

Abstract
Genetic subgrouping of gliomas has been emphasized recently, particularly after the finding of isocitrate dehydrogenase 1 (IDH1) mutations. In a previous study, we investigated whole-chromosome copy number aberrations (CNAs) of gliomas and have described genetic subgrouping based on CNAs and IDH1 mutations. Subsequently, we classified gliomas using simple polymerase chain reaction (PCR)-based methods to improve the availability of genetic subgrouping. We selected IDH1/2 and TP53 as markers and analyzed 237 adult supratentorial gliomas using Sanger sequencing. Using these markers, we classified gliomas into three subgroups that were strongly associated with patient prognoses. These included IDH mutant gliomas without TP53 mutations, IDH mutant gliomas with TP53 mutations, and IDH wild-type gliomas. IDH mutant gliomas without TP53 mutations, which mostly corresponded to gliomas carrying 1p19q co-deletions, showed lower recurrence rates than the other 2 groups. In the other high-recurrence groups, the median progression-free survival (PFS) and overall survival (OS) of patients with IDH mutant gliomas with TP53 mutations were significantly longer than those of patients with IDH wild-type gliomas. Notably, most IDH mutant gliomas with TP53 mutations had at least one of the CNAs +7q, +8q, −9p, and −11p. Moreover, IDH mutant gliomas with at least one of these CNAs had a significantly worse prognosis than did other IDH mutant gliomas. PCR-based mutation analyses of IDH and TP53 were sufficient for simple genetic diagnosis of glioma that were strongly associated with prognosis of patients and enabled us to detect negative CNAs in IDH mutant gliomas.
Introduction
Gliomas are currently classified according to their histological appearance, and the associated malignancy is defined by the World Health Organization (WHO) grading system. In cases of high grade gliomas, patients tend to show high recurrence rates and a worse prognosis. However, in some cases, the clinical course does not reflect the histological classification, warranting the use of genetic diagnoses and subgroups. We previously reported that adult supratentorial gliomas could be classified into genetic subgroups on the basis of their copy number aberrations (CNAs) using comparative genomic hybridization (CGH) and suggested that gliomas with +7q and 1p/19q co-deletions may have a better prognosis than those with −9p, −10q, and +7 CNAs [1].
The clinical significance of isocitrate dehydrogenase 1 (IDH1) point mutation in gliomas was first reported in 2008, the overall survival (OS) of glioblastoma patients with IDH1-mutated glioblastoma was demonstrated to be significantly longer than that of patients with wild-type IDH1 glioblastoma [2]. Various subsequent studies confirmed the prognostic importance of IDH1 mutations [3–6]. Therefore, we combined a CGH analysis with the IDH1 mutation status to propose the genetic subgrouping of gliomas [5]. The data demonstrated that IDH1 mutant gliomas with −1p/19q and +7q CNAs are associated with a better prognosis than that associated with IDH1 wild-type gliomas.
Although these genetic subgroups were clinically informative, copy number-independent and simplified methods are desirable for genetic classification in clinical use. Therefore, in the present study we aimed to identify simpler and more widely available methods by which gliomas could be diagnosed at many clinical institutions. We focused on Sanger sequencing to address this problem and selected IDH1/2 and TP53 as markers for polymerase chain reaction (PCR) analyses. IDH2 mutations were first detected in gliomas by Yan et al. [3]; similar to IDH1 mutations, IDH2 mutation were associated with a better prognosis, although these mutations occurred at considerable lower frequency. Moreover, TP53 mutations are often detected in astrocytic tumors [7] and it has been shown that these are mutually exclusive with 1p/19q co-deleted gliomas [8]. Therefore, we hypothesized that most IDH mutant gliomas without TP53 mutations carry 1p/19q co-deletions.
Given the increase in CNAs with tumor regrowth or progression to high grade gliomas, according to our CGH analyses, the identification of common and specific CNAs for each genetic subgroup should facilitate an oncological understanding of gliomas. Although we previously reported that 1p/19q co-deletions and +7q are frequently detected in IDH mutant gliomas [5] according to our CGH data, only 68% of IDH mutant gliomas harbored 1p/19q co-deletions and/or +7q. Therefore, in this study, we also aimed to identify common CNAs in IDH mutant gliomas, particularly those harboring TP53 mutation.
