

Abstract
A high rate of relapse is a defining characteristic of substance use disorder for which few treatments are available. Exposure to environmental cues associated with previous drug use can elicit relapse by causing the involuntary retrieval of deeply engrained associative memories that trigger a strong motivation to seek out drugs. Our lab is focused on identifying and disrupting mechanisms that support these powerful consolidated memories, with the goal of developing therapeutics. A particularly promising mechanism is regulation of synaptic dynamics by actin polymerization within dendritic spines. Emerging evidence indicates that memory is supported by structural and functional plasticity dendritic spines, for which actin polymerization is critical, and that prior drug use increases both spine and actin dynamics. Indeed we have found that inhibiting amygdala (AMY) actin polymerization immediately or twenty-four hours prior to testing disrupted methamphetamine (METH)-associated memories, but not food reward or fear memories. Furthermore, METH training increased AMY spine density which was reversed by actin depolymerization treatment. Actin dynamics were also shifted to a more dynamic state by METH training. While promising, actin polymerization inhibitors are not a viable therapeutic, as a multitude of peripheral process (e.g. cardiac function) rely on dynamic actin. For this reason, we have shifted our focus upstream of actin polymerization to nonmuscle myosin II. We and others have demonstrated that myosin IIb imparts a mechanical force that triggers spine actin polymerization in response to synaptic stimulation. Similar to an actin depolymerizing compound, pre-test inhibition of myosin II ATPase activity in the AMY produced a rapid and lasting disruption of drug-seeking behavior. While many questions remain, these findings indicate that myosin II represents a potential therapeutic avenue to target the actin cytoskeleton and disrupt the powerful, extinction-resistant memories capable of triggering relapse.
Keywords: addiction, substance use disorder, memory, treatment
Memory in SUD Relapse
Substance use disorder (SUD) is a chronic, relapsing disorder with limited treatment options. Although somewhat effective replacement therapies exist for opiate and nicotine dependence, there are no FDA-approved pharmacotherapies for stimulant abuse or relapse. Many of the challenges facing the development of effective SUD pharmacotherapies stem from the complexity of the disorder, which is characterized by diverse, interrelated emotional and physical aspects. One particularly recalcitrant issue associated with SUD is the deeply engrained and highly motivating associations for drug-associated stimuli that develop during drug use. Reminders of these associations can trigger immediate and overwhelming motivation, both conscious and unconscious, to seek the drug. Perhaps most troubling, former substance users are susceptible to these relapse triggers even after seemingly successful rehabilitation and prolonged drug-free periods. Triggers of these memories are numerous and often unconscious or abstract (e.g. craving for cocaine at the sight of a snow-capped mountain). This makes them difficult to predict and treat with current behavioral modification strategies, such as exposure therapy, that rely on active retrieval of each trigger, followed by manipulation of the memory. Retrieval-dependent treatment strategies include attempts to target extinction of the drug-associated memory (i.e. exposure therapy), by accelerating learning of a new, non-reward contingency for the associated stimuli, or reconsolidation, by preventing the return of the associative memory to storage after retrieval [1–5].
Taken together, this suggests that it might be more efficacious to devise a retrieval-independent approach. However, this is not without its own challenges. Retrieval-dependent strategies exist because the process of active recall allows for the selective targeting of the memory of interest, rather than a broad induction of amnesia. In order to overcome this barrier, a mechanism unique to the storage of a drug-associated memory would need to be identified. By turning to the physical site of memory storage in the brain, dendritic spines, we recently stumbled upon such a mechanism [6].
Why Actin: Memory
Dendritic spines are small, but highly dynamic post-synaptic structures on dendrites that receive the majority of the brain’s excitatory input (Fig. 1). There is a growing consensus that dendritic spines serve as a structural storage site for memories. They enable input-specific biochemical and electrical isolation of synapses, which facilitates signal integration and information storage at the time of memory formation. Furthermore, the successful formation of a long-term memory is dependent on the ability of spines to undergo volumetric and functional changes [7, 8]. Calcium influx into a spine, following either long-term potentiation (LTP) stimulation or glutamate receptor activation, leads to spine enlargement and synapse stabilization (Fig. 2). This structural and functional plasticity is regulated by polymerization of actin, a cytoskeletal protein found in abundance within spines [9] [10–17]. Actin polymerization is the highly dynamic process by which actin monomers (G-actin) become linked into complex, branched filaments (F-actin) of the cytoskeleton, allowing dendritic spines to change size and shape on the order of seconds following stimulation [18]. Importantly, disruption of F-actin dynamics at the time of learning prevents the formation of long-term memory. Current studies report that these learning-associated F-actin dynamics are under tight temporal control, such that actin stabilizes just as rapidly as the polymerization was triggered [16, 19–22]. As a result, inhibiting actin polymerization just minutes after learning no longer interferes with long-term memory [19].
Figure 1.
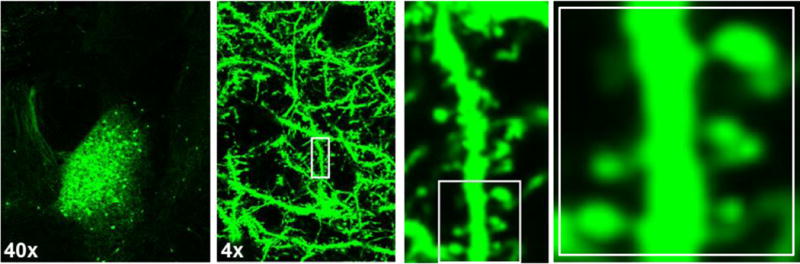
Thy1-GFP(m) expression in dendrites and spine processes of the basolateral amygdala complex imaged at 4× and 40×.
Figure 2.
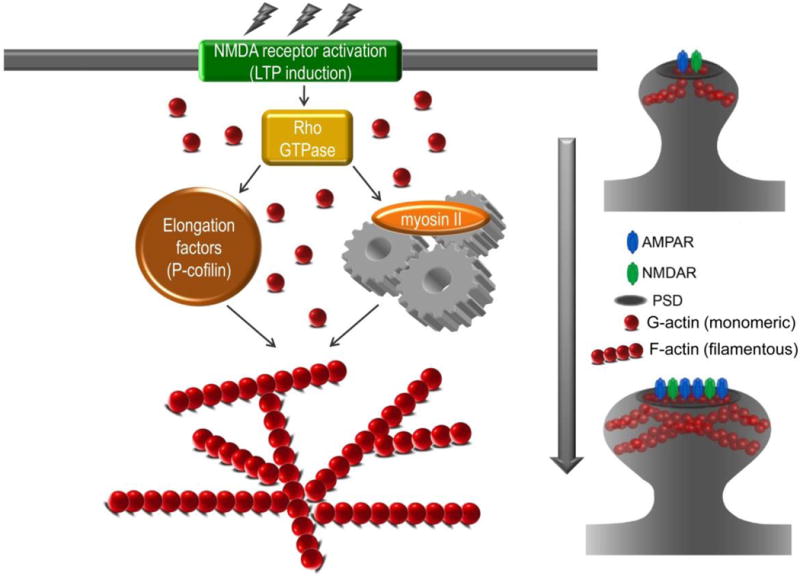
Memory formation is dependent on structural and functional synaptic plasticty. Following learning or LTP induction, NMDA receptor activation initiates actin polymerization, or enlongation, which drives dendritic spine enlargement and stablization.
Why Actin: Drugs of Abuse
SUD is considered by many to be a memory disorder [23, 24]. As discussed above, drug users form long-lasting and highly motivating associative memories for environmental stimuli present at the time of drug use. In humans, non-human primates and rodents, encountering drug-associated stimuli following withdrawal can elicit feelings of intense craving and/or drug seeking, with concomitant activation of a number of brain areas, including the prefrontal cortex (PFC), nucleus accumbens (NAc) and amygdala (AMY) [25–32]. Drug users frequently report that these feelings of craving or motivation for the drug intensify as abstinence periods progress, usually peaking after a month of abstinence [33]. Interestingly, the first 30 days of withdrawal in animal models is marked by dynamic changes in NAc L-DOPA (L-3,4-dihydroxyphenylalanine) and dopamine levels, resulting in a more sensitized state following a month of withdrawal, as compared to one week. Such temporal dopamine effects may support enhanced craving and/or drug seeking with time [34–36]. Furthermore, structural plasticity accompanies long withdrawal periods from cocaine or amphetamine, as evidenced by spine density increases in the PFC and NAc [37]. Morphine exposure, on the other hand, decreases NAc and cortical spine density [38]. Recent evidence indicates that these drug-associated changes in spine density correspond to associative drug conditioning rather than non-associative drug sensitization [39]. Indeed, prior cocaine self-administration training increased spine density and F-actin dynamics in the NAc and spine density in the neocortex [40–44]. While these studies investigated drug-associated synaptic and actin dynamics following a long period of withdrawal, there is evidence that some of these changes also take place after shorter withdrawal periods [6, 45]. Indeed, withdrawal is slowly emerging as a highly dynamic period that promotes drug seeking by intensifying drug-environment associative memories, in part, through actin-mediated structural plasticity.
Why Actin: Bridging the Gap between Memory and SUD
Given the critical role of the NAc and ventral tegmental area (VTA) in reward seeking and dopamine release, it is, perhaps, not surprising that stimulants trigger synaptic structural plasticity in these regions. However, the amygdala (AMY), a structure often associated with anxiety and both positive and negative emotionally arousing memories, also undergoes stimulant-induced structural plasticity [6, 46–49]. Interestingly, these AMY changes appear to rely upon the stimulant being delivered in a novel environment, such that an association forms. AMY-dependent associative memories are major motivators of drug seeking, driving both reward seeking and avoidance of withdrawal symptoms [50–53]. Moreover, expression of aversive withdrawal memories is supported by AMY actin polymerization [54]. Taken together, these findings suggest that drugs of abuse are capable of altering F-actin dynamics throughout the brain.
We recently reported a surprising role for F-actin dynamics in a subregion of the AMY, the basolateral amydala complex (BLC), in supporting memories associated with the highly addictive stimulant, methamphetamine (METH) [6]. We found that directly depolymerizing BLC actin with a naturally occurring toxin, Latrunculin A (LatA), prior to retrieval produced a long-lasting disruption of METH-associated conditioned place preference (CPP) memories, but not memories for food reward. Unexpectedly, this manipulation was equally effective whether administered minutes before testing, or in the home cage an entire day before testing. This indicates that LatA’s effect on the drug-associated memory does not require a retrieval session. Rather, the actin depolymerizer targets the memory in storage. Furthermore, depolymerized actin also blocked reinstatement of context-induced METH self-administration, an animal model of drug seeking with strong face validity. Analysis of spine density revealed an increase in BLC spine density with METH training that was reversed by LatA. LatA’s mechanism of action is to sequester actin monomers, preventing their incorporation into F-actin filaments. Thus, the drug is ineffective when actin is stable, as occurs shortly after a typical learning event (e.g. fear and food reward memories). Given the unique ability of LatA to disrupt drug-associated memories days after learning in the absence of retrieval, we hypothesized that the actin cytoskeleton fails to stabilize, remaining in an active, cycling state that is susceptible to depolymerization.
To begin to address this, we recently analyzed the ratio of actin monomers (G-actin) to filaments (F-actin) two days after METH CPP training. In other cell types, higher rates of actin cycling are associated with the presence of more actin monomers (44). Consistent with this, we observed a shift towards monomeric (G-) actin in the BLC of METH, but not saline, CPP-trained animals (Fig. 3; METH: F(1,7) = 11.29; P < 0.05; Saline: F(1,7) = 1.19; P > 0.05), with no difference in total actin (Saline: 31879.8 + 2772.3, METH: 31409.8 + 2403.8; F(1,7) = 0.02; P > 0.05). This evidence, in conjunction with the ability of actin depolymerization to disrupt drug seeking further suggests that the actin cytoskeleton supporting a METH-associated memory may be in a constitutively active state of cycling, rather than being triggered by the act of memory retrieval.
Figure 3.

METH-associated memories are associated with a dynamic actin cytoskeleton during the maintenance phase. Animals underwent Saline or METH CPP training then the ratio of monomeric (G-) and filamentous (F-) actin the BLC was assessed. METH CPP potentiated BLC G-actin compared to F-actin.
Is Actin Druggable?
The ability of a single actin depolymerization treatment to produce a long-lasting disruption of METH-associated memories independent of retrieval initially seems like a promising therapeutic target for the selective disruption of relapse-inducing triggers. Indeed, actin is also a seemingly promising target for other indications, such as cancer. However, the actin isoform implicated in adult neuronal structural plasticity, β-actin, is ubiquitously expressed throughout the body and critical to a multitude of processes in the periphery (e.g. cell division, cardiac function) [13, 55–58]. Indeed, the body’s fundamental reliance on actin polymerization is why pharmaceutical companies have limited their pursuit of actin-targeting drugs to indications calling for topical treatment, such as glaucoma. Because an actin depolymerizing agent is not a viable therapeutic option, we are actively investigating upstream regulators of the synaptic actin cytoskeleton. Nonmuscle myosin II (NMII), which is highly expressed in neuronal tissue, is one such potential target.
Why Myosin
The 143 member myosin family is made up of seventeen different classes of molecular motors with tissue-specific properties. NMII is the most common class of myosin found in neuronal tissue, with three distinct isoforms of the heavy chain, which bears the actin-binding site and ATPase activity (NMIIA (Myh9), NMIIB (Myh10) and NMIIC (Myh14)) [59, 60]. It is well established that NMII regulates F-actin dynamics in growth cones of developing neurons by driving retrograde actin flow [16, 61]. To do this, NMII’s ATPase motor protein binds to actin filaments, causing them to contract and sever. Subsequently, this NMII-driven action replenishes the pool of G-actin monomers, thereby facilitating actin polymerization in growth cones [61–63]. Recently, Rex et al. (2010) reported that the NMIIB ATPase motor is also a critical regulator of structural plasticity following LTP induction and NMDA receptor stimulation, through direct action on dendritic spine actin [16]. Similar to the results of these NMIIB genetic manipulations, pharmacologic inhibition via Blebbistatin (Blebb), disrupts LTP stability [16, 64]. Rex et al. (2010) went on to show that myosin II activity and NMIIB, in particular, is required for hippocampus-dependent memory formation [16]. In a follow up study, Gavin et al. (2011) established that NMIIB is also required for amygdala-dependent memory formation [20].
Taken together, these studies establish that myosin II activity reorganizes the actin cytoskeleton to support the stable expression of LTP, as well as the formation of memories reliant upon the hippocampus and amygdala. In light of this, we have begun to test the hypothesis that NMIIB is uniquely required for the stabilization of drug-associated memories, such that it can be targeted in lieu of direct depolymerization of actin. Indeed, we recently reported that administration of Blebb directly into the BLC prior to testing disrupted METH CPP [6].
Unfortunately, the only available NMII inhibitor is Blebb and it has a number of properties in need of improvement [65]. For instance, not only does Blebb lack NMII isoform selectivity, but it also hits skeletal, smooth and, perhaps, cardiac muscle myosin IIs. However, in silico assessments indicate that Blebb should have high brain penetrance and it has been shown to be tolerated by rodents when delivered intravenously [66]. Given that Blebb was found through a relatively small-scale screen, the potential for improving Blebb through derivitization and/or identifying novel NMII inhibitors remains high [65].
Future Questions
While NMII represents a potential target for safely targeting central nervous system (CNS) actin polymerization, a multitude of questions remain. At the forefront are questions related to what regulatory mechanisms could be responsible for the constitutively cycling actin that is unique to the storage of METH-associated memories. While memories of fear, food reward and METH are all known to be long-lasting, evidence suggests that they are supported by different states of F-actin, cycling versus stable. We and others have previously demonstrated that fear learning triggers a brief window of F-actin dynamics [16, 20]. This limits the efӿcacy of actin depolymerizing agents to a brief window surrounding learning. One possible contributor to this unique feature of cycling F-actin associated with METH memories is withdrawal-induced release of neuromodulators, such as dopamine or brain-derived neurotrophic factor (BDNF), into the AMY. Both are capable of regulating functional and structural plasticity [67–69] and importantly, they are both released into the BLC during periods of cocaine withdrawal [70, 71]. Therefore, it is possible that the actin cytoskeleton supporting METH-associated memory initially stabilizes, but is later reactivated upon forced abstinence from METH at the completion of training.
As stated previously, drugs of abuse activate brain regions involved in memory processes, including the AMY, hippocampus, NAc, PFC and the dorsal striatum. This naturally leads to the question of actin dynamics in other regions of the brain. Is actively cycling actin unique to the AMY, or is it occurring in other memory centers of the brain? Further, are memories associated with other drug classes similarly susceptible to actin depolymerization? It is known that withdrawal from cocaine and development of aversive associations with morphine withdrawal both involve actin polymerization [44, 54, 72]. However, very little is known about F-actin dynamics associated with drug-related memories in these regions. There is evidence, as detailed above, that many drugs of abuse are capable of altering spine density. For instance, studies performed by Robinson and Kolb show increases in spine density in the NAc and PFC following cocaine and amphetamine injections, while spine density decreases after morphine treatment [37, 38]. Indeed, the persistent, neurobehavioral effects of repeated exposure to these drugs may stem from their ability to reorganize the actin cytoskeleton of dendritic spines. Although it is not yet known if actin has a prolonged period of cycling in these regions after exposure to drugs of abuse, as in the AMY, one can speculate that memories associated with any drugs capable of altering actin dynamics may be susceptible to selective, retrieval-independent disruption.
In seeking a potential SUD therapeutic, one must consider that users seeking treatment will have different drug consumption histories. This raises the important question of how drug history, such as amount of drug use, period of use and polydrug use, might affect the efficacy of targeting the actin cytoskeleton. One way to address this is through what is known as extended access self-administration, in which animals are given six or more hours of daily access to self-administer the drug of choice, rather than the standard two hours [73, 74]. Ferrario et al (2005) demonstrated that following both limited and extended access to cocaine, there was an increase in dendritic spine density in NAc and the medial PFC, with a further enhancement in spine density in the NAc with extended access [75]. This data suggests that an increase in drug intake, as seen with the extended access model, may have a similar, but perhaps even more substantial, impact on synaptic reorganization as the limited access protocol. We have previously shown a correlation between spine density and the strength of a METH-associated memory, but that memory strength does not affect the efficiency with which actin depolymerization disrupts METH-associated memories [6]. Thus, we have reason to believe that memories formed through extended access to METH will be similarly susceptible to actin depolymerization. Clearly, more research in this area is still needed.
In designing any therapeutic, safety is a primary concern. In the case of targeting drug-associated memories to mitigate the risk of SUD relapse, we have directed our search for a pharmacotherapy upstream of actin. Our initial results indicate that NMII inhibition in the BLC produces similar results to that of direct actin depolymerization with LatA. Indeed, myosin II represents an attractive therapeutic target because its distribution is more tissue-specific, with the NMII isoforms being abundantly expressed within the CNS, including the AMY. Further, systemic inhibition with Blebb is tolerated by rodents [66]. An additional benefit of our discovery, from a safety perspective, is that it appears a single manipulation of the actin cytoskeleton is efficacious. This precludes the need for chronic treatment, mitigating the associated safety and off-target concerns.
EXPERIMENTAL PROCEDURES
Animals
Adult male Sprague-Dawley rats (300–350g) were obtained from Charles River Laboratories and adult male C57BL/6. All animals were housed under a 12:12 light/dark cycle, with food and water ad libitum. All procedures were performed in accordance with the Scripps Research Institute Animal Care and Use Committee and national regulations and policies. All animals were handled for 3–5 days prior to the start of behavioral conditioning.
Behavioral Procedures
Conditioned Place Preference
Animals were trained using an unbiased conditioned place preference (CPP) procedure. The box consisted of three chambers (Med Associates) – two of equal size and one smaller, center chamber. The chambers differed in terms of their visual, tactile and olfactory cues.
The CPP protocol consisted of three phases: Pretesting, training and testing. During the pretest, rats were given free access to the apparatus for 15 min and the time spent in each chamber was recorded. During training, Methamphetamine hydrochloride (Sigma-Aldrich) was delivered IP (1mg/kg at 1ml/kg) to rats prior to placement in the CS+ chamber for 30 min (counterbalanced between chambers within groups). On alternating sessions, rats were injected with an equivalent volume of 0.9% saline and confined to the opposite chamber for 30 min. Animals received five pairings with METH and saline during twice daily training sessions (am and pm), such that rats received METH (1mg/kg) and saline each day. Time of day for METH treatment was counterbalanced within groups. Tissue was collected 48 hrs after the last day of training.
G/F Actin Assay
G- and F-actin were measured using an in vivo assay kit for actin polymerization (Cytoskeleton). Briefly, fresh BLC tissue punches were rapidly obtained and immediately placed in warmed (37°C) LAS2 buffer for homogenization. The homogenized tissue lysate was kept at 37°C during the entire fractionation process to preserve the state of F-actin. Following a 10 min incubation for cell lysis, the lysates were centrifuged at 2,000 rpm for 5 min. The supernatant was then transferred into centrifuge tubes and spun in a Beckman Coulter Ultracentrifuge with a SW-55 rotor at 55,000 rpm for 1hr. Immediately following fractionation, the tubes were placed on ice. The pellets (F-actin containing fraction) were resuspended to the same volume as the supernatant (G-actin containing fraction) in ice-cold Milli-Q water containing 10uM Cytochalasin D and left on ice for 1hr to dissociate the actin pellet. The fractions were stored at −20 for Western blot analysis.
Equal volumes of each fraction, as well as total actin, were mixed with SDS-PAGE buffer, heated for 5 mins at 95°C and then loaded onto a 12% SDS-PAGE gel and transferred to a PVDF membrane. The blots were blocked for 1hr at room temperature in 5% non-fat dry milk-TBST (Tris-buffered saline with 0.1% Tween20) and exposed to anti-actin primary antibody (Cytoskeleton, 1:1000) overnight at 4°C. After washes, blots were incubated in secondary antibody (Promega, anti-rabbit IgG HRP-conjugated, 1:5000) for 1hr at room temperature. Protein expression was assessed by chemiluminescence and exposure to Biomax Light film (Eastman Kodak Co). Image J software (NIH) was used to quantify band intensities. Relative protein expression was calculated by normalizing the integrated band density values for G- and F-actin fractions to the values determined for total actin.
Statistical Analysis
One-way analysis of variance and repeated measures ANOVAs were used to analyze all behavioral data, with post hoc tests performed when appropriate.
ABBREVIATIONS
- AMY
Amygdala
- BDNF
Brain-derived neurotrophic factor
- BLC
Basolateral amygdala complex
- Blebb
Blebbistatin
- CPP
Conditioned place preference
- CNS
Central nervous system
- L-DOPA
L-3,4-dihydroxyphenylalanine
- LatA
Latrunculin A
- LTP
Long-term potentiation
- METH
Methamphetamine
- NAc
Nucleus accumbens
- PFC
Prefrontal cortex
- SUD
Substance use disorder
- VTA
Ventral tegmental area
References
- 1.Lee JLC, Di Ciano P, Thomas KL, Everitt BJ. Disrupting Reconsolidation of Drug Memories Reduces Cocaine-Seeking Behavior. Neuron. 2005;47:795–801. doi: 10.1016/j.neuron.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Malvaez M, Sanchis-Segura C, Vo D, Lattal KM, Wood MA. Modulation of Chromatin Modification Facilitates Extinction of Cocaine-Induced Conditioned Place Preference. Biological Psychiatry. 2010;67:36–43. doi: 10.1016/j.biopsych.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller CA, Marshall JF. Molecular Substrates for Retrieval and Reconsolidation of Cocaine-Associated Contextual Memory. Neuron. 2005;47:873–84. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Wells AM, Arguello AA, Xie X, Blanton MA, Lasseter HC, Reittinger AM, et al. Extracellular Signal-regulated Kinase in the Basolateral Amygdala, but not the Nucleus Accumbens Core, is Critical for Context-response-cocaine Memory Reconsolidation in Rats. Neuropsychopharmacology. 2012 doi: 10.1038/npp.2012.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xue Y-X, Luo Y-X, Wu P, Shi H-S, Xue L-F, Chen C, et al. A Memory Retrieval-Extinction Procedure to Prevent Drug Craving and Relapse. Science. 2012;336:241–5. doi: 10.1126/science.1215070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young EJ, Aceti M, Griggs EM, Fuchs RA, Zigmond Z, Rumbaugh G, et al. Selective, retrieval-independent disruption of methamphetamine-associated memory by actin depolymerization. Biol Psychiatry. 2014;75:96–104. doi: 10.1016/j.biopsych.2013.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai CSW, Franke TF, Gan W-B. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature. 2012;483:87–91. doi: 10.1038/nature10792. [DOI] [PubMed] [Google Scholar]
- 8.Yang G, Pan F, Gan W-B. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–4. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J. Structural dynamics of dendritic spines in memory and cognition. Trends in Neurosciences. 2010;33:121–9. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Hotulainen P, Hoogenraad CC. Actin in dendritic spines: connecting dynamics to function. The Journal of Cell Biology. 2010;189:619–29. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure–stability–function relationships of dendritic spines. Trends in Neurosciences. 2003;26:360–8. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- 12.Kim C-H, Lisman JE. A Role of Actin Filament in Synaptic Transmission and Long-Term Potentiation. The Journal of Neuroscience. 1999;19:4314–24. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proceedings of the National Academy of Sciences. 2000;97:6856–61. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta Stimulation Polymerizes Actin in Dendritic Spines of Hippocampus. The Journal of Neuroscience. 2005;25:2062–9. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch G, Rex CS, Gall CM. LTP consolidation: Substrates, explanatory power, and functional significance. Neuropharmacology. 2007;52:12–23. doi: 10.1016/j.neuropharm.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 16.Rex CS, Gavin CF, Rubio MD, Kramar EA, Chen LY, Jia Y, et al. Myosin IIb Regulates Actin Dynamics during Synaptic Plasticity and Memory Formation. Neuron. 2010;67:603–17. doi: 10.1016/j.neuron.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smart FM, Halpain S. Regulation of dendritic spine stability. Hippocampus. 2000;10:542–54. doi: 10.1002/1098-1063(2000)10:5<542::AID-HIPO4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 18.Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–46. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- 19.Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct Roles of Hippocampal De Novo Protein Synthesis and Actin Rearrangement in Extinction of Contextual Fear. The Journal of Neuroscience. 2004;24:1962–6. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gavin CF, Rubio MD, Young E, Miller C, Rumbaugh G. Myosin II motor activity in the lateral amygdala is required for fear memory consolidation. Learning & Memory. 2012;19:9–14. doi: 10.1101/lm.024042.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantzur L, Joels G, Lamprecht R. Actin polymerization in lateral amygdala is essential for fear memory formation. Neurobiology of Learning and Memory. 2009;91:85–8. doi: 10.1016/j.nlm.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Rehberg K, Bergado-Acosta JR, Koch JC, Stork O. Disruption of fear memory consolidation and reconsolidation by actin filament arrest in the basolateral amygdala. Neurobiology of Learning and Memory. 2010;94:117–26. doi: 10.1016/j.nlm.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–98. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 24.Milton AL, Everitt BJ. The persistence of maladaptive memory: Addiction, drug memories and anti-relapse treatments. Neuroscience & Biobehavioral Reviews. 2012;36:1119–39. doi: 10.1016/j.neubiorev.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Childress A, Ehrman R, McLellan AT, O’Brien C. Conditioned craving and arousal in cocaine addiction: a preliminary report. NIDA Res Monogr. 1988;81:74–80. [PubMed] [Google Scholar]
- 26.Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. Limbic activation during cue-induced cocaine craving. The American journal of psychiatry. 1999;156:11–8. doi: 10.1176/ajp.156.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuchs RA, Evans KA, Ledford CC, Parker MP, Case JM, Mehta RH, et al. The Role of the Dorsomedial Prefrontal Cortex, Basolateral Amygdala, and Dorsal Hippocampus in Contextual Reinstatement of Cocaine Seeking in Rats. Neuropsychopharmacology. 2004;30:296–309. doi: 10.1038/sj.npp.1300579. [DOI] [PubMed] [Google Scholar]
- 28.Koob GF. Dynamics of Neuronal Circuits in Addiction: Reward, Antireward, and Emotional Memory. Pharmacopsychiatry. 2009;42:S32–S41. doi: 10.1055/s-0029-1216356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lasseter HC, Wells AM, Xie X, Fuchs RA. Interaction of the Basolateral Amygdala and Orbitofrontal Cortex is Critical for Drug Context-Induced Reinstatement of Cocaine-Seeking Behavior in Rats. Neuropsychopharmacology. 2011;36:711–20. doi: 10.1038/npp.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller CA, Marshall JF. Altered Prelimbic Cortex Output during Cue-Elicited Drug Seeking. The Journal of Neuroscience. 2004;24:6889–97. doi: 10.1523/JNEUROSCI.1685-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller CA, Marshall JF. Altered Fos expression in neural pathways underlying cue-elicited drug seeking in the rat. European Journal of Neuroscience. 2005;21:1385–93. doi: 10.1111/j.1460-9568.2005.03974.x. [DOI] [PubMed] [Google Scholar]
- 32.Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, Robbins TW. Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2008;363:3125–35. doi: 10.1098/rstb.2008.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pickens CL, Airavaara M, Theberge F, Fanous S, Hope BT, Shaham Y. Neurobiology of the incubation of drug craving. Trends Neurosci. 2011;34:411–20. doi: 10.1016/j.tins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulson PE, Robinson TE. Amphetamine-induced time-dependent sensitization of dopamine neurotransmission in the dorsal and ventral striatum: a microdialysis study in behaving rats. Synapse. 1995;19:56–65. doi: 10.1002/syn.890190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulson PE, Robinson TE. Regional differences in the effects of amphetamine withdrawal on dopamine dynamics in the striatum. Analysis of circadian patterns using automated on-line microdialysis. Neuropsychopharmacology. 1996;14:325–37. doi: 10.1016/0893-133X(95)00141-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson TE, Camp DM. Long-lasting effects of escalating doses of d-amphetamine on brain monoamines, amphetamine-induced stereotyped behavior and spontaneous nocturnal locomotion. Pharmacology, biochemistry, and behavior. 1987;26:821–7. doi: 10.1016/0091-3057(87)90616-2. [DOI] [PubMed] [Google Scholar]
- 37.Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. European Journal of Neuroscience. 1999;11:1598–604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- 38.Robinson TE, Kolb B. Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse. 1999;33:160–2. doi: 10.1002/(SICI)1098-2396(199908)33:2<160::AID-SYN6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 39.Singer BF, Tanabe LM, Gorny G, Jake-Matthews C, Li Y, Kolb B, et al. Amphetamine-Induced Changes in Dendritic Morphology in Rat Forebrain Correspond to Associative Drug Conditioning Rather than Nonassociative Drug Sensitization. Biological Psychiatry. 2009;65:835–40. doi: 10.1016/j.biopsych.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee KW, Kim Y, Kim AM, Helmin K, Nairn AC, Greengard P. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci U S A. 2006;103:3399–404. doi: 10.1073/pnas.0511244103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson TE, Gorny G, Mitton E, Kolb B. Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse. 2001;39:257–66. doi: 10.1002/1098-2396(20010301)39:3<257::AID-SYN1007>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 42.Shen H-w, Toda S, Moussawi K, Bouknight A, Zahm DS, Kalivas PW. Altered Dendritic Spine Plasticity in Cocaine-Withdrawn Rats. J Neurosci. 2009;29:2876–84. doi: 10.1523/JNEUROSCI.5638-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toda S, Shen H, Kalivas PW. Inhibition of actin polymerization prevents cocaine-induced changes in spine morphology in the nucleus accumbens. Neurotox Res. 2010;18:410–5. doi: 10.1007/s12640-010-9193-z. [DOI] [PubMed] [Google Scholar]
- 44.Toda S, Shen H-W, Peters J, Cagle S, Kalivas PW. Cocaine Increases Actin Cycling: Effects in the Reinstatement Model of Drug Seeking. The Journal of Neuroscience. 2006;26:1579–87. doi: 10.1523/JNEUROSCI.4132-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dobi A, Seabold GK, Christensen CH, Bock R, Alvarez VA. Cocaine-Induced Plasticity in the Nucleus Accumbens Is Cell Specific and Develops without Prolonged Withdrawal. The Journal of Neuroscience. 2011;31:1895–904. doi: 10.1523/JNEUROSCI.5375-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rademacher DJ, Rosenkranz JA, Morshedi MM, Sullivan EM, Meredith GE. Amphetamine-Associated Contextual Learning Is Accompanied by Structural and Functional Plasticity in the Basolateral Amygdala. The Journal of Neuroscience. 2010;30:4676–86. doi: 10.1523/JNEUROSCI.6165-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Young EJ, Williams CL. Valence dependent asymmetric release of norepinephrine in the basolateral amygdala. Behavioral neuroscience. 2010;124:633–44. doi: 10.1037/a0020885. [DOI] [PubMed] [Google Scholar]
- 48.Baxter MG, Murray EA. The amygdala and reward. Nature reviews Neuroscience. 2002;3:563–73. doi: 10.1038/nrn875. [DOI] [PubMed] [Google Scholar]
- 49.Lalumiere RT. Optogenetic dissection of amygdala functioning. Frontiers in behavioral neuroscience. 2014;8:107. doi: 10.3389/fnbeh.2014.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corbit LH, Leung BK, Balleine BW. The role of the amygdala-striatal pathway in the acquisition and performance of goal-directed instrumental actions. J Neurosci. 2013;33:17682–90. doi: 10.1523/JNEUROSCI.3271-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feltenstein MW, See RE. NMDA receptor blockade in the basolateral amygdala disrupts consolidation of stimulus-reward memory and extinction learning during reinstatement of cocaine-seeking in an animal model of relapse. Neurobiol Learn Mem. 2007;88:435–44. doi: 10.1016/j.nlm.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haghparast A, Shamsizadeh A, Samandari R, Omranifard A, Vaziri A, Razavi Y. Cannabinoid receptors in the basolateral amygdala are involved in the potentiation of morphine rewarding properties in the acquisition, but not expression of conditioned place preference in rats. Brain research. 2014;1565:28–36. doi: 10.1016/j.brainres.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 53.Pelloux Y, Murray JE, Everitt BJ. Differential roles of the prefrontal cortical subregions and basolateral amygdala in compulsive cocaine seeking and relapse after voluntary abstinence in rats. Eur J Neurosci. 2013 doi: 10.1111/ejn.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y, Zhou Q-X, Hou Y-Y, Lu B, Yu C, Chen J, et al. Actin Polymerization-Dependent Increase in Synaptic Arc/Arg3.1 Expression in the Amygdala Is Crucial for the Expression of Aversive Memory Associated with Drug Withdrawal. The Journal of Neuroscience. 2012;32:12005–17. doi: 10.1523/JNEUROSCI.0871-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron. 2008;57:719–29. doi: 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 56.Kaech S, Fischer M, Doll T, Matus A. Isoform specificity in the relationship of actin to dendritic spines. J Neurosci. 1997;17:9565–72. doi: 10.1523/JNEUROSCI.17-24-09565.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kramar EA. Integrin-driven actin polymerization consolidates long-term potentiation. Proceedings of the National Academy of Sciences. 2006;103:5579–84. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Micheva KD, Vallée A, Beaulieu C, Herman IM, Leclerc N. β-Actin is confined to structures having high capacity of remodelling in developing and adult rat cerebellum. European Journal of Neuroscience. 1998;10:3785–98. doi: 10.1046/j.1460-9568.1998.00391.x. [DOI] [PubMed] [Google Scholar]
- 59.Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5:1158–70. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 60.Cheng XT, Hayashi K, Shirao T. Non-muscle myosin IIB-like immunoreactivity is present at the drebrin-binding cytoskeleton in neurons. Neurosci Res. 2000;36:167–73. doi: 10.1016/s0168-0102(99)00123-6. [DOI] [PubMed] [Google Scholar]
- 61.Medeiros NA, Burnette DT, Forscher P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat Cell Biol. 2006;8:216–26. doi: 10.1038/ncb1367. [DOI] [PubMed] [Google Scholar]
- 62.Brown J, Bridgman PC. Role of myosin II in axon outgrowth. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2003;51:421–8. doi: 10.1177/002215540305100403. [DOI] [PubMed] [Google Scholar]
- 63.Rosenfeld SS, Xing J, Chen LQ, Sweeney HL. Myosin IIb is unconventionally conventional. J Biol Chem. 2003;278:27449–55. doi: 10.1074/jbc.M302555200. [DOI] [PubMed] [Google Scholar]
- 64.Mizui T, Sekino Y, Yamazaki H, Ishizuka Y, Takahashi H, Kojima N, et al. Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PloS one. 2014;9:e85367. doi: 10.1371/journal.pone.0085367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, et al. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743–7. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- 66.Si J, Ge Y, Zhuang S, Gong R. Inhibiting nonmuscle myosin II impedes inflammatory infiltration and ameliorates progressive renal disease. Laboratory investigation; a journal of technical methods and pathology. 2010;90:448–58. doi: 10.1038/labinvest.2009.142. [DOI] [PubMed] [Google Scholar]
- 67.Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–66. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rex CS, Lin CY, Kramar EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–29. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimada A, Mason CA, Morrison ME. TrkB signaling modulates spine density and morphology independent of dendrite structure in cultured neonatal Purkinje cells. J Neurosci. 1998;18:8559–70. doi: 10.1523/JNEUROSCI.18-21-08559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–7. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tran-Nguyen LT, Fuchs RA, Coffey GP, Baker DA, O’Dell LE, Neisewander JL. Time-dependent changes in cocaine-seeking behavior and extracellular dopamine levels in the amygdala during cocaine withdrawal. Neuropsychopharmacology. 1998;19:48–59. doi: 10.1016/S0893-133X(97)00205-4. [DOI] [PubMed] [Google Scholar]
- 72.Hou YY, Lu B, Li M, Liu Y, Chen J, Chi ZQ, et al. Involvement of actin rearrangements within the amygdala and the dorsal hippocampus in aversive memories of drug withdrawal in acute morphine-dependent rats. J Neurosci. 2009;29:12244–54. doi: 10.1523/JNEUROSCI.1970-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science. 1998;282:298–300. doi: 10.1126/science.282.5387.298. [DOI] [PubMed] [Google Scholar]
- 74.Bozarth MA, Wise RA. Toxicity associated with long-term intravenous heroin and cocaine self-administration in the rat. Jama. 1985;254:81–3. [PubMed] [Google Scholar]
- 75.Ferrario CR, Gorny G, Crombag HS, Li Y, Kolb B, Robinson TE. Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol Psychiatry. 2005;58:751–9. doi: 10.1016/j.biopsych.2005.04.046. [DOI] [PubMed] [Google Scholar]