

Abstract

We report a novel approach to synthesize carfentanil amide analogues utilizing the isocyanide-based four-component Ugi multicomponent reaction. A small library of bis-amide analogues of carfentanil was created using N-alkylpiperidones, aniline, propionic acid, and various aliphatic isocyanides. Our lead compound showed high affinity for mu (MOR) and delta opioid receptors (DOR) with no appreciable affinity for kappa (KOR) receptors in radioligand binding assays. The compound was found to be a mixed MOR agonist/partial DOR agonist in [35S]GTPγS functional assays, and it showed moderate analgesic potency in vivo. The compound showed no visible signs of physical dependence or constipation in mice. In addition, it produced less respiratory depression than morphine. Most mixed MOR/DOR opioids reported in the literature are peptides and thereby systemically inactive. Our approach utilizing a multicomponent reaction has the promise to deliver potent and efficacious small-molecule analgesics with potential clinical utility.
Keywords: Ugi reaction, multicomponent reactions, mu-delta, opioid analgesics, carfentanil
Opioids are the most widely used drugs for the treatment of moderate to severe, chronic pain. The most commonly used compound of this class is the epoxymorphinan alkaloid morphine.1 Morphine (1) and its semisynthetic analogues based on the same scaffold exhibit their analgesic properties through the activation of the three major opioid receptors: mu (MOR), delta (DOR), and kappa (KOR).2 Unfortunately, the activation of these receptors, particularly MOR, also causes significant side effects,3 including respiratory depression, constipation, tolerance, physical dependence, and substance abuse.4 A great increase in analgesic potency is achieved by using aryl anilido piperidines including fentanyl (2) and carfentanil (3); however, the problem of severe side effects remains unsolved by this approach (Figure 1).5
Figure 1.

Structures of commonly used opioid analgesics and the proposed carfentanil amides.
One possible way to overcome MOR-mediated adverse effects is to synthesize mixed partial agonists or compounds with mixed MOR agonist/DOR antagonist properties.6 Previous studies have shown that tolerance and dependence to morphine can be reversed by DOR antagonists without sacrificing analgesic potency.7 Co-administration of DOR agonists with MOR agonists increases the potency and efficacy of MOR agonists as well.8 Similarly, partial activation of multiple opioid receptors by a single ligand could produce analgesia without MOR-mediated side-effects. Most of the previously reported MOR/DOR mixed agonists are peptides, which have somewhat limited relevance from a clinical utility viewpoint.9 Systemically active peptidomimetics and nonpeptide small molecules have also been reported with a similar pharmacological profile.10 An example of a mixed agonist small molecule is SoRI 9409, which showed a preferable side effect profile to morphine. However, SoRI 9409 produced limited antinociception in thermal pain models, thereby limiting its potential therapeutic value.11
We substituted the ester moiety with an amide to synthesize carfentanil amide analogues (Figure 1, R = alkyl, carbocycle) and determined if they were analgesics with improved side effect profiles compared to morphine. The only compound that has been reported in this series is a primary amide (R = H).12 To the best of our knowledge, there is no precedence in the literature for the synthesis and systematic pharmacological characterization of further carfentanil amides. Our inspiration to use carfentanil as a template to synthesize mixed MOR/DOR ligands came from a paper from the Portoghese group on fentanyl (structurally closely related to carfentanil). In rhesus monkeys, fentanyl analgesia was attenuated by the selective DOR antagonist naltrindole, implicating a role for DOR in mediating analgesia of this essentially MOR-selective compound.13 To assemble the carfentanil scaffold in one step, we turned our attention to multicomponent reactions. Multi-component reactions (MCRs) allow for rapid synthesis of drug-like compound libraries by combining three or more reagents into a single product in one step.14 Recently, we have reported a novel MCR between 2-aminophenol, ketones and isocyanides to generate a diverse library of heterocyclic drug-like scaffolds.15 In the present work, four-component Ugi reactions were carried out between N-alkylpiperidones, aniline, propionic acid and an array of aliphatic isocyanides. The reaction has previously been used to synthesize a carfentanil precursor16 and bivalent ligands.17 We herein report the synthesis and pharmacological characterization of novel carfentanil amide analogues. The synthesized derivatives were characterized using receptor binding and analgesia assays. The lead compound N-cycloheptyl-1-phenylethyl-4-(N-phenylpropionamido)-piperidine-4-carboxamide (7) was subjected to detailed pharmacological studies. This analgesic is a mixed MOR agonist/DOR partial agonist that does not produce physical dependence or constipation in mice.
RESULTS AND DISCUSSION
Chemistry
A series of carfentanil amides (4–13) were synthesized using the well-known Ugi reaction from commercially available starting materials (Scheme 1). In this particular work, we varied the amide substituent of carfentanil amides using various commercially available linear, branched and cyclic isocyanides. A ketone with N-cyclopropylmethyl substituent was also employed in the same manner. The N-cyclopropylmethyl group is primarily responsible for the MOR-antagonistic nature of the clinically used epoxymorphinan antagonist naltrexone. The desired carfentanil amides were isolated in moderate to good yields. The use of Ugi reactions to access carfentanil-based scaffolds makes diversification library-friendly because of the commercial availability of numerous carboxylic acids, amines, and isocyanides.
Scheme 1.