In the present study, we analyzed IDH1/2 and TP53 mutations in adult supratentorial gliomas via direct sequencing and characterized these malignancies using PCR-based genetic subgrouping, achieving greater prognostic accuracy than that achieved with pathological classifications. In addition, we confirmed that most IDH mutant gliomas with TP53 mutations contained at least one of the CNAs +7q, +8q, −9p, and −11p. Because IDH mutant gliomas with TP53 mutations showed high recurrence rates, we suggest that these CNAs are negative prognostic factors for patients with IDH mutant gliomas.
Materials and Methods
Patients, samples, and DNA preparation
We analyzed 237 adult supratentorial glioma samples that had been surgically resected at Keio University from 1994 until 2004 and at Fujita Health University from 2001 until 2015. The histological diagnoses included (anaplastic) astrocytomas, (anaplastic) oligodendrogliomas, (anaplastic) oligoastrocytomas, and glioblastomas. Some patients underwent ≥2 surgeries during their clinical course, thereby contributing to multiple glioma samples. The samples were evaluated by neuropathologists and were classified according to the WHO criteria. Tumor samples were available as frozen tissues and/or as formalin-fixed paraffin-embedded (FFPE) samples. DNA was extracted from freshly frozen tissue using DNeasy blood and tissue kits (QIAGEN) and from FFPE samples with DNA FFPE tissue kits (QIAGEN) or REPLI-g kits (QIAGEN). DNA quality was assessed via absorptiometric analyses. This study was approved by the Ethics Committee of the Fujita Health University (Approval number: 11–106). Written informed consent was obtained from each patient.
CGH
The CGH analysis was conducted as described by Hirose et al. [1]. Tumor tissues were removed from FFPE samples according to pathological appearance or MIB-1 density and tumor DNA was amplified via degenerate oligonucleotide-primed PCR (DOP-PCR). DNA from peripheral blood lymphocytes was obtained from healthy donors and was used as a control. DNA from these samples was labeled with biotin–deoxyuridine triphosphate (Roche) after amplification. Subsequently, labeled DNA from tumors and normal tissues was hybridized to normal metaphase spreads. After unhybridized probes were washed away, the spreads were counterstained with 4,6-diamino-2-phenylindole and the fluorescence intensity ratios for each chromosome were assessed using CytoVision software (Applied Imaging).
As described previously, total chromosomal gains and partial gains, such as +7 and +7q, were interpreted as different CNAs [5]; +7 was interpreted as a typical copy number change for IDH wild-type gliomas and +7q was often detected in IDH mutant gliomas. Because gliomas with and without IDH mutations are thought to be evolved through different lineages [5], we assumed that the total and partial chromosomal gains would reflect different processes. However, we considered total loss and partial losses (such as −10 and −10q) to be identical CNAs, although they were frequently detected in IDH wild-type gliomas that did not show differences in prognosis or histology. Accordingly, we categorized −10 and −10q as −10q.
Mutation analysis
Sanger sequencing was used to detect IDH1/2 and TP53 mutations in the samples. We analyzed the sequence of codon 132 for IDH1 and codon 172 for IDH2. In previous studies, most TP53 missense mutation hotspots were found in exons 5–8 [9–11], and missense mutations in the DNA-binding domains affected the prognosis of patients with breast carcinoma [12]; therefore, we investigated exons 5–8 in TP53 mutation analyses. The primers used in our study were selected according to previous studies [10, 13–15], and sequence analyses were conducted using ABI 3100 apparatus (Applied Biosystems).
Statistical analysis
The primary endpoint in this study was progression-free survival (PFS), which is defined as the period from the date of first surgery until the confirmation of tumor regrowth via magnetic resonance imaging (MRI) or symptomatic deterioration. Prognoses were calculated according to PFS and OS, another important factor with respect to a patient’s prognosis. OS was defined as the period from the date of first surgery until the date of death. Kaplan–Meier curves were generated in cases involving first surgery. Cox log-rank tests were used for group comparison. A multivariate logistic regression analysis was conducted to examine correlation between recurrence within 3 years and clinical factors, histology, or specific CNAs. We selected 3 years as the cut-off for recurrence since the median PFS of IDH mutant gliomas ranged from 42 to 51 months according to our patient data and a previous study [16] and excluded cases if follow-up months were less than 36 months. We defined subtotal resection (STR) as a tumor resection volume of >90%.
Results
Comparison of the PCR-based genetic classification and histological classification
The histological diagnosis of the 237 adult supratentorial gliomas evaluated in this study included astrocytomas, oligodendrogliomas, oligoastrocytomas, and glioblastomas; IDH1/2 and TP53 mutation statuses were determined via direct sequencing (S1 Fig). Among the 113 IDH mutant gliomas, 42 harbored TP53 mutations and 42 did not. The remaining 29 samples were from biopsies or were very old; thus those samples provided DNA of insufficient quantity or quality for analyses. Because TP53 mutations did not affect the prognosis of IDH wild-type gliomas according to our study, we classified gliomas as IDH mutant gliomas without TP53 mutations, IDH mutant gliomas with TP53 mutations, and IDH wild-type gliomas.
Fig 1 shows the prognoses of patients grouped according to histology or genetics. Table present patients background information, including gender, age at diagnosis, recurrence rates, median PFS, and median OS, for each histological and genetic subgroup, respectively. As this was a retrospective study, the study cases had not undergone adjuvant therapy according to strict regimen. Patients with IDH wild-type gliomas were significantly older than those with IDH mutant gliomas (p < 0.05). Those harboring IDH mutant gliomas without TP53 mutations had a lower recurrence rate relative to the other two subgroups (p < 0.05). Although IDH mutant gliomas with TP53 mutations and IDH wild-type gliomas were both associated with high recurrence rates, the median PFS in the latter group was significantly shorter than that in the former group (Table 1B and Fig 1C; hazard ratio = 0.229; 95% confidence interval [CI]: 0.142–0.368; p < 0.0001). In these two genetic subgroups, the median OS of patients with IDH wild-type gliomas was also significantly shorter than that of patients with IDH and TP53 mutant gliomas (Table 1B and Fig 1D; hazard ratio = 0.270; 95% CI: 0.155–0.460; p < 0.0001). On the other hand, the median OSs for grade III and IV gliomas were relatively similar (10 and 6 months, respectively) although the difference between grade III and IV gliomas were statistically significant. These results suggests that PCR-based genetic classification provides more precise clinical information, which includes recurrence rates and PFS, than that provided by histological classification.
Fig 1. Kaplan–Meier curves of progression-free survival (PFS) according to subgroups.
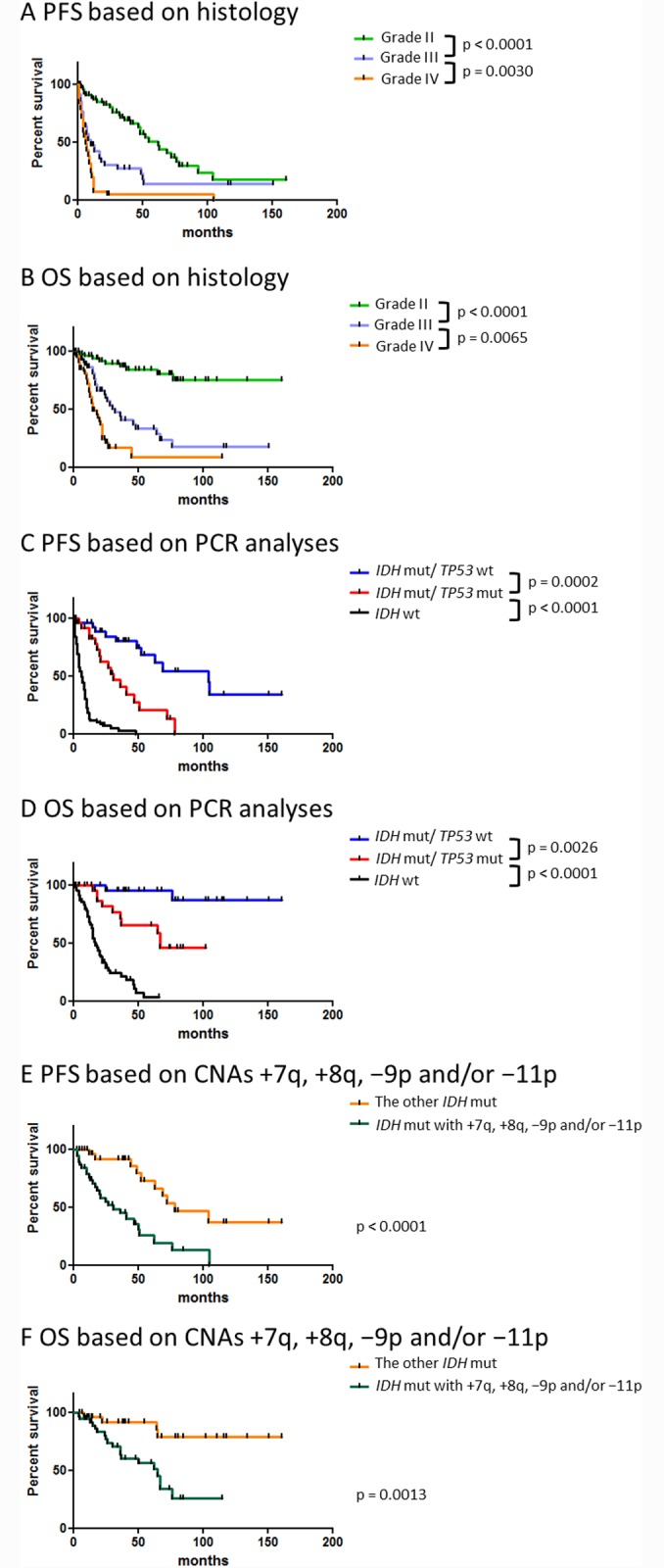
A comparison of PFS and overall survival (OS) according to (A and B, respectively) pathological (n = 171) and (C and D, respectively) genetic classification (n = 158). Kaplan–Meier curves comparing PFS (E) and OS (F) associated with IDH mutant gliomas harboring CNAs +7q, +8q, −9p, and/or −11p with the PFS and OS of other IDH mutant gliomas (n = 73). Only patients who underwent an initial surgical intervention were included in these analyses. Abbreviations: mut; mutation, wt; wild-type.
Table 1. Background of patients who underwent initial surgical intervention.
| A | ||||
| WHO grade II (n = 63) | WHO grade III (n = 48) | WHO grade IV (n = 60) | ||
| Female | 30 | 20 | 25 | |
| Age at diagnosis | 42.6 | 54.0 | 62.4 | |
| IDH mutation | 53 | 30 | 2 | |
| Prognosis | ||||
| Recurrent | 28 | 33 | 50 | |
| Dropped | 12 | 9 | 7 | |
| Following | 23 | 6 | 3 | |
| Median PFS (mo) | 62 | 10 | 6 | |
| Median OS (mo) | Undefined | 32 | 15 | |
| B | ||||
| IDH mutant gliomas without TP53 mutation (n = 32) | IDH mutant gliomas with TP53 mutation (n = 29) | IDH wild-type gliomas (n = 97) | ||
| Female | 14 | 16 | 43 | |
| Age at diagnosis | 41.3 | 43.5 | 62.1 | |
| Histology | ||||
| Oligo Grade II | 19 | 9 | 3 | |
| Oligo Grade III | 8 | 3 | 3 | |
| Astro Grade II | 3 | 9 | 5 | |
| Astro Grade III | 1 | 7 | 14 | |
| Astro Grade IV | 1 | 0 | 55 | |
| Not defined/others | 0 | 1 | 17 | |
| Additional therapy | ||||
| None | 8 | 10 | 10 | |
| Radiotherapy only | 4 | 2 | 9 | |
| Chemotherapy only | 14 | 5 | 12 | |
| Combined | 5 | 12 | 62 | |
| Others or unknown | 1 | 0 | 4 | |
| Prognosis | ||||
| Recurrent | 11 | 18 | 81 | |
| Dropped | 3 | 2 | 10 | |
| Following | 18 | 9 | 6 | |
| Median PFS (mo) | 104 | 31 | 6 | |
| Median OS (mo) | Undefined | 67 | 17 | |
A comparison of patient backgrounds according to histological classification (A) and genetic classification (B). In this table, oligoastrocytomas were classified as oligodendroglial tumor.
Results of CGH analysis in IDH mutant gliomas
In a previous study, we reported a high frequency of +7 and −10q CNAs among patients with IDH1 wild-type gliomas [5]. In this study, we analyzed whole-chromosome gains and losses and identified the CNAs frequently observed in IDH mutant gliomas with and without TP53 mutations (Fig 2). Notably, −1p was uniquely observed in IDH mutant gliomas without TP53 mutations and was always accompanied by −19q. Moreover, the CNAs −4q, +7, −14q, and −19q were mainly detected in IDH mutant gliomas without TP53 mutations. However, +7q and −9p were more frequently found in IDH mutant gliomas with TP53 mutations than in other IDH mutant gliomas; +8q, −11p, and +12p were almost exclusively detected in IDH mutant gliