Nsmce2‐deficient animals. To this end, we took advantage of a murine ES cell line in which one Nsmce2 allele had been altered by insertion of a promoterless Gene Trap (clone AA0032; Sanger Institute Genetrap Resource). The mutation (Nsmce2 GT) generates a chimeric cDNA containing the first 88 amino acids of NSMCE2 followed by a β‐geo coding sequence and thus lacks the SUMO ligase domain (Fig 2A and B). By using inverse PCR, we mapped the insertion at intron 3/4 of Nsmce2, which enabled genotyping. No Nsmce2 GT/GT mice were born, and we also failed to detect homozygous mutant embryos at 13.5 or 10.5 dpc (Appendix Table S1). In contrast, 2.5 dpc Nsmce2 GT/GT embryos were obtained at Mendelian ratios. However, upon 36 h of in vitro culture, a number of embryos rapidly degenerated, which were found to be Nsmce2 GT/GT (Fig 2C and D). To determine the cause of death, we cultured 2.5 dpc embryos for 12 h to prevent their full degeneration. Nsmce2 GT/GT embryos were identified by the absence of NSMCE2, as measured by immunofluorescence (Fig 2E). Whereas cells from wild‐type (wt) embryos were of similar shape and size and mitotic figures were rarely found, mutant embryos accumulated irregularly sized or multilobulated nuclei, as well as cells with condensed chromosomes, all of which would be consistent with defects in chromosome segregation. Supporting this view, some cells from 2.5 dpc Nsmce2 GT/GT morulae presented an abnormal size (Fig 2C), consistent with an imbalanced segregation during the first embryonic cycles. Altogether, our data suggest that the early embryonic lethality found on Nsmce2‐deficient mice is due to segregation abnormalities that limit cell division during the first developmental stages.
Figure 2. NSMCE2 is essential for early embryonic development.
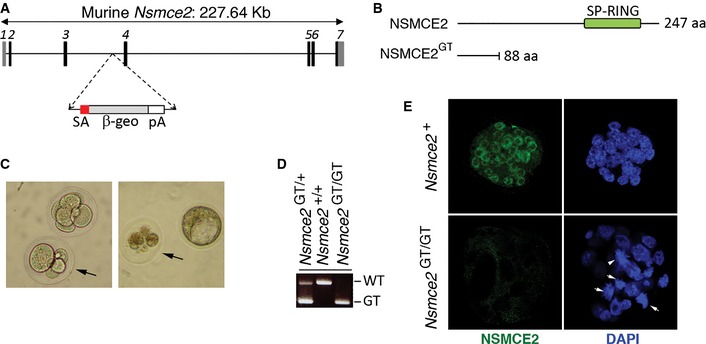
- Scheme illustrating the Nsmce2 genetrap allele (Nsmce2 GT) used in this study.
- Comparison of the wt and mutant NSMCE2 protein. Note that the trapped protein lacks most of the C‐terminal region including the SUMO ligase domain.
- Brightfield microscopy of 2.5 dpc embryos obtained from crosses of Nsmce2 GT /+ mice. The right panel is of the same 2 embryos shown on the left panel, 36 h after being cultured in vitro. The arrow indicates embryos that showed asymmetric cell sizes when isolated, which degenerated upon culture. These embryos were identified as Nsmce2 GT / GT through genotyping.
- Examples of the three genotypes found at Mendelian frequencies (see Appendix Table S1) on 2.5 dpc embryos.
- Examples of the type of nuclei found on Nsmce2 GT / GT and Nsmce2 + (+/+ or +/ GT) 2.5 dpc embryos, 12 h after in vitro culture. Mutant embryos were identified due to the absence of NSMCE2 signal (green). DAPI was used to stain DNA (blue). White arrows indicate examples of cells with condensed chromosomes or irregular shapes, which are hardly seen in wild type embryos.
The only known activity of NSMCE2 is a SUMO E3 ligase conferred by its C‐terminal Siz/PIAS‐RING (SP‐RING) domain. To evaluate the role of the NSMCE2 SUMO ligase activity in mammals, we generated a knock‐in mutant allele in which C185S and H187A mutations were introduced on the SP‐RING domain (Nsmce2 SD) (Fig 3A and B). In vitro NSMCE2 auto‐SUMOylation assays using wild‐type and mutant NSMCE2 protein (cloned directly by RT–PCR from the corresponding mouse strains) confirmed the inactivating nature of the mutations (Appendix Fig S3). The mutation did not alter NSMCE2 protein levels, the stability of the SMC5/6 complex, nor its capacity to form MMS‐ or MMC‐induced foci (Fig 3C and D; Appendix Fig S4). Moreover, and in contrast to NSMCE2 deficiency, Nsmce2 SD/SD animals were viable and fertile and showed no alteration of their lifespan (Fig 3E and F). Whereas in vitro assays using overexpressed NSMCE2 and SUMO detected some residual activity of the mutant protein upon SUMO1 (but not SUMO2) overexpression, this could derive from the endogenous wild‐type NSMCE2 that is present on the HEK293T cells used for these assays. In addition, the mutation introduced in these mice was first described and has been widely used in yeast as a SUMOylation‐deficient model (Andrews et al, 2005; Branzei et al, 2006; Pebernard et al, 2008b; Takahashi et al, 2008; Kegel et al, 2011). Hence, and whereas we cannot formally discard that some residual activity remains on Nsmce2 SD/SD cells, our data reveal that the SUMO ligase activity of NSMCE2 is largely dispensable in mice.
Figure 3. NSMCE2 SUMO ligase activity is largely dispensable in mice.
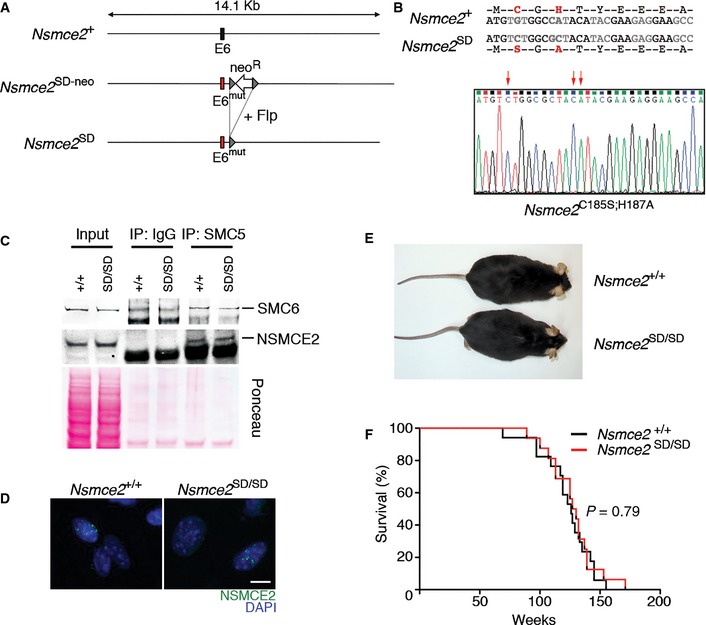
- Scheme illustrating the Nsmce2‐mutant allele (Nsmce2 SD) used in this study, which was generated through introducing the mutations on exon 6 (see Materials and Methods).
- Aminoacid and nucleotide sequences of the Nsmce2‐mutant allele. The SUMO ligase‐inactivating mutation used C185S; H187A (SD), which is the same originally described in yeast (Andrews et al, 2005) and which has been used as a SUMOylation‐deficient model in multiple studies. The nucleotide sequence derives directly from sequencing DNA from Nsmce2 SD / SD mice.
- Immunoprecipitation of SMC5 (and IgG, as a control) from a 48‐h culture of B lymphocytes of wt and Nsmce2 SD / SD animals. NSMCE2 and SMC6 levels on the immunoprecipitates were analyzed by WB. The image of the Ponceau staining is shown as a loading control.
- IF of NSMCE2 foci in MMS‐treated Nsmce2 +/+ and Nsmce2 SD / SD MEFs. Scale bar, 2.5 μm.
- Representative picture of 6‐month‐old Nsmce2 +/+ and Nsmce2 SD / SD littermates.
- Kaplan–Meyer survival curves of Nsmce2 +/+ and Nsmce2 SD / SD mice. The P‐value was calculated with the Mantel–Cox long‐rank test.
Nsmce2 is a haploinsufficient tumor suppressor
In contrast to Nsmce2 GT/GT, Nsmce2 heterozygous mice were viable and fertile with no apparent phenotype. However, Nsmce2 GT/+ mice showed a decreased lifespan, which was associated with a higher incidence of tumors (Fig 4A and B). PCR analyses on tumors (n = 7) from Nsmce2 heterozygous animals revealed that they always kept a wild‐type Nsmce2 copy, suggesting that a reduced dosage of NSMCE2 is responsible for this phenotype. In order to investigate the impact of Nsmce2 haploinsufficiency, we analyzed the behavior of Nsmce2 GT/+ primary cells. Nsmce2 heterozygous MEFs presented an ~50% reduction in NSMCE2 protein levels (Fig 4C). This reduction correlated with higher rates of spontaneous mitotic recombination as measured by sister chromatid exchanges (SCE) (Fig 4D). In addition, Nsmce2 GT/+ MEFs showed evidences of improper chromosome segregation such as a higher incidence of micronuclei or polynucleated cells (Fig 4E and F). Hence, Nsmce2 is a haploinsufficient tumor suppressor in mice, which is associated with increased rates of mitotic recombination and chromosome mis‐segregation on heterozygous cells.
Figure 4. NSMCE2 is a haploinsufficient tumor suppressor.
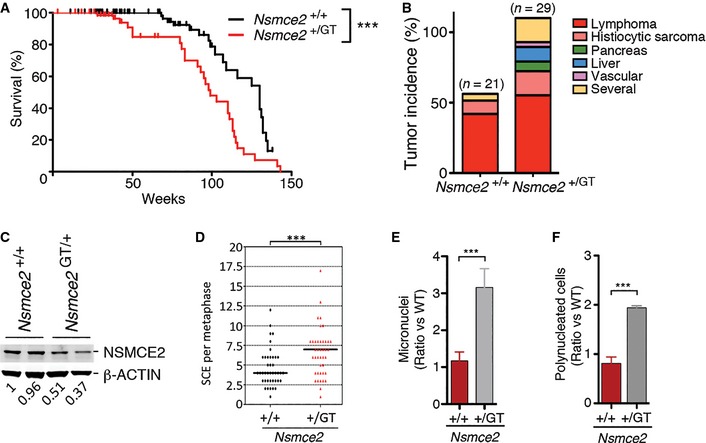
-
AKaplan–Meyer survival curves of Nsmce2 +/+ and Nsmce2 +/ GT mice. The P‐value was calculated with the Mantel–Cox long‐rank test. ***P < 0.001.
-
BTumor incidence found on Nsmce2 +/+ and Nsmce2 +/ GT mice at the time of death (i.e. moribund mice).
-
CNSMCE2 levels evaluated by Western blot in 2 independent pairs of Nsmce2 +/+ and Nsmce2 +/ GT MEFs. β‐actin was used as a loading control. Numbers shown bellow indicate the relative amounts of NSMCE2 in each sample.
-
DSister chromatid exchange (SCE) events found per metaphase on Nsmce2 +/+ and Nsmce2 +/ GT MEFs. ***P < 0.001.
-
E, FRelative percentages of micronuclei (E) and polynucleated cells (F) on cultures of Nsmce2 +/+ and Nsmce2 +/ GT MEFs. Data are representative of three independent experiments. Error bars indicate SD. ***P < 0.001.
NSMCE2 suppresses recombination and facilitates segregation in MEFs
To overcome the limitations imposed by the essential nature of NSMCE2, we developed an Nsmce2 conditional allele where exon 3 is flanked by loxP sites (Nsmce2 lox) (Fig 5A). We first used this allele in combination with a 4‐hydroxytamoxifen (4‐OHT)‐inducible Cre recombinase that is under the control of the ubiquitin promoter (Ub.CreERT2) (Ruzankina et al, 2007). To study the consequences of Nsmce2 nullyzygosity, Ub.CreERT2/Nsmce2 lox/lox MEFs were generated. Upon exposure to 4‐OHT, Nsmce2 lox/lox MEFs lost the expression of NSMCE2 (Fig 5B), and this was found to be essential at the cellular level as seen by clonogenic assays (Fig 5C). In agreement with Western blot data, immunofluorescence (IF) analyses revealed the loss of MMS‐induced NSMCE2 foci on 4‐OHT‐treated Ub.CreERT2/Nsmce2 lox/lox MEFs (Fig 5D). Interestingly, NSMCE2‐depleted cells presented large amounts of BRCA1 foci, indicative of increased recombination. Consistently, NSMCE2‐deficient MEFs presented a significant increase in SCE levels even in the absence of exogenous agents (Fig 5E and F). Moreover, and similar to observations in BLM‐deficient cells (Wechsler et al, 2011), the increased SCE rates observed on NSMCE‐deficient cells were dependent on the MUS81 nuclease (Appendix Fig S5). Given that NSMCE2 was previously proposed to promote, rather than suppress, recombination at telomeres (Potts & Yu, 2007), we specifically evaluated inter‐telomeric recombination rates (T‐SCE) using chromosome orientation FISH. In agreement with the overall increase in recombination, NSMCE2‐deficient cells presented increased rates of T‐SCE (Fig 5G) which, however, did not led into any significant change on overall telomere lengths (Fig 5H and I). Finally, and in agreement with the segregation defects found on Nsmce2 GT/GT embryos, NSMCE2‐deficient MEFs presented a significant accumulation of micronuclei and intercellular DNA bridges as well as cells with major segregation defects (Fig 5J; Appendix Fig S6). In contrast to the roles of NSMCE2 in segregation and recombination, high‐throughput microscopy analyses failed to detect any obvious impact of NSMCE2‐deficiency on the repair of DSB, as measured by the kinetics of appearance and disappearance of IR‐induced 53BP1 foci (Appendix Fig S7). Altogether, these data further support that NSMCE2 plays an essential role in limiting recombination and facilitating chromosome segregation in mammalian cells.
Figure 5. NSMCE2 suppresses recombination and micronuclei formation.
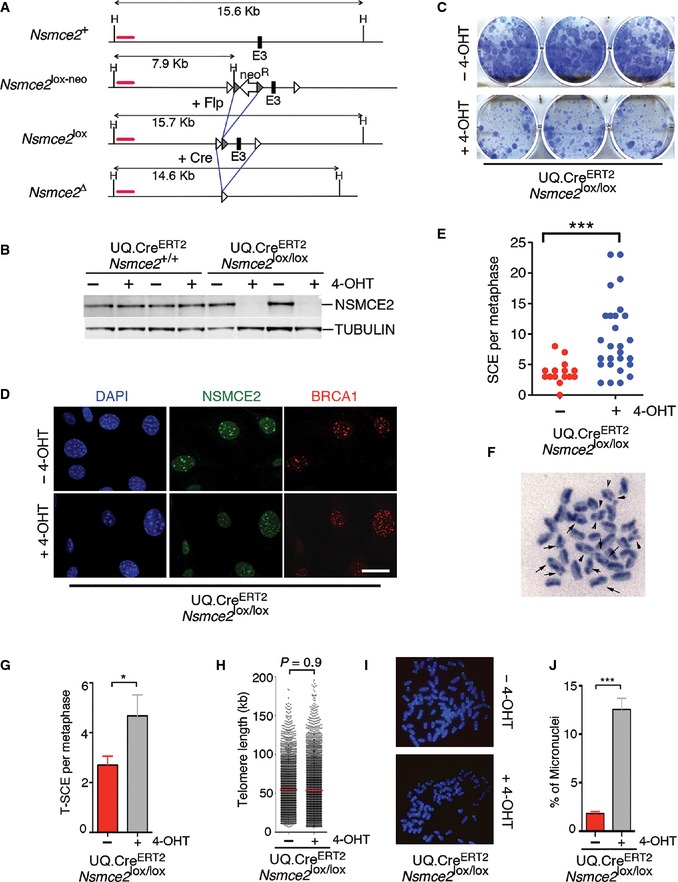
- Scheme illustrating the Nsmce2 conditional allele (Nsmce2 lox) used in this study. A cross with Flp recombinase‐expressing mice eliminated the FRT‐PGK‐neo‐FRT cassette present on the initial allele (Nsmce2 lox‐neo) (see Materials and Methods). In the conditional allele, exon 3 is flanked by loxP sites allowing for its deletion in the presence of Cre recombinase.
- NSMCE2 levels assessed by WB in UQ.CreERT 2 transgenic Nsmce2 +/+ and Nsmce2 lox/lox littermate MEFs, 24 h after exposure to 4‐OHT. Tubulin levels are shown as a loading control.
- Clonogenic assay on immortalized UQ.CreERT 2/Nsmce2 lox/lox MEFs. The picture shows the colonies detected after 10 days of growth in the presence or absence of 4‐OHT, upon staining with crystal violet.
- NSMCE2 and BRCA1 foci in MMS‐treated (1 mM for 1 h; 24‐h recovery) UQ.CreERT 2/Nsmce2 lox/lox MEFs, 48 h after being treated (or not) with 4‐OHT. Scale bar, 5 ?m.
- SCE events per metaphase on UQ.CreERT 2/Nsmce2 lox/lox MEFs grown in the presence of 4‐OHT for 72 h. ***P < 0.001.
- Example of SCE events (arrows) found on Nsmce2‐deleted MEFs.
- SCE events at telomeres on UQ.CreERT 2/Nsmce2 lox/lox MEFs grown in the presence of 4‐OHT for 72 h, as detected by CO‐FISH (see Materials and Methods). Data are representative of three independent experiments. Error bars indicate SD. *P < 0.05.
- Telomere lengths of UQ.CreERT 2 Nsmce2 lox/lox MEFs treated or not with 4‐OHT for 72 h.
- Example of the intensity of telomere signals (yellow) used for (H).
- Percentage of cells with micronuclei found on cultures of UQ.CreERT 2/Nsmce2 lox/lox MEFs treated or not with 4‐OHT for 72 h. Data are representative of three independent experiments. Error bars indicate SD. ***P < 0.001.
Nsmce2 deletion in adult mice leads to accelerated aging
To investigate the consequences of NSMCE2 deficiency in an entire organism, Nsmce2 +/+ and Nsmce2 lox/lox animals carrying the UQ.CreERT2 transgene were exposed to 4‐OHT. First, 14.5 dpc embryos were treated in utero for three consecutive days by intraperitoneal injections of 4‐OHT on pregnant mothers. Nsmce2 lox/lox embryos showed intrauterine dwarfism that was accompanied by an overall loss of cellularity, more notorious on some specific organs such as the thymus (Fig 6A; Appendix Fig S8). Interestingly, NSMCE2 deletion on the developing thymus led to a significant accumulation of cells in mitosis, which once again could be consistent with segregation defects (Appendix Fig S8). However, given that the tamoxifen treatment prevents delivery, no born mice could be obtained regardless of the genotype. To analyze the impact of NSMCE2 deficiency on live mice, mice were fed with a 4‐OHT‐containing diet starting at weaning. The strategy reduced NSMCE2 expression on all tissues analyzed (Appendix Fig S9), an effect that was less pronounced on organs with higher turnover such as the spleen, probably reflecting a competitive disadvantage of NSMCE2‐depleted cells. At the organism level, Nsmce2 deletion led to a progressive and generalized progeroid syndrome to which animals succumbed within less than a year (Fig 6B; Appendix Fig S10; Movies EV1 and EV2). In addition, NSMCE2 deletion led to a number of pathologies that are also found on Bloom patients including altered pigmentation (Fig 6C), a reduced percentage of fat (Fig 6D), and progressive anemia (Fig 6E). Furthermore, NSMCE2 deficiency also led to a dramatic accumulation of micronuclei on proliferating tissues such as the intestinal crypts (Fig 6F; Appendix Fig S11). In summary, NSMCE2 deletion in adult mice leads to a generalized progeroid syndrome that presents several of the cellular and phenotypic hallmarks of Bloom's syndrome.
Figure 6. Impact of NSMCE2 deletion in embryos and adult mice.
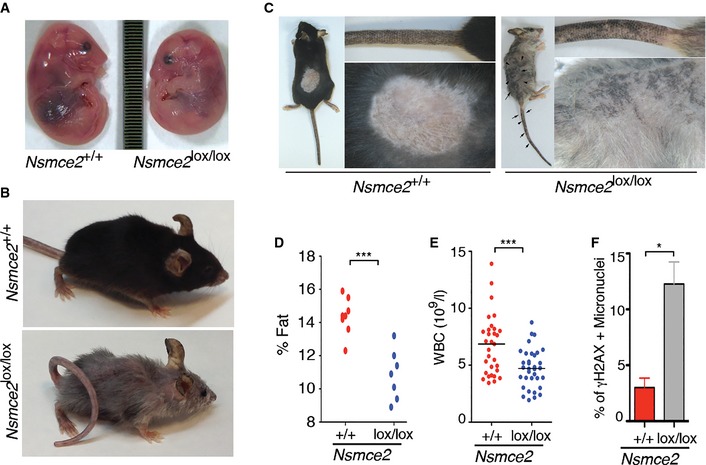
- Intrauterine dwarfism observed upon NSMCE2 deletion on 17.5 dpc embryos. Pregnant females were treated at 14.5 dpc for 3 days with intraperitoneal injections of 4‐OHT. The image represents the difference in size observed between Nsmce2 +/+ and Nsmce2 lox/lox embryos carrying the UQ.CreERT 2 transgene (Nsmce2 +/+ and Nsmce2 lox/lox, from now on).
- Representative picture of Nsmce2 +/+ and Nsmce2 lox/lox animals fed for 40 weeks with a 4‐OHT‐containing diet (see also Movies EV1 and EV2).
- Images illustrating the presence of altered pigmentation problems that arise on Nsmce2 lox/lox mice.
- Percentage of fat against total body mass on Nsmce2 +/+ and Nsmce2 lox/lox animals. ***P < 0.001.
- White blood cell counts (WBC) from Nsmce2 +/+ and Nsmce2 lox/lox animals. ***P < 0.001.
- Percentage of γH2AX‐positive micronuclei found on the intestinal epithelia of Nsmce2 +/+ and Nsmce2 lox/lox animals (see Appendix Fig S11). Data are representative of three independent experiments. Error bars indicate SD. *P < 0.05.
Independent roles of BLM and NSMCE2 in B lymphocytes
Given the similarities between NSMCE2 and BLM deficiency, we finally explored the potential relationship between these two pathways. To do so, and due to the essential nature of Blm and Nsmce2 in mice, we simultaneously deleted both genes in B lymphocytes with the help of a CD19.Cre allele (Rickert et al, 1997). Given that splenic B cells are in G0, lymphocyte cultures provide a unique system where to study DNA replication in a synchronously dividing population. Like previously observed on Nsmce2 SD/SD cells (Fig 3), NSMCE2 deletion in B cells did not abrogate the formation of SMC5/6 complexes (Appendix Fig S12). Interestingly, whereas like MEFs or early embryos NSMCE2‐deficient B cells showed evidences of mis‐segregation, DNA fiber analyses revealed that DNA replication was not affected by NSMCE2 loss (Fig 7). Accordingly, NSMCE2 (or BLM) deficiency did not have a profound impact on spleen B‐cell percentages and mutant B cells were able to grow ex vivo. However, the combined deletion of Blm and Nsmce2 led to a severe synthetic lethal interaction evidenced by the reduced spleen size of double‐mutant mice (Fig 8A) and the acute loss of B cells on double‐mutant spleens (Fig 8B). Importantly, B cells lacking both NSMCE2 and BLM presented an additional increase in spontaneous SCE levels when compared to the individual mutants (Fig 8C). Moreover, double‐mutant B cells presented a remarkable increase in cell size, which was detectable by flow cytometry (Fig 8D) and even by visual inspection of the cultures (Fig 8E). This increase in cell size coincided with the presence of large and irregular multilobulated nuclei on Blm/Nsmce2 double‐mutant cells, indicative of severe segregation defects. Collectively, these data suggest that NSMCE2 and BLM suppress mis‐segregation and recombination through independent pathways. Further supporting this concept, MMS‐induced BLM and NSMCE2 foci do not colocalize (Appendix Fig S13). Altogether, we propose that NSMCE2 and BLM independently facilitate chromosome segregation and limit recombination in mammalian cells, with each complex recognizing a different subset of substrates.
Figure 7. NSMCE2 is dispensable for DNA replication.
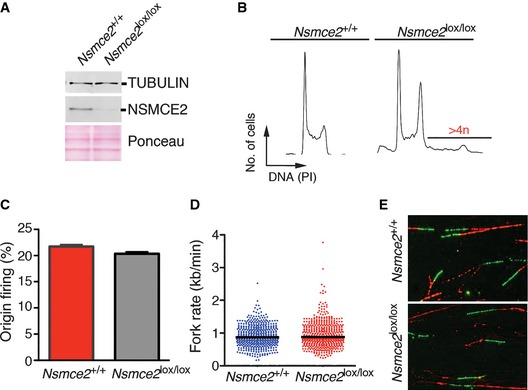
-
AWB illustrating the depletion of NSMCE2 in B cells isolated from 2‐month‐old CD19+/Cre animals of the indicated genotypes, 48 h after being stimulated in vitro with lipopolysaccharide (LPS). Tubulin was used as a loading control.
-
BRepresentative FACS analysis from the B‐cell cultures mentioned in (A). DNA content was measured with propidium iodide (PI). Note the presence of cells with > 4n DNA content in NSMCE2‐deleted cells.
-
C, DFrequency of origin firing (C) and fork rate (D) were analyzed by DNA fiber analyses (see Materials and Methods) from fibers prepared from B‐cell cultures explained in (A). No significant differences were observed between Nsmce2 +/+ and Nsmce2 lox/lox B cells. Data are representative of two independent experiments. Error bars indicate SD. Between 200 and 300 tracks were measured to estimate fork rate and 300–500 for the calculation of origin firing frequencies.
-
ERepresentative images of the DNA fibers used for the analysis explained in (C, D). CldU (red) and IdU (green) channels are shown.
Figure 8. Synthetic lethality between NSMCE2 and BLM in B cells.
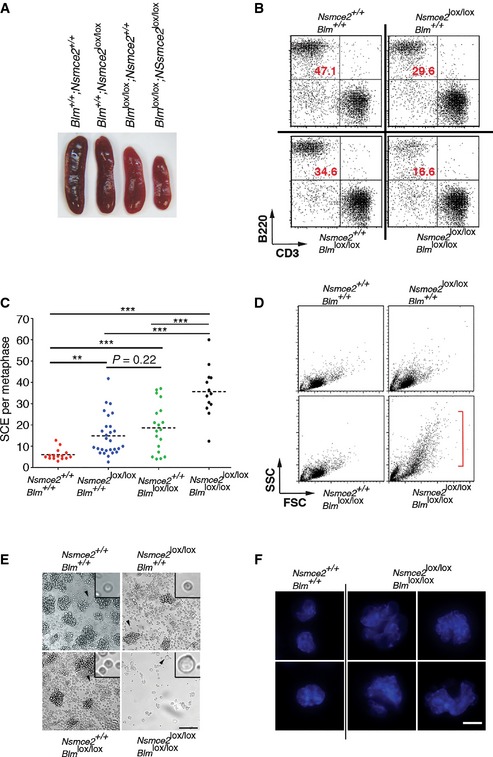
- Representative pictures of the spleens from 2‐month‐old CD19+/Cre animals of the indicated genotypes.
- Numbers of B and T cells from 2‐month‐old CD19+/Cre animals of the indicated genotypes evaluated by flow cytometry (B220: B‐cell marker; CD3: T‐cell marker).
- SCE events per metaphase on B cells obtained from CD19+/Cre animals of the indicated genotypes, 72 h after being stimulated in vitro with LPS. **P < 0.01, ***P < 0.001.
- Analysis of forward (FSC) and side (SSC) scatter parameters of B cells obtained from CD19+/Cre animals of the indicated genotypes, 48 h after being stimulated in vitro with LPS. The emergence of bigger cells is noticeable on cells lacking both BLM and NSMCE2.
- Representative images of B‐cell cultures of the indicated genotypes 48 h after being stimulated in vitro with LPS. Insets provide a magnified view of an individual cell (arrowhead) for better comparison of the observed differences in cell size. Scale bar, 100 μm.
- Examples of the types of nuclei found on Blm and Nsmce2 double‐mutant B cells (compared with wild‐type B cells), in cells obtained from (E). DAPI was used to stain DNA. Whereas wt nuclei are regular in size, double‐mutant cells invariably presented enlarged, multilobulated, and irregular nuclei. Scale bar, 2.5 μm.
Discussion
On the essential roles of the SMC5/6 complex
All subunits of the SMC5/6 complex are essential in yeast (Zhao & Blobel, 2005). Likewise, a recent work revealed that SMC6 is essential for mouse development (Ju et al, 2013). The only example where vertebrate cells can tolerate deletion of a complex member is the chicken B‐cell line DT40, where SMC5‐ or NSE2‐deficient cells can be generated (Stephan et al, 2011a). This might reflect species‐specific roles or the fact that lymphoid cells are specially tolerant to the deletion of essential genes related to replication‐borne genomic instability such as Brca1 (Bunting et al, 2010). Consistently, Nsmce2 or Blm‐deleted mouse B lymphocytes could also be grown ex vivo. Nevertheless, B cells only undergo a few division cycles ex vivo and the pleiotropic phenotypes observed on NSMCE2‐deleted adult mice argue for an overall essential role of this pathway in mammalian cells. In addition, the finding that Nsmce2 heterozygous tumors retain the wild‐type copy and that Nscme2 is also essential for the growth of immortalized MEFs argues that the pathway might also be essential for cancer cells. What is then the basis for this essential role?
On one hand, some studies have suggested that the SMC5/6 complex plays a role in promoting recombinational repair, which was based on early findings that detected the recruitment of SMC6 to a nuclease‐induced DNA break (De Piccoli et al, 2006). More recent data have also detected the complex at recombination sites during yeast meiosis (Lilienthal et al, 2013; Xaver et al, 2013). Along these lines, siRNA‐mediated depletion of SMC5/6 complex members in human cells has been shown to diminish inter‐sister recombination rates, particularly at telomeres (Potts et al, 2006; Potts & Yu, 2007; Wu et al, 2012). In contrast to this view in promoting recombination, a careful analysis of the recombinatorial repair of meiotic breaks in yeast SMC5/6 mutants shows normal DSB repair kinetics (Lilienthal et al, 2013). Likewise, our analysis of 53BP1 foci shows no major deficiency in overall DNA repair kinetics on NSMCE2‐deficient cells. On the contrary, rather than an absence of recombination between sister chromatids, we consistently observed a significant increase in SCE rates on Nsmce2‐mutant cells, including at telomeres. This increase in recombination is in agreement with all previous data using genetically modified models including yeast, chicken, or mouse (Prakash & Prakash, 1977; Stephan et al, 2011a; Kliszczak et al, 2012; Ju et al, 2013). Whereas the complex might be dispensable for the initial steps of HR, we cannot rule out that it might be required for resolving inter‐sister chromatid links that might arise as products of recombination (i.e. Holiday Junctions). Accordingly, RAD51 deletion alleviates the growth defects of SMC5/6 mutants exposed to genotoxic agents (Chen et al, 2009; Chavez et al, 2011). Nevertheless, and regardless of a role in recombinational repair, HR is not essential in yeast and thus such an activity cannot account for the essential functions of the SMC5/6 complex.
In addition to HR, additional studies in yeast have revealed a role of the SMC5/6 complex in the resolution and/or disolution of joint DNA molecules (JM) (Torres‐Rosell et al, 2005; Branzei et al, 2006; Farmer et al, 2011; Copsey et al, 2013; Lilienthal et al, 2013; Xaver et al, 2013). Supporting the role of entangled DNA molecules, early works revealed a synthetic lethal interaction between SMC5/6 and topoisomerase mutants in yeast (Verkade et al, 1999). Moreover, the fact that catalytic inhibitors of TOP2A induce NSMCE2 foci (this study), together with a previous report showing that SMC6 colocalizes with TOP2A during meiosis (Gomez et al, 2013), further supports that the complex is recruited to substrates different to DNA breaks. This disentangling activity of SMC5/6 is also essential for the full completion of S‐phase, and similar to topoisomerase I mutants, SMC5/6 mutants have difficulties in completing DNA replication (Torres‐Rosell et al, 2007; Kegel et al, 2011). Whereas our DNA fiber analyses discards a general role of SMC5/6 on DNA replication, we cannot exclude that a more restricted role at certain loci (i.e. repetitive sequences) might facilitate the completion of DNA replication also in mammals, which could explain the segregation defects observed on Nsmce2‐deficient cells. In contrast to its proposed roles on HR, a role of the complex on the resolution of JM could explain all the phenotypes that we have observed on NSMCE2‐deficient cells. The accumulation of JM has two outcomes. First, some JM are resolved through the action of nucleases and subsequent recombination, which is the basis for SCE in mammalian cells. Second, if JM persist into mitosis, they constitute a barrier for chromosome segregation that can ultimately lead to mitotic catastrophe. The increased MUS81‐dependent SCE levels and severe chromosome segregation defects found on NSMCE2‐deleted MEFs and adult tissues support this view. In what regards to the role of SUMOylation, and in agreement with our findings, whereas Mms21 is essential for chromosome segregation during yeast meiosis, its SUMO ligase activity is not (Xaver et al, 2013). Whereas at this point we cannot discard that some residual SUMO ligase activity persists in Nsmce2SD/SD cells, the limited role of NSMCE2‐dependent SUMOylation could also simply reflect redundancy by alternative SUMO ligases. Consistently, the mild phenotypes of yeast Mms21 SUMOylation deficient strains on DNA damage‐induced SUMOylation or in suppressing recombination are greatly exacerbated upon deletion of additional SUMO ligases (Cremona et al, 2012; Albuquerque et al, 2013). Of note, whereas the SUMO ligase activity of NSMCE2 is dispensable for mouse lifespan and overall health in unchallenged conditions, it remains to be seen whether it becomes limiting in the context of DNA damage. Regardless of SUMOylation, a role on the dissolution of JM is consistent with the phenotypes observed on NSMCE2‐deficient cells and can account for the essential role of this complex in mice.
NSMCE2 operates independently of BLM
In mammals, JM elimination is mediated either by a pathway initiated by BLM (disolution), or through the action of mitotic structure‐specific nucleases such as MUS81 or GEN1 (resolution) (Mankouri et al, 2013). In yeast, many of the phenotypes observed on SMC5/6 mutants phenocopy those observed in sgs1 deficient strains, which has led to the suggestion that the complex could operate in JM disolution. Consistently, and similar to Sgs1, reexpression of Smc6 specifically in mitosis promotes the disolution of JM that accumulates on smc6‐deficient yeast cells (Bermudez‐Lopez et al, 2010). Despite the similarities between BLM and NSMCE2, early studies failed to link Smc5/6 to Sgs1 (Branzei et al, 2006). Moreover, and in agreement with our data, sgs1 deletion is synthetic sick with Mms21 deficiency in yeast (Chen et al, 2009), and double mutants present an increased accumulation of JM in mitosis (Torres‐Rosell et al, 2005) or meiosis (Copsey et al, 2013; Xaver et al, 2013). Finally, a recent work revealed that SMC6 knockdown in human cells leads to increased BLM foci (Gallego‐Paez et al, 2014). Together with our findings on NSMCE2‐ and BLM‐deleted B cells, these data support that NSMCE2 and BLM independently suppress recombination and facilitate segregation in mammalian cells.
One possibility to explain the independent roles of NSMCE2 and BLM would be that whereas both perform similar functions, they operate on different structures. Interestingly, whereas BLM foci are scattered throughout the nucleus, NSMCE2 foci form almost invariably around pericentromeric heterochromatin (Figs 1, 3 and 5; Appendix Figs S2 and S13). The same pericentromeric pattern has also been recently described for SMC6 during human spermatogenesis (Verver et al, 2014). This localization might suggest a preferential role of the SMC5/6 complex at repeats, given that centromeric sequences are the largest repeat of the mammalian genome. Consistently, several reports have observed a preferential localization of the Smc5/6 complex at repeat sequences in yeast and the complex is essential for the segregation of ribosomal DNA in S. cerevisiae, this being the largest repeat in the yeast genome (Torres‐Rosell et al, 2005, 2007; Zhao & Blobel, 2005; Lindroos et al, 2006; Chavez et al, 2010; Noel & Wellinger, 2011). Altogether, we propose that BLM and the SMC5/6 complex might perform similar functions, but with a preference toward structures that arise at repeated sequences in the case of SMC5/6.
Physiological impact of NSMCE2 deficiency
Whereas work in model organisms has suggested an important role for the SMC5/6 complex in safeguarding genomic integrity, the impact of this pathway on a mammalian organism remained largely unknown. Our work here reveals that whereas mild deficiencies on the complex might promote cancer development, more penetrant mutations might lead to accelerated aging. This transition from cancer to aging depending on the severity of the mutation has been previously observed on other genome integrity‐related pathways such as nucleotide excision repair or replication stress (Lopez‐Contreras & Fernandez‐Capetillo, 2010; Diderich et al, 2011). As to how a deficiency in NSMCE2 might promote cancer, one possibility is that this is a consequence of chromosomal rearrangements arising due to increased recombination rates. Supporting this view, Mms21 suppresses gross chromosomal rearrangements in S. cerevisiae (Albuquerque et al, 2013), and ChIP‐seq analysis of mouse B cells exposed to hydroxyurea revealed that SMC5 maps to regions frequently rearranged in human leukemias (Barlow et al, 2013). Interestingly, a recent report has revealed fusions involving NSMCE2 in acute myeloid leukemias (Chinen et al, 2014). The impact of these fusions on NSMCE2 activity remains unknown. Regarding the haploinsufficiency of NSMCE2, and even if our antibodies failed to detect the expression of a truncated NSMCE2 variant in Nsmce2 +/GT cells, we cannot discard that our genetrap allele does express a smaller version of NSMCE2, which could have an effect on the observed phenotypes. Nevertheless, from the comparison between the findings made in Nsmce2 +/GT versus nullyzygosity, it is clear that while milder deficiencies in the complex might promote cancer, more profound deficiencies will have severe detrimental effects at the organism level affecting multiple organs.
In what regards to aging, we can envision two non‐mutually exclusive mechanisms for the observed progeria. On one hand, NSMCE2 deletion might lead to the elimination of the mutant cells followed by a subsequent compensatory proliferation from non‐deleted wild‐type cells. This mechanism, previously proposed for the accelerated aging that arises upon deletion of the ATR kinase in adult mice (Ruzankina et al, 2007), would be supported by the lower reduction in NSMCE2 levels that is observed in organs with high turnover such as the spleen. Alternatively, or in addition, NSMCE2 deletion might promote the accumulation of abnormal cells due to inefficient chromosomal segregation, particularly in tissues with lower renewal rates, which could at some point compromise the functionality of the organ. Further studies on the organs of NSMCE2 depleted adult organs should be clarifying in this regard.
Finally, besides cancer, and regardless of the overall progeria, the use of NSMCE2‐deleted adult mice revealed a subset of phenotypes that are also found on BLM patients such as a reduced percentage of fat, altered pigmentation, and progressive anemia which, like in the case of BS cells, were associated with a significant increase in micronuclei on mutant tissues. Noteworthy, a recent report revealed the discovery of two human patients presenting reduced levels of NSMCE2 (Payne et al, 2014). Both human patients also presented dwarfism, altered pigmentation, and increased micronuclei, and whereas no obvious progeroid symptoms were reported, one of them died at 33 years of age from a sudden cardiovascular event. Of note, one of the mutations reported in humans also affected the SUMO ligase activity of NSMCE2. However, this mutation also severely reduced NSMCE2 levels so that the symptoms could be more related to the hypomorphism than to the SUMO ligase activity. Likewise, it is possible that the more severe disease we observe in mice versus what has been reported in human patients relates to the differences between nullyzygosity and hypomorphism. In any case, our work here has shed new light into the roles of the SMC5/6 complex in mammals and revealed that SUMOylation‐independent activities of NSMCE2 suppress recombination and micronuclei through a mechanism that operates independently of BLM and which is critical to prevent the onset of cancer and aging in mice.
Materials and Methods
Mouse models
The Nsmce2 genetrap strain was generated using the gene trap ES clone AA0032 from the Sanger Institute. Nsmce2 SD knock‐in mice were generated by gene targeting, using a vector generated by recombineering (Genebridges, Dresden, Germany). Briefly, we introduced the mutations encoding C195S and H197A that inactivate the SUMO ligase activity of NSMCE2 (Andrews et al, 2005) into a targeting plasmid containing an FRT‐PGK‐neo‐FRT cassette. The neomycin resistance cassette was subsequently eliminated by crossing NSMCE2SD‐frt‐neo‐frt/+ mice with a strain expressing Flp recombinase from the β‐actin promoter (Rodriguez et al, 2000). Finally, the targeting vector for the generation of Nsmce2 conditional knockout mice was also obtained from Genebridges (Dresden, Germany). The targeting vector flanked exon 3 with loxP sequences and also contained an FRT‐PGK‐neo‐FRT that enabled selection in ES cells and which was later deleted as explained above. Blmlox/lox, CD19.Cre, UQ.CreERT2, and Mus81 −/− mice have been previously described (Rickert et al, 1997; McPherson et al, 2004; Chester et al, 2006; Ruzankina et al, 2007). For Nsmce2 deletion in UQ.CreERT2 mice, animals were fed with a diet containing 400 mg of tamoxifen per kg (Harlan Tekland, Madison, USA). For intrauterine treatments, 14.5 dpc females were injected intraperitoneally for 3 consecutive days with 4‐OHT dissolved in corn oil, where embryos were isolated for further analyses. The percentage of fat was determined by dual energy X‐ray absorptiometry (DEXA) (Lunar PIXImus Densitometer, GE Medical Systems). All mice were kept under standard conditions at serum‐pathogen free facility of the Spanish National Cancer Centre in a mixed C57BL/6‐129/Sv background. All mouse work was performed in accordance with the Guidelines for Humane Endpoints for Animals Used in Biomedical Research and under the supervision of the Ethics Committee for Animal Research of the Instituto de Salud Carlos III.
Immunofluorescence and immunoblotting
Mouse spermatocyte IF was performed as described (Fernandez‐Capetillo et al, 2003). Briefly, spermatocyte spreads were treated for 3 min in 2% PFA and 0.03% SDS, washed in PFA 2%, and permeabilized for 1 min with 0.4% Photolo, Kodak. Preparations were subsequently stained following standard procedures using a blocking buffer containing 10% goat serum, 3% BSA, and 0.05% Triton X‐100. For embryo IF, mouse morulae were fixed with PTEMF buffer (0.2% Triton X‐100, 20 mM PIPES pH 6.8; 1 mM MgCl2; 10 mM EGTA, 4% PFA) for 10 min at RT in a Microtest Plate (Greiner Bio‐One). Fixed morulae were next blocked in 3% BSA/PBS for 30 min and stained using standard procedures. For the detection of NSMCE2, BLM, and BRCA1 foci, a protocol that washes out the fraction of nucleoplasmic proteins previous to fixation was used as described before (Celeste et al, 2003). High‐throughput microscopy was performed by growing MEFs in μCLEAR bottom 96‐well plates (Greiner Bio‐One). Images were automatically acquired from each well with an Opera High‐Content Screening System (PerkinElmer) at 40× magnification and non‐saturating conditions. Images were segmented using the DAPI staining to generate masks matching cell nuclei from which the corresponding signals were calculated. Recombinant full‐length human NSMCE2 fused to GST was used to generate either rabbit polyclonal (VivoTecnia, Spain) or mouse monoclonal (Monoclonal Antibodies Unit, CNIO, Madrid) antibodies. The rabbit polyclonal antibody showed reactivity with human and mouse NSMCE2, whereas the monoclonal antibody only recognizes the human protein. Antibodies were purified using Affi‐Gel columns (Bio‐Rad) previously coated with immobilized GST‐NSMCE2 protein. γH2AX (Upstate Biotechnology, 05‐636), 53BP1 (Novus Biologicals, 100‐304A2), β‐ACTIN (Sigma, A2228), TUBULIN (Sigma, T9023), and SCP3 (Abcam, ab15093); BRCA1 (provided by Andre Nussenzweig); SMC5 (Biothyl, A300‐236A); and SMC6 (provided by Alan Lehmann) were used. Western blot analyses were performed using the LICOR platform (Biosciences).
Immunohistochemistry
Embryos or adult organs were fixed in formalin and embedded in paraffin for subsequent processing. Consecutive 2.5‐μm sections were treated with citrate for antigen recovery and processed for IHC with antibodies to NSMCE2 (described above), KI67, γH2AX, or activated caspase 3 (Upstate Biotechnology). Hematoxylin was used to counterstain. IHC samples were scanned with a MIRAX digitalized system (Zeiss).
MEF and B‐cell analyses
MEFs were isolated from 12.5 dpc embryos using standard procedures. For B‐cell cultures, resting splenic B lymphocytes were isolated from 8‐ to 12‐week‐old WT or mutant spleens with anti‐CD43 microbeads (anti‐Ly48; Miltenyi Biotech) and cultured with standard protocols. For SCE analyses, cells were cultured in the presence of 5′‐bromo‐2′‐deoxyuridine (BrdU; Sigma) at a final concentration of 10 μM and then allowed to replicate their DNA once for 24 h. Colcemide was added at 0.1 μg/ml during the last 5 h. Metaphases were prepared using a standard hypotonic/Carnoy procedure and subsequently treated with UV (355 nm) and Hoechst to counterstain nascent DNA strands. Slides were finally stained with a 1% Leishman solution diluted 2/3 in Gurr's buffer. Analysis of telomere lengths using Q‐FISH (Samper et al, 2000) or T‐SCE events using chromosome orientation FISH (CO‐FISH) (Gonzalo et al, 2006) was performed as described. NSMCE2lox/lox.Ub.Cre.ERT2+/T MEFs were treated with 1 μM 4‐hydroxytamoxifen (4‐OHT, H7904, Sigma) 48 h before the correspondent assay.
NSMCE2 auto‐SUMOylation assay
FLP‐In™‐293 cells over‐expressing NSMCE2WT or NSMCE2C185S/H187A were transfected with plasmids containing SUMO1‐GFP or FLAG‐SUMO2 cDNAs using Lipofectamine 2000 (Invitrogen). Forty‐eight hours post‐transfection, cells were harvested and processed as following. For SUMO1‐GFP, lysates were treated according to the instructions of the μMACS GFP Isolation kit (Miltenyi Biotec). In the case of FLAG‐SUMO2, cells were lysed in protein lysate buffer (50 mM Tris–HCl pH 7.4; 150 mM NaCl; 1 mM EDTA; 1 mM EGTA; 270 mM sucrose; 1% Triton X‐100) and supplemented with a mix of protease inhibitors (Sigma‐Aldrich), Benzonase® (Merck), and 20 mM N‐ethylmaleimide (Sigma‐Aldrich). Precleared lysates were subsequently immunoprecipitated with magnetic anti‐FLAG beads (Sigma‐Aldrich) for 4 h at 4°C. After three washes in lysis buffer, immunoprecipitated samples were denatured in Nupage buffer and loaded on gradient 4–12% SDS–PAGE gels (Invitrogen). GFP (Roche), NSMCE2 (described above), Tubulin, and FLAG (Sigma) antibodies were used.
DNA fiber analysis
Exponentially growing cells were pulse‐labeled with 50 μM CldU (20 min) followed by 250 μM IdU (20 min). Labeled cells were collected, and DNA fibers were spread in buffer containing 0.5% SDS, 200 mM Tris pH 7.4, and 50 mM EDTA. For immunodetection of labeled tracks, fibers were incubated with primary antibodies (for CldU, rat anti‐BrdU; for IdU, mouse anti‐BrdU) and developed with the corresponding secondary antibodies conjugated to Alexa dyes. Mouse anti‐ssDNA antibody was used to assess fiber integrity. Slides were examined with a Leica DM6000 B microscope, as described previously (Mouron et al, 2013). The conversion factor used was 1 μm = 2.59 kb (Jackson & Pombo, 1998). In each assay, 200–300 tracks were measured to estimate fork rate and 300–500 tracks were analyzed to estimate the frequency of origin firing [first label origins (green‐red‐green) are shown as percentage of all red‐labeled tracks (CldU)] (Petermann et al, 2010).
Author contributions
AJ and PG‐M participated in the generation and analyses of NSMCE2 mouse models. FS participated on the analysis of BLM and NSMCE2 interaction and helped on the analysis of segregation, NSMCE2 foci, and DNA replication experiments. ET helped on the analysis of SUMOylation‐deficient mutants and performed SCE analyses on Nsmce2 and Mus81 double‐mutant MEFs. EL helped with biochemistry and analysis of Nsmce2‐mutant cells. MM helped on the characterization of Nsmce2‐mutant MEFs and B cells. SR and JM performed DNA fiber analyses. PM and MAB performed telomere analyses. OF‐C supervised the study and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Movie EV2
Review Process File
Acknowledgements
The authors want to thank Jordi Torres and Mark O'Driscoll for comments on the manuscript. Work in OF laboratory related to this project was supported by Fundación Botín, by Banco Santander through its Santander Universities Global Division and by grants from MINECO (SAF2011‐23753 and SAF2014‐57791‐REDC), Howard Hughes Medical Institute, and the European Research Council (ERC‐617840). Work in JM laboratory was funded by a grant from MINECO (BFU2013‐49153P).
The EMBO Journal (2015) 34: 2604–2619
References
- Albuquerque CP, Wang G, Lee NS, Kolodner RD, Putnam CD, Zhou H (2013) Distinct SUMO ligases cooperate with Esc2 and Slx5 to suppress duplication‐mediated genome rearrangements. PLoS Genet 9: e1003670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews EA, Palecek J, Sergeant J, Taylor E, Lehmann AR, Watts FZ (2005) Nse2, a component of the Smc5‐6 complex, is a SUMO ligase required for the response to DNA damage. Mol Cell Biol 25: 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez‐Cruz G, Sun HW, McKinnon P, Wright G, Casellas R, Robbiani DF, Staudt L, Fernandez‐Capetillo O, Nussenzweig A (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell 152: 620–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez‐Lopez M, Ceschia A, de Piccoli G, Colomina N, Pasero P, Aragon L, Torres‐Rosell J (2010) The Smc5/6 complex is required for dissolution of DNA‐mediated sister chromatid linkages. Nucleic Acids Res 38: 6502–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Sollier J, Liberi G, Zhao X, Maeda D, Seki M, Enomoto T, Ohta K, Foiani M (2006) Ubc9‐ and mms21‐mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 127: 509–522 [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez‐Capetillo O, Cao L, Xu X, Deng CX, Finkel T, Nussenzweig M, Stark JM, Nussenzweig A (2010) 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell 141: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Fernandez‐Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5: 675–679 [DOI] [PubMed] [Google Scholar]
- Chan KL, North PS, Hickson ID (2007) BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 26: 3397–3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Palmai‐Pallag T, Ying S, Hickson ID (2009) Replication stress induces sister‐chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11: 753–760 [DOI] [PubMed] [Google Scholar]
- Chavez A, George V, Agrawal V, Johnson FB (2010) Sumoylation and the structural maintenance of chromosomes (Smc) 5/6 complex slow senescence through recombination intermediate resolution. J Biol Chem 285: 11922–11930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Agrawal V, Johnson FB (2011) Homologous recombination‐dependent rescue of deficiency in the structural maintenance of chromosomes (Smc) 5/6 complex. J Biol Chem 286: 5119–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Choi K, Szakal B, Arenz J, Duan X, Ye H, Branzei D, Zhao X (2009) Interplay between the Smc5/6 complex and the Mph1 helicase in recombinational repair. Proc Natl Acad Sci USA 106: 21252–21257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester N, Babbe H, Pinkas J, Manning C, Leder P (2006) Mutation of the murine Bloom's syndrome gene produces global genome destabilization. Mol Cell Biol 26: 6713–6726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen Y, Sakamoto N, Nagoshi H, Taki T, Maegawa S, Tatekawa S, Tsukamoto T, Mizutani S, Shimura Y, Yamamoto‐Sugitani M, Kobayashi T, Matsumoto Y, Horiike S, Kuroda J, Taniwaki M (2014) 8q24 amplified segments involve novel fusion genes between NSMCE2 and long noncoding RNAs in acute myelogenous leukemia. J Hematol Oncol 7: 68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu WK, Hickson ID (2009) RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer 9: 644–654 [DOI] [PubMed] [Google Scholar]
- Copsey A, Tang S, Jordan PW, Blitzblau HG, Newcombe S, Chan AC, Newnham L, Li Z, Gray S, Herbert AD, Arumugam P, Hochwagen A, Hunter N, Hoffmann E (2013) Smc5/6 coordinates formation and resolution of joint molecules with chromosome morphology to ensure meiotic divisions. PLoS Genet 9: e1004071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona CA, Sarangi P, Yang Y, Hang LE, Rahman S, Zhao X (2012) Extensive DNA damage‐induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol Cell 45: 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Piccoli G, Cortes‐Ledesma F, Ira G, Torres‐Rosell J, Uhle S, Farmer S, Hwang JY, Machin F, Ceschia A, McAleenan A, Cordon‐Preciado V, Clemente‐Blanco A, Vilella‐Mitjana F, Ullal P, Jarmuz A, Leitao B, Bressan D, Dotiwala F, Papusha A, Zhao X et al (2006) Smc5‐Smc6 mediate DNA double‐strand‐break repair by promoting sister‐chromatid recombination. Nat Cell Biol 8: 1032–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Piccoli G, Torres‐Rosell J, Aragon L (2009) The unnamed complex: what do we know about Smc5‐Smc6? Chromosome Res 17: 251–263 [DOI] [PubMed] [Google Scholar]
- Diderich K, Alanazi M, Hoeijmakers JH (2011) Premature aging and cancer in nucleotide excision repair‐disorders. DNA Repair 10: 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JM, Gao J, Wang J, Yang M, Potts PR (2010) MAGE‐RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell 39: 963–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer S, San‐Segundo PA, Aragon L (2011) The Smc5‐Smc6 complex is required to remove chromosome junctions in meiosis. PLoS ONE 6: e20948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini‐Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A (2003) H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell 4: 497–508 [DOI] [PubMed] [Google Scholar]
- Fousteri MI, Lehmann AR (2000) A novel SMC protein complex in Schizosaccharomyces pombe contains the Rad18 DNA repair protein. EMBO J 19: 1691–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego‐Paez LM, Tanaka H, Bando M, Takahashi M, Nozaki N, Nakato R, Shirahige K, Hirota T (2014) Smc5/6‐mediated regulation of replication progression contributes to chromosome assembly during mitosis in human cells. Mol Biol Cell 25: 302–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez R, Jordan PW, Viera A, Alsheimer M, Fukuda T, Jessberger R, Llano E, Pendas AM, Handel MA, Suja JA (2013) Dynamic localization of SMC5/6 complex proteins during mammalian meiosis and mitosis suggests functions in distinct chromosome processes. J Cell Sci 126: 4239–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA (2006) DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 8: 416–424 [DOI] [PubMed] [Google Scholar]
- Jackson DA, Pombo A (1998) Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140: 1285–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju L, Wing J, Taylor E, Brandt R, Slijepcevic P, Horsch M, Rathkolb B, Racz I, Becker L, Hans W, Adler T, Beckers J, Rozman J, Klingenspor M, Wolf E, Zimmer A, Klopstock T, Busch DH, Gailus‐Durner V, Fuchs H et al (2013) SMC6 is an essential gene in mice, but a hypomorphic mutant in the ATPase domain has a mild phenotype with a range of subtle abnormalities. DNA Repair 12: 356–366 [DOI] [PubMed] [Google Scholar]
- Kegel A, Betts‐Lindroos H, Kanno T, Jeppsson K, Strom L, Katou Y, Itoh T, Shirahige K, Sjogren C (2011) Chromosome length influences replication‐induced topological stress. Nature 471: 392–396 [DOI] [PubMed] [Google Scholar]
- Kliszczak M, Stephan AK, Flanagan AM, Morrison CG (2012) SUMO ligase activity of vertebrate Mms21/Nse2 is required for efficient DNA repair but not for Smc5/6 complex stability. DNA Repair 11: 799–810 [DOI] [PubMed] [Google Scholar]
- Lehmann AR, Walicka M, Griffiths DJ, Murray JM, Watts FZ, McCready S, Carr AM (1995) The rad18 gene of Schizosaccharomyces pombe defines a new subgroup of the SMC superfamily involved in DNA repair. Mol Cell Biol 15: 7067–7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilienthal I, Kanno T, Sjogren C (2013) Inhibition of the Smc5/6 complex during meiosis perturbs joint molecule formation and resolution without significantly changing crossover or non‐crossover levels. PLoS Genet 9: e1003898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroos HB, Strom L, Itoh T, Katou Y, Shirahige K, Sjogren C (2006) Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol Cell 22: 755–767 [DOI] [PubMed] [Google Scholar]
- Lopez‐Contreras AJ, Fernandez‐Capetillo O (2010) The ATR barrier to replication‐born DNA damage. DNA Repair 9: 1249–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Hirano T (2005) Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev 19: 1269–1287 [DOI] [PubMed] [Google Scholar]
- Mahadevaiah SK, Bourc'his D, de Rooij DG, Bestor TH, Turner JM, Burgoyne PS (2008) Extensive meiotic asynapsis in mice antagonises meiotic silencing of unsynapsed chromatin and consequently disrupts meiotic sex chromosome inactivation. J Cell Biol 182: 263–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Huttner D, Hickson ID (2013) How unfinished business from S‐phase affects mitosis and beyond. EMBO J 32: 2661–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald WH, Pavlova Y, Yates JR III, Boddy MN (2003) Novel essential DNA repair proteins Nse1 and Nse2 are subunits of the fission yeast Smc5‐Smc6 complex. J Biol Chem 278: 45460–45467 [DOI] [PubMed] [Google Scholar]
- McPherson JP, Lemmers B, Chahwan R, Pamidi A, Migon E, Matysiak‐Zablocki E, Moynahan ME, Essers J, Hanada K, Poonepalli A, Sanchez‐Sweatman O, Khokha R, Kanaar R, Jasin M, Hande MP, Hakem R (2004) Involvement of mammalian Mus81 in genome integrity and tumor suppression. Science 304: 1822–1826 [DOI] [PubMed] [Google Scholar]
- Morikawa H, Morishita T, Kawane S, Iwasaki H, Carr AM, Shinagawa H (2004) Rad62 protein functionally and physically associates with the smc5/smc6 protein complex and is required for chromosome integrity and recombination repair in fission yeast. Mol Cell Biol 24: 9401–9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouron S, Rodriguez‐Acebes S, Martinez‐Jimenez MI, Garcia‐Gomez S, Chocron S, Blanco L, Mendez J (2013) Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20: 1383–1389 [DOI] [PubMed] [Google Scholar]
- Murray JM, Carr AM (2008) Smc5/6: a link between DNA repair and unidirectional replication? Nat Rev Mol Cell Biol 9: 177–182 [DOI] [PubMed] [Google Scholar]
- Naim V, Rosselli F (2009) The FANC pathway and BLM collaborate during mitosis to prevent micro‐nucleation and chromosome abnormalities. Nat Cell Biol 11: 761–768 [DOI] [PubMed] [Google Scholar]
- Nasim A, Smith BP (1975) Genetic control of radiation sensitivity in Schizosaccharomyces pombe . Genetics 79: 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K, Haering CH (2005) The structure and function of SMC and kleisin complexes. Annu Rev Biochem 74: 595–648 [DOI] [PubMed] [Google Scholar]
- Noel JF, Wellinger RJ (2011) Abrupt telomere losses and reduced end‐resection can explain accelerated senescence of Smc5/6 mutants lacking telomerase. DNA Repair 10: 271–282 [DOI] [PubMed] [Google Scholar]
- Outwin EA, Irmisch A, Murray JM, O'Connell MJ (2009) Smc5‐Smc6‐dependent removal of cohesin from mitotic chromosomes. Mol Cell Biol 29: 4363–4375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek J, Vidot S, Feng M, Doherty AJ, Lehmann AR (2006) The Smc5‐Smc6 DNA repair complex. bridging of the Smc5‐Smc6 heads by the KLEISIN, Nse4, and non‐Kleisin subunits. J Biol Chem 281: 36952–36959 [DOI] [PubMed] [Google Scholar]
- Payne F, Colnaghi R, Rocha N, Seth A, Harris J, Carpenter G, Bottomley WE, Wheeler E, Wong S, Saudek V, Savage D, O'Rahilly S, Carel JC, Barroso I, O'Driscoll M, Semple R (2014) Hypomorphism in human NSMCE2 linked to primordial dwarfism and insulin resistance. J Clin Invest 124: 4028–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebernard S, McDonald WH, Pavlova Y, Yates JR III, Boddy MN (2004) Nse1, Nse2, and a novel subunit of the Smc5‐Smc6 complex, Nse3, play a crucial role in meiosis. Mol Biol Cell 15: 4866–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebernard S, Perry JJ, Tainer JA, Boddy MN (2008a) Nse1 RING‐like domain supports functions of the Smc5‐Smc6 holocomplex in genome stability. Mol Biol Cell 19: 4099–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN (2008b) Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J 27: 3011–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, Helleday T (2010) Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci USA 107: 16090–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts PR, Yu H (2005) Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol Cell Biol 25: 7021–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts PR, Porteus MH, Yu H (2006) Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double‐strand breaks. EMBO J 25: 3377–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts PR, Yu H (2007) The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere‐binding proteins. Nat Struct Mol Biol 14: 581–590 [DOI] [PubMed] [Google Scholar]
- Prakash S, Prakash L (1977) Increased spontaneous mitotic segregation in MMS‐sensitive mutants of Saccharomyces cerevisiae . Genetics 87: 229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert RC, Roes J, Rajewsky K (1997) B lymphocyte‐specific, Cre‐mediated mutagenesis in mice. Nucleic Acids Res 25: 1317–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM (2000) High‐efficiency deleter mice show that FLPe is an alternative to Cre‐loxP. Nat Genet 25: 139–140 [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double‐strand breaks in vivo. J Cell Biol 146: 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon‐Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ (2007) Deletion of the developmentally essential gene ATR in adult mice leads to age‐related phenotypes and stem cell loss. Cell Stem Cell 1: 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA (2000) Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G‐strand overhang. EMBO Rep 1: 244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM (1997a) Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 90: 425–435 [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM (1997b) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88: 265–275 [DOI] [PubMed] [Google Scholar]
- Sergeant J, Taylor E, Palecek J, Fousteri M, Andrews EA, Sweeney S, Shinagawa H, Watts FZ, Lehmann AR (2005) Composition and architecture of the Schizosaccharomyces pombe Rad18 (Smc5‐6) complex. Mol Cell Biol 25: 172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AK, Kliszczak M, Dodson H, Cooley C, Morrison CG (2011a) Roles of vertebrate Smc5 in sister chromatid cohesion and homologous recombinational repair. Mol Cell Biol 31: 1369–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AK, Kliszczak M, Morrison CG (2011b) The Nse2/Mms21 SUMO ligase of the Smc5/6 complex in the maintenance of genome stability. FEBS Lett 585: 2907–2913 [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Dulev S, Liu X, Hiller NJ, Zhao X, Strunnikov A (2008) Cooperation of sumoylated chromosomal proteins in rDNA maintenance. PLoS Genet 4: e1000215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro S, Walter J, Shinohara A, Kamada N, Cremer T (2000) Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J Cell Biol 150: 283–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EM, Moghraby JS, Lees JH, Smit B, Moens PB, Lehmann AR (2001) Characterization of a novel human SMC heterodimer homologous to the Schizosaccharomyces pombe Rad18/Spr18 complex. Mol Biol Cell 12: 1583–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EM, Copsey AC, Hudson JJ, Vidot S, Lehmann AR (2008) Identification of the proteins, including MAGEG1, that make up the human SMC5‐6 protein complex. Mol Cell Biol 28: 1197–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres‐Rosell J, Machin F, Farmer S, Jarmuz A, Eydmann T, Dalgaard JZ, Aragon L (2005) SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat Cell Biol 7: 412–419 [DOI] [PubMed] [Google Scholar]
- Torres‐Rosell J, De Piccoli G, Cordon‐Preciado V, Farmer S, Jarmuz A, Machin F, Pasero P, Lisby M, Haber JE, Aragon L (2007) Anaphase onset before complete DNA replication with intact checkpoint responses. Science 315: 1411–1415 [DOI] [PubMed] [Google Scholar]
- Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O'Connell MJ (1999) Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell 10: 2905–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verver DE, Langedijk NS, Jordan PW, Repping S, Hamer G (2014) The SMC5/6 Complex is involved in crucial processes during human spermatogenesis. Biol Reprod 91: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler T, Newman S, West SC (2011) Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471: 642–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Kong X, Ji Z, Zeng W, Potts PR, Yokomori K, Yu H (2012) Scc1 sumoylation by Mms21 promotes sister chromatid recombination through counteracting Wapl. Genes Dev 26: 1473–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xaver M, Huang L, Chen D, Klein F (2013) Smc5/6‐mms21 prevents and eliminates inappropriate recombination intermediates in meiosis. PLoS Genet 9: e1004067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Blobel G (2005) A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci USA 102: 4777–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Movie EV1
Movie EV2
Review Process File