Synthesis of Carfentanil Amides 4–13
Pharmacology
All synthesized compounds were characterized in in vitro radioligand binding assays in cell lines stably transfected with murine MOR, DOR, and KOR. Analogs with N-cyclopropylmethyl (12, 13) displayed low affinity across all opioid receptors (Ki > 100 nM), whereas analogues with N-phenylethyl substituents (4–11) showed moderate to high affinity. As expected, all carfentanil amides competed MOR with high affinity (Ki < 10 nM). Most analogues had low affinity (Ki > 100 nM) for KOR except 5 and 6 with cyclopropyl and cyclohexyl groups at the isocyanide-derived amide moiety, respectively. Three compounds, 6 (R2 = cyclohexyl), 7 (R2 = cycloheptyl), and 11 (R2 = adamantyl) had DOR affinity of less than 10 nM. We were interested in studying the pharmacology of compounds with affinity at MOR and DOR. Therefore, 7 was selected for further pharmacological evaluation because its affinity for DOR was the highest among all synthesized compounds (Table 1).
Table 1.
Summary of in Vitro and in Silico Modeled Receptor Binding and in Vivo Tail-Flick Analgesiaa
|
K
i
|
||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | MOR-CHO (nM) |
MOR in silico (nM) |
DOR-CHO (nM) |
DOR in silico (nM) |
KOR-CHO (nM) |
in vivo ED50 (mg/kg)c |
| 4 | phenylethyl | cyclopropyl | 10.3 ± 5.1 | 13.79 | >100 | 54.73 | 87.6 ± 29 | 0.78 ± 0.26 |
| 5 | phenylethyl | cyclopentyl | 29.4 ± 15 | 4.29 | 90.7 ± 23 | 50.58 | >100 | 9.92 ± 0.08 |
| 6 | phenylethyl | cyclohexyl | 0.84 ± 0.34 | 0.66 | 2.65 ± 0.32 | 44.60 | 0.44 ± 0.05 | 3.10 ± 0.19 |
| 7 | phenylethyl | cycloheptyl | 2.66 ± 1.3 | 3.80 | 8.90 ± 7.7 | 7.90 | >100 | 10.0 ± 0.00 |
| 8 | phenylethyl | butyl | 21.1 ± 11 | 13.38 | 87.9 ± 4.8 | 244.5 | >100 | >10 |
| 9 | phenylethyl | t-butyl | 2.73 ± 2.2 | 3.65 | 71.2 ± 8.7 | 251.0 | >100 | 1.09 ± 0.05 |
| 10 | phenylethyl | isoamyl | 27.0 ± 20 | 24.15 | 27.0 ± 3.6 | 120.4 | >100 | >10 |
| 11 | phenylethyl | adamantyl | 25.0 ± 9.8 | 0.88 | 8.83 ± 0.63 | 7.65 | >100 | >10 |
| 12 | CPMb | cyclohexyl | >100 | >100 | >100 | >10 | ||
| 13 | CPMb | t-butyl | >100 | >100 | >100 | >10 | ||
| DAMGO | 3.34 ± 0.43d | |||||||
| morphine | 4.6 ± 1.81d | 5d | ||||||
| U50,488H | 0.73 ± 0.32d | |||||||
| DPDPE | 1.39 ± 0.67d | |||||||
Competition studies were performed with the indicated compounds against 125I-BNtxA (0.1 nM) in membranes from CHO cells stably expressing the indicated cloned mouse opioid receptors. Ki values were calculated from the IC50 values18 and represent the means ± SEM of at least three independent replications. In silico Inhibitory constants were calculated from the binding free energies obtained from docking (vide infra) according to the following equation: ΔH = RT ln Ki. 125IBNtxA KD values for MOR, KOR, and DOR sites were 0.11, 0.03, and 0.24, respectively.
CPM = cyclopropylmethyl.
Analgesia was determined using the radiant heat tail-flick technique on CD-1 mice as described in the Methods section.
Values from literature ref 25.
In vitro [35S]GTPγS functional assays were carried out on 7, and it was found to be a full agonist at MOR (EC50 = 158.7 ± 33 nM, % stimulation = 90.3 ± 0.72) compared with the prototypic MOR agonist DAMGO at 100 nM, and a partial agonist at DOR (EC50 = 42.8 ± 12 nM, % stimulation = 62.3 ± 9.8) compared with the prototypic DOR agonist DPDPE at 100 nM. 7 is about 15-fold less potent an agonist than DAMGO (EC50 = 10.16 ± 2.5 nM) and 1.75-fold less potent than DPDPE (EC50= 24.61 ± 7.7 nM) (Table 2).
Table 2.
Opioid Receptor Efficacy of Compound 7a
| EC50 (nM) |
Emax (% stimulation) |
|||
|---|---|---|---|---|
| compd | MOR | DOR | MOR | DOR |
| 7 | 158.7 ± 33.85 | 42.8 ± 11.56 | 90.3 ± 0.7 | 62.3 ± 9.82 |
| DPDPE | ndb | 24.61 ± 7.7 | ||
| DAMGO | 10.16 ± 2.5 | ndb | ||
Efficacy data were obtained using agonist induced stimulation of [35S]GTPγS binding assay. Efficacy is represented as EC50 (nM) and percent maximal stimulation relative to standard agonist DAMGO (MOR), DPDPE (DOR), or U50,488H (KOR) at 100 nM. All values are expressed as the mean ± SEM of three separate assays performed in triplicate.
Not determined.
All compounds were also evaluated in tail-flick analgesia assays with the drug given subcutaneously in CD1 mice. Some compounds were inactive, whereas 4–7 and 9 showed analgesia at the highest given dose of 10 mg/kg. The lack of in vivo analgesic response to 12 and 13 was not surprising given that these analogues did not possess any appreciable binding affinity to opioid receptors. Three compounds in the series (4, 6, and 9) were more potent than morphine (ED50 ~ 5 mg/kg, sc).19 The analgesic ED50 values of 5 and our compound of interest 7 (ED50 = 10 mg/kg, sc) was about 2-fold lower than that of morphine (Table 1, Figure 2A). 7 was next characterized in in vivo antagonism assays. The analgesia of 7 was partially blocked by the MOR selective antagonist beta-FNA (40 mg/kg, sc) and DOR selective antagonist naltrindole (NTI, 20 mg/kg, sc), suggesting a role of both MOR and DOR in mediating the analgesia. This is consistent with our in vitro [35S]GTPγS functional assay results (Figure 2B).
Figure 2.

Pharmacology of 7. (A) Analgesia: Cumulative dose–response curves were carried out on groups of mice (n = 10) with 7 at the indicated doses (sc) and analgesia tested 30 min later at peak effect. The ED50 value was 10 ± 0 mg/kg in CD1 mice by using the radiant heat tail-flick assay. (B) Sensitivity of 7 to opioid antagonists: Groups of mice (n = 10) received a fixed dose of 7 (15 mg/kg, sc) alone or with β-FNA (40 mg/kg, sc) or NTI (20 mg/kg, sc). β-FNA was given 24h before 7 whereas NTI was given 15 min before 7. Tail flick analgesia was measured 30 min after dosing with 7. Similar results were observed in two independent replications. 7 analgesia is partially antagonized by both β-FNA and NTI (ANOVA followed by Bonferroni multiple comparison test (p < 0.05). (C) Respiratory rate. Animals were randomly assigned to receive saline (n = 5), 7 (40 mg/kg, n = 5), or morphine (20 mg/kg, n = 5). Each animal’s baseline average breath rate was measured every 5 min for 25 min before drug injection, and breath rates after drug injection are expressed as a percent of baseline. 7 did not depress respiratory rate and was not significantly different from saline at any time point, whereas morphine decreased respiratory depression in comparison with both saline and 7 (p < 0.05) as determined by repeated-measures ANOVA followed by Bonferroni multiple-comparison test. (D) Physical dependence. Groups of mice (n ≥ 10) received either morphine (10 mg/kg sc) or 7 (1 mg/kg sc) until they showed complete tolerance. They were then challenged with naloxone. Naloxone precipitated a profound withdrawal syndrome in the morphine-treated animals, as shown by the number of jumps per 15 min, which was significantly greater than that in the morphine or 7 controls (i.e., given no antagonist) or in 7 mice given naloxone. Mice chronically administered 7 showed no significant difference from controls when challenged by naloxone (1 mg/kg sc). (E) Gastrointestinal (GI) transit. Groups of mice (n = 10) received saline, morphine (5 mg/kg), or 7 (20 and 50 mg/kg) before receiving an oral dose of 0.2 mL of charcoal meal (2.5% gum tragacanth in 10% activated charcoal in water) by gavage. Animals were sacrificed 30 min later, and the distance traveled by charcoal was measured. 7 did not lower GI transit significantly compared with saline (P < 0.05), and the effect was significantly lower than that of morphine (P < 0.05) as determined by ANOVA followed by Tukey’s multiple-comparison test.
We next looked at the side-effect profile of 7 in mouse models of respiratory depression (RD) and physical dependence. At doses 4× ED50 (40 mg/kg, sc) 7 did show some signs of RD, although it was significantly lower than RD caused by the same relative dose of morphine (Figure 2C). Chronic administration of traditional opioids leads to both tolerance and physical dependence. Daily administration of morphine (10 mg/kg sc, 2× ED50) produced a diminishing analgesic response with no analgesia by day 5. These chronically morphine-treated mice were both tolerant and physically dependent. Naloxone precipitated a profound withdrawal syndrome in morphine treated-mice. Chronic dosing of 7 also produced tolerance. However, 7-tolerant mice challenged with naloxone demonstrated fewer withdrawal symptoms. Figure 2D indicates they jumped fewer times than morphine treated mice. In addition, there were no signs of diarrhea in 7-tolerance mice challenged with naloxone. Another serious side-effect associated with clinically used mu analgesics is constipation. At doses 2× ED50 (20 mg/kg) and 5× ED50 (50 mg/kg) 7 showed no signs of constipation, while morphine caused constipation at its ED50 dose (Figure 2E). Thus, according to mouse models of GI transit and physical dependence. the full MOR agonist and partial DOR agonist 7 may be useful in negating multiple major side-effects seen with classical MOR analgesics such as morphine. We hope to optimize the structure of this carfentanil amide scaffold to maintain receptor affinities and MOR agonism, while reducing the DOR efficacy to attain a MOR agonist/DOR antagonist based pharmacophore to further attenuate respiratory depression. The utilization of Ugi chemistry to diversify the amine and carboxylic acid ends with commercially available reagents makes further derivatives readily accessible.
In Silico Receptor Binding
Docking of carfentanil amides 4–11 to the MOR and the in silico predictions of inhibitory constants (Table 1) were most successful when the original crystal structure was used as the target. Dockings to the experimentally derived DOR structure were unsuccessful, as only unrealistic or high binding free energy receptor ligand complexes were obtained. Docking to a molecular dynamics (MD) simulation-derived DOR model however resulted in accurate reproduction of experimental receptor binding data. This suggests that changes in the receptor conformation introduced by crystal packing and antagonist binding could be different for MOR and DOR. Blind docking results showed that compounds 4–11 also bind to the same cavity where alkaloid-type ligands20 as well as peptides21 were found to bind (Figure 3). This contradicts a previous hypothesis which states that chemically different ligand types have separate binding sites. However, binding orientations of each ligand type were found to be different both in the case of MOR and DOR ligands. Because of its exceptional properties demonstrated in the pharmacological assays, binding orientation and interactions of 7 with MOR and DOR were analyzed in more detail. Compound 7 was found to form more contacts with the binding pockets than the alkaloid antagonists showed in the crystal structures. Three of the MOR side-chains which were found to interact with the ligand in the crystal structure do not form contacts with 7 in the docked complexes; however four new contacts are formed with other amino acids (Figure 4A). In the case of 7 binding to the DOR, two native contacts were missing and six new contacts were observed compared to the crystal complex (Figure 4B). Therefore, binding and functional properties of different ligands may not necessarily involve more than one binding site.22 Instead, ligand-specific interactions23 may trigger (or arrest) conformational changes of the receptor upon binding.
Figure 3.
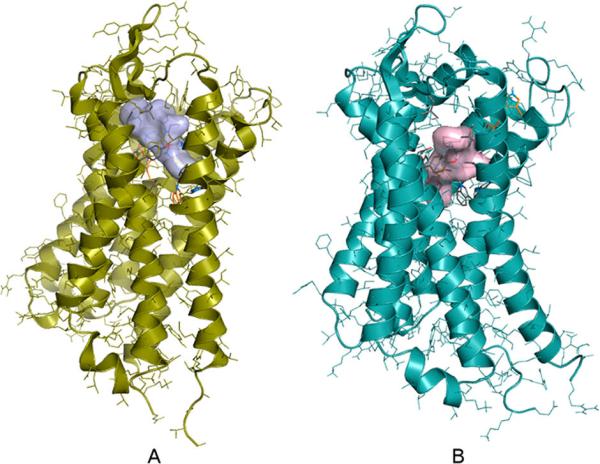
Docked complexes of 7 and MOR (A); 7 and DOR (B). N- and C-terminal tails are omitted for clarity. Compounds were blind-docked to full sequence MOR and DOR receptor models derived from experimental structures (PDB codes: 4DKL and 4EJ4, respectively) and molecular dynamics simulations. Dockings were performed using the Autodock 4.2 software, having the side chains of the binding site residues and all ligand torsions kept flexible. Inhibitory constants were calculated from the binding free energies obtained from docking according to the following equation: ΔH = RT ln Ki.
Figure 4.

Amino acid side chains of MOR (A) and DOR (B) that participate in interactions with 7. Side chains observed in the crystal structures to take part in receptor–ligand interactions but do not form contacts with 7 in the docked complexes are depicted in orange. Nonpolar hydrogens are omitted for clarity.
CONCLUSIONS
Our hypothesis was to synthesize a compound with high affinity for MOR and DOR and low affinity for KOR using a robust, library-friendly method. Ten compounds based on carfentanil using the Ugi multicomponent reaction were synthesized. Our lead was found to be a full agonist at MOR and partial agonist at DOR. It showed moderate analgesic affinity compared with morphine, sc. This compound showed some respiratory depression. However, it produced no physical dependence or inhibition of GI transit in mouse models. To our knowledge, this is the first time an opioid scaffold with mixed MOR/DOR profile has been synthesized using the Ugi MCR. While there are plenty of examples of dual MOR/DOR agonists and MOR agonism-DOR antagonism based ligands in the literature,24 in vivo side-effect profile evaluation of full MOR agonist partial DOR agonist compounds has not been reported previously. It seems likely that mixed MOR/DOR agonists can negate at least two of the important side-effects of morphine, namely physical dependence and constipation. Future diversification of analogues will aim to study the SAR at the amine and carboxylic acid end using the chemistry presented in this manuscript. The utilization of Ugi chemistry to diversify the amine and carboxylic acid residues with commercially available substrates in a library friendly manner makes this approach even more attractive and readily accessible.
METHODS
All chemicals were purchased from Sigma-Aldrich Chemicals and Alfa Aesar, and were used without further purification. Reaction mixtures were purified by Silica Flash chromatography on E. Merck 230–400 mesh silica gel 60 using a Teledyne ISCO CombiFlash Rf instrument with UV detection at 280 and 254 nm. RediSep Rf silica gel normal phase columns were used with a gradient of 0–10% MeOH in DCM. The yields reported are isolated yields. IR spectra were recorded on a Bruker Optics Tensor 27 FTIR spectrometer with peaks reported in cm−1. NMR spectra were recorded on Bruker Avance III 500 and Avance III 600 systems with DCH CryoProbe instruments. NMR spectra were processed with MestReNova software (ver. 6.1.1.). Chemical shifts are reported in parts per million (ppm) relative to residual solvent peaks rounded to the nearest 0.01 for proton and 0.1 for carbon (CDCl3 1H: 7.26, 13C: 77.3; CD3OD 1H: 3.31, 13C: 49.0; DMSO-d6 13C: 39.5). Peak multiplicity is reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants (J) are expressed in Hz. Mass spectra were obtained at the MSKCC Analytical Core Facility on a Waters Acuity SQD LC MS instrument by electrospray (ESI) ionization. High resolution mass spectra were obtained on a Waters Acuity Premiere XE TOF LC-MS instrument by electrospray ionization. Accurate masses are reported for the molecular ion [M + H]+. A reversed-phase HPLC using a PerkinElmer LC pump series 200 and a 785A UV/vis detector (214 nM) was used. A Varian microsorb MV 100-5 reversed-phase column (5 μm × 4.6 mm × 250 mm) with the mobile phases being 0.1% TFA in water and 0.1% TFA in ACN with a gradient elution at a flow rate of 1 mL/min was used.
Chemical Synthesis
General Procedure for the Ugi Multi-component Reaction (Synthesis of 4–13)
To a solution of aniline (40.1 μL, 0.44 mmol) in methanol (2.2 mL) were added isocyanide (0.44 mmol, 1 equiv), substituted 4-piperidone (0.44 mmol, 1 equiv), and propionic acid (32.89 μL, 0.44 mmol, 1 equiv) and stirred at 55 °C for 18 h. Solvent was removed under reduced pressure. The reaction mixture was purified by silica gel flash chromatography (0–15% MeOH in DCM).
Receptor-Binding Assays
Competition-binding assays in CHO cells stably expressing MOR, DOR or KOR were performed at 25 °C in potassium phosphate buffer (50 mM; pH 7.4), with the inclusion of MgSO4 (5 mM) in the MORassays. All competition assays were carried out using 125I-BNtxA as described.25 Specific binding was defined as the difference between total binding and nonspecific binding, determined in the presence of levallorphan (8 μM). Protein concentrations were between 30 and 40 μg/mL, and incubation times were 90 min. Protein concentration was determined as described by Lowry et al.26 using bovine serum albumin as the standard.
Tail Flick Analgesia Assays
Male CD-1 mice (25–35 g; Charles River Breeding Laboratories, Wilmington, MA) were maintained on a 12-h light/dark cycle with Purina rodent chow and water available ad libitum. Mice were housed in groups of five until testing. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center. Analgesia was determined using the radiant heat tail-flick technique27 using a machine from Ugo Basile (model number 37360). The intensity was set to achieve a baseline between 2 and 3 s. The latency to withdraw the tail from a focused light stimulus was measured electronically using a photocell. Baseline latencies (2.0–3.0 s) were determined before experimental treatments for all animals. Post-treatment tail-flick latencies were determined as indicated for each experiment, and a maximal latency of 10 s for tail-flick was used to minimize tissue damage. Analgesia was defined quantally as a doubling, or greater, of the baseline latency. Similar results were obtained analyzing the data in a graded response manner as percent of maximum possible effect [(observed latency – baseline latency)/(maximal latency – baseline latency)]. Analgesic ED50 values and confidence limits were determined using nonlinear regression analysis GraphPad Prism (San Diego, CA). Drugs were given subcutaneously and cumulative dose–response experiments carried out with at least two independent assays with each group (n = 10). The combined results presented as the ED50 with SEM of replicates presented.
[35S]GTPγS-Binding Assay
[35S]GTPγS binding was performed on membranes prepared from transfected cells in the presence and absence of the indicated opioid for 60 min at 30 °C in the assay buffer (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 10 mM NaCl) containing 0.05 nM [35S]GTPγS and 30 uM GDP, as previously reported.26 After the incubation, the reaction was filtered through glass-fiber filters (Whatman Schleicher & Schuell, Keene, NH) and washed three times with 3 mL of ice-cold 50 mM Tris-HCl, pH 7.4, on a semiautomatic cell harvester. Filters were transferred into vials with 3 mL of Liquiscent (National Diagnostics, Atlanta, GA), and the radioactivity in vials was determined by scintillation spectroscopy in a Tri-Carb 2900TR counter (PerkinElmer Life and Analytical Sciences). Basal binding was determined in the presence of GDP and the absence of drug. Maximum stimulation was determine in the presence of 100 nM DAMGO, DPDPE, and U50,488h for MOR, DOR, and KOR, respectively.
Respiratory Depression Assay
Respiratory rate was assessed in awake, freely moving, adult male CD1 mice with the MouseOx pulse oximeter system (Starr Life Sciences), as previously reported.27 Each animal was habituated to the device for 30 min and then tested. A 5 s average breath rate was assessed at 5 min intervals. A baseline for each animal was obtained over a 25 min period before drug injection, and testing began at 15 min postinjection and continued for a period of 35 min. Groups of mice (n = 5) were treated subcutaneously with either saline or morphine (20 mg/kg) or 7 (40 mg/kg). Morphine and 7 were given at doses approximately four times its analgesic ED50. Groups were compared with repeated-measures ANOVA followed by Bonferroni multiple-comparison test.
Gastrointestinal Transit
Groups of mice (n = 10) received saline, morphine (5 mg/kg), or 7 (20 and 50 mg/kg) before receiving an oral dose of 0.2 mL of charcoal meal (2.5% gum tragacanth in 10% activated charcoal in water) by gavage. Animals were sacrificed 30 min later, and the distance traveled by charcoal was measured. 7 did not lower transit significantly compared with saline (P > 0.05) as determined by ANOVA followed by Tukey’s multiple-comparison test.
Receptor Docking
Full sequence target structures of the human MOR and DOR receptors for docking studies were built using the recently deposited crystal strucures of the homologuous murine opioid receptors30 as templates (PDB codes: 4DKL and 4EJ4, respectively). Homology modeling of the transmembrane region and intra- and extracellular loops was performed using the Modeler 9.11 software package. The missing intracellular loop and N- and C-terminal tails were built using the loop module of Modeler. One hundred structures were generated for both receptors and ranked by the modeler energy function. The best ranking models of each receptor were then subjected to 200 ns molecular dynamics simulations to obtain relaxed, equlibrated, ligand-free inactive structures of the receptors, exempt of strains occurrently introduced by crystal lattice forces and/or induced fit binding of antagonists.
Molecular dynamics (MD) simulations (200 ns long) in the NPγT ensemble and explicit, hydrated DOPC membrane bilayer environment31 were performed using the Gromacs 4.5.4 software package and the Amber ff02 and gAFF force fields. The temperature, pressure and surface tension were set to 310 K, 1 and 440 bar nm, respectively. The time step was set to 2 fs and nonbonded interactions were calculated using the PME method with all cutoff values set at 12 Å. The resultant trajectories were analyzed by clustering to identify dominantly occurring spatial arranegements of the amino acids which were shown to interact with ligands in the crystal structures. Clustering was performed using the g_cluster utility and the gromos method32 with 1 Å of RMSD similarity cutoff, fitting all heavy atoms of the binding site residues. The geometry of the transmembrane region of unliganded DOR was found to change more compared to that of the MOR during the course of MD simulations indicating a more intense reverse rearrangement of DOR upon ligand removal. For each receptor, representative structures of the five most populated structural families, as well as the original crystal structure, complemented with the missing loops were used for docking studies.
Dockings were performed with the Autodock 4.2 software, where the side chains of the binding site residues were kept flexible and all ligand torsions were allowed. Compounds 4–11 were docked using the Lamarckian genetic algorithm in a grid volume large enough to cover the whole receptor region accessible from the extracellular side. The grid spacing was set to 0.375 Å and 1000 dockings were done for all receptor models. To check the validity of the applied methods and receptor models, well characterized, selective alkaloid and peptide agonists of both receptors were docked for comparison. Inhibitory constants were calculated from the binding free energies obtained from docking according to the following equation: ΔH = RT ln Ki.
Supplementary Material
Acknowledgments
Funding
This work was supported by research grants from the National Institute on Drug Abuse (DA034106) to S.M. and (DA06241) to G.W.P., National Science Foundation Graduate Research Fellowship (DGE-1257284) to G.F.M. Research of A.B. was supported by the European Union and the State of Hungary, cofinanced by the European Social Fund in the framework of TÁMOP 4.2.4. A/2-11-1-2012-0001 “National Excellence Program”.
Notes
The authors declare no competing financial interest.
Chemical analysis of compounds and NMR spectra. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.5b00137.
REFERENCES
- (1).Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Med. Res. Rev. 2004;24(2):182–212. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- (2).Trescot AM, Datta S, Lee M, Hans H. Opioid pharmacology. Pain Phys. 2008;11:S133–S153. [PubMed] [Google Scholar]
- (3).Benyamin R, Trescot AM, Datta S, Buenaventura R, Adlaka R, Sehgal N, Glaser SE, Vallejo R. Opioid complications and side effects. Pain Phys. 2008;11:S105–S120. [PubMed] [Google Scholar]
- (4).Compton WM, Volkow ND. Major increases in opioid analgesic abuse in the United States: Concerns and strategies. Drug Alcohol Depend. 2006;81(2):103–107. doi: 10.1016/j.drugalcdep.2005.05.009. [DOI] [PubMed] [Google Scholar]
- (5) (a).Corbett AD, Henderson G, McKnight AT, Paterson SJ. 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br. J. Pharmacol. 2006;147(Suppl 1):S153–S162. doi: 10.1038/sj.bjp.0706435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vardanyan RS, Hruby VJ. Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Med. Chem. 2014;6(4):385–412. doi: 10.4155/fmc.13.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Mosberg HI, Yeomans L, Anand JP, Porter V, Sobczyk-Kojiro K, Traynor JR, Jutkiewicz EM. Development of a bioavailable mu opioid receptor (MOPr) agonist, delta opioid receptor (DOPr) antagonist peptide that evokes antinociception without development of acute tolerance. J. Med. Chem. 2014;57(7):3148–53. doi: 10.1021/jm5002088. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ananthan S. Opioid ligands with mixed mu/delta opioid receptor interactions: an emerging approach to novel analgesics. AAPS J. 2006;8(1):E118–25. doi: 10.1208/aapsj080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Ananthan S, Kezar HS, Carter RL, Saini SK, Rice KC, Wells JL, Davis P, Xu H, Dersch CM, Bilsky EJ, Porreca F, Rothman RB. Synthesis, Opioid Receptor Binding, and Biological Activities of Naltrexone-Derived Pyrido- and Pyrimidomorphinans. J. Med. Chem. 1999;42(18):3527–3538. doi: 10.1021/jm990039i. [DOI] [PubMed] [Google Scholar]; (b) Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TMD, Lemieux C, Chung NN, Coderre TJ. The Opioid μ Agonist/δ Antagonist DIPP-NH2[Ψ] Produces a Potent Analgesic Effect, No Physical Dependence, and Less Tolerance than Morphine in Rats. J. Med. Chem. 1999;42(18):3520–3526. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- (8) (a).Porreca F, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI. Modulation of mu-mediated antinociception in the mouse involves opioid delta-2 receptors. J. Pharmacol. Exp. Ther. 1992;263(1):147–152. [PubMed] [Google Scholar]; (b) He L, Lee NM. Delta opioid receptor enhancement of mu opioid receptor-induced antinociception in spinal cord. J. Pharmacol. Exp. Ther. 1998;285(3):1181–1186. [PubMed] [Google Scholar]; (c) Horan P, Tallarida RJ, Haaseth RC, Matsunaga TO, Hruby VJ, Porreca F. Antinociceptive interactions of opioid delta receptor agonists with morphine in mice: supra- and sub-additivity. Life Sci. 1992;50(20):1535–41. doi: 10.1016/0024-3205(92)90144-e. [DOI] [PubMed] [Google Scholar]; (d) Vaught JL, Takemori AE. Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J. Pharmacol. Exp. Ther. 1979;208(1):86–90. [PubMed] [Google Scholar]
- (9) (a).Purington LC, Pogozheva ID, Traynor JR, Mosberg HI. Pentapeptides displaying mu opioid receptor agonist and delta opioid receptor partial agonist/antagonist properties. J. Med. Chem. 2009;52(23):7724–31. doi: 10.1021/jm9007483. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lowery JJ, Raymond TJ, Giuvelis D, Bidlack JM, Polt R, Bilsky EJ. In vivo characterization of MMP-2200, a mixed delta/mu opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011;336(3):767–78. doi: 10.1124/jpet.110.172866. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Do Carmo GP, Polt R, Bilsky EJ, Rice KC, Negus SS. Behavioral Pharmacology of the μ/δ Opioid Glycopeptide MMP2200 in Rhesus Monkeys. J. Pharmacol. Exp. Ther. 2008;326(3):939–948. doi: 10.1124/jpet.108.138180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10) (a).Balboni G, Salvadori S, Trapella C, Knapp BI, Bidlack JM, Lazarus LH, Peng X, Neumeyer JL. Evolution of the Bifunctional Lead mu Agonist/delta Antagonist Containing the Dmt-Tic Opioid Pharmacophore. ACS Chem. Neurosci. 2010;1(2):155–164. doi: 10.1021/cn900025j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Balboni G, Guerrini R, Salvadori S, Bianchi C, Rizzi D, Bryant SD, Lazarus LH. Evaluation of the Dmt-Tic pharmacophore: conversion of a potent delta-opioid receptor antagonist into a potent delta agonist and ligands with mixed properties. J. Med. Chem. 2002;45(3):713–20. doi: 10.1021/jm010449i. [DOI] [PubMed] [Google Scholar]; (c) Bishop MJ, Garrido DM, Boswell GE, Collins MA, Harris PA, McNutt RW, O'Neill SJ, Wei K, Chang KJ. 3-(alphaR)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-hydroxybenzyl)-N- alkyl-N-arylbenzamides: potent, non-peptidic agonists of both the micro and delta opioid receptors. J. Med. Chem. 2003;46(4):623–633. doi: 10.1021/jm020395s. [DOI] [PubMed] [Google Scholar]; (d) Coats SJ, Schulz MJ, Carson JR, Codd EE, Hlasta DJ, Pitis PM, Stone DJ, Jr, Zhang S-P, Colburn RW, Dax SL. Parallel methods for the preparation and SAR exploration of N-ethyl-4-[(8-alkyl-8-azabicyclo[3.2.1]oct-3-ylidene)-aryl-methyl]-benzamides, powerful mu and delta opioid agonists. Bioorg. Med. Chem. Lett. 2004;14(22):5493–5498. doi: 10.1016/j.bmcl.2004.09.004. [DOI] [PubMed] [Google Scholar]; (e) Mosberg HI, Yeomans L, Harland AA, Bender AM, Sobczyk-Kojiro K, Anand JP, Clark MJ, Jutkiewicz EM, Traynor JR. Opioid peptidomimetics: leads for the design of bioavailable mixed efficacy mu opioid receptor (MOR) agonist/delta opioid receptor (DOR) antagonist ligands. J. Med. Chem. 2013;56(5):2139–49. doi: 10.1021/jm400050y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wells JL, Bartlett JL, Ananthan S, Bilsky EJ. In vivo pharmacological characterization of SoRI 9409, a nonpeptidic opioid mu-agonist/delta-antagonist that produces limited antinociceptive tolerance and attenuates morphine physical dependence. J. Pharmacol. Exp. Ther. 2001;297(2):597–605. [PubMed] [Google Scholar]
- (12) (a).Srulevitch DB, Lien EJ. Design, synthesis and SAR of analgesics. Prog. Clin. Biol. Res. 1989;291:377–81. [PubMed] [Google Scholar]; (b) Srulevitch DB, Lien EJ. 4-Phenylamidopiperidines: synthesis, pharmacological testing and SAR analysis. Acta Pharmaceut. Jugosl. 1991;41:89–106. [Google Scholar]
- (13).Yekkirala AS, Banks ML, Lunzer MM, Negus SS, Rice KC, Portoghese PS. Clinically Employed Opioid Analgesics Produce Antinociception via μ-δ Opioid Receptor Heteromers in Rhesus Monkeys. ACS Chem. Neurosci. 2012;3(9):720–727. doi: 10.1021/cn300049m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hulme C, Gore V. “Multi-component reactions: emerging chemistry in drug discovery” ‘from xylocain to crixivan’. Curr. Med. Chem. 2003;10(1):51–80. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]
- (15).Varadi A, Palmer TC, Notis PR, Redel-Traub GN, Afonin D, Subrath JJ, Pasternak GW, Hu C, Sharma I, Majumdar S. Three-component coupling approach for the synthesis of diverse heterocycles utilizing reactive nitrilium trapping. Org. Lett. 2014;16(6):1668–71. doi: 10.1021/ol500328t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Malaquin S, Jida M, Gesquiere J-C, Deprez-Poulain R, Deprez B, Laconde G. Ugi reaction for the synthesis of 4-aminopiperidine-4-carboxylic acid derivatives. Application to the synthesis of carfentanil and remifentanil. Tetrahedron Lett. 2010;51(22):2983–2985. [Google Scholar]
- (17) (a).Pentel PR, Portoghese PS, Pravetoni M, Naour MCPL. Compositions and methods of treating opioid addiction. US20140093525A1 U.S. Patent. 2014 Apr 3;; (b) Portoghese P, Eyup A. Analgesic conjugates. WO2014124317A1 Patent. 2014 Aug 14;
- (18).Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- (19).Kolesnikov YA, Pick CG, Ciszewska G, Pasternak GW. Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 1993;90(11):5162–5166. doi: 10.1073/pnas.90.11.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20) (a).Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the δ-opioid receptor bound to naltrindole. Nature. 2012;485(7398):400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the [micro]-opioid receptor bound to a morphinan antagonist. Nature. 2012;485(7398):321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shim J, Coop A, MacKerell AD. Molecular Details of the Activation of the μ Opioid Receptor. J. Phys. Chem. B. 2013;117(26):7907–7917. doi: 10.1021/jp404238n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang W, Shahrestanifar M, Jin J, Howells R. Studies on m and d opioid receptor selectivity utilizing chimeric and site-mutagenized receptors. Proc. Natl. Acad. Sci. U. S. A. 1995;92:12436–12440. doi: 10.1073/pnas.92.26.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bot G, Blake AD, Li S, Reisine T. Mutagenesis of a single amino acid in the rat m-opioid receptor discriminates ligand binding. J. Neurochem. 1998;70:358–365. doi: 10.1046/j.1471-4159.1998.70010358.x. [DOI] [PubMed] [Google Scholar]
- (24).Váradi A, Hosztafi S, Le Rouzic V, Tóth G, Urai Á, Noszál B, Pasternak GW, Grinnell SG, Majumdar S. Novel 6β-acylaminomorphinans with analgesic activity. Eur. J. Med. Chem. 2013;69:786–789. doi: 10.1016/j.ejmech.2013.09.031. (0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Majumdar S, Burgman M, Haselton N, Grinnell S, Ocampo J, Pasternak AR, Pasternak GW. Generation of novel radiolabeled opiates through site-selective iodination. Bioorg. Med. Chem. Lett. 2011;21(13):4001–4004. doi: 10.1016/j.bmcl.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- (27).D’Amour FE, Smith DL. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941;72(1):74–79. [Google Scholar]
- (28).Bolan EA, Pan YX, Pasternak GW. Functional analysis of MOR-1 splice variants of the mouse mu opioid receptor gene Oprm. Synapse (Hoboken, NJ, U. S.) 2004;51(1):11–8. doi: 10.1002/syn.10277. [DOI] [PubMed] [Google Scholar]
- (29).Majumdar S, Grinnell S, Le Rouzic V, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan YX, Pasternak GW. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc. Natl. Acad. Sci. U. S. A. 2011;108(49):19776–19783. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30) (a).Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485(7398):400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Schäfer B, Orbán E, Borics A, Huszár K, Nyeste A, Welker E, Tömböly C. Preparation of Semisynthetic Lipoproteins with Fluorescent Cholesterol Anchor and Their Introduction to the Cell Membrane with Minimal Disruption of the Membrane. Bioconjugate Chem. 2013;24(10):1684–1697. doi: 10.1021/bc4002135. [DOI] [PubMed] [Google Scholar]
- (32).Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, Mark AE. Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed. 1999;38(1–2):236–240. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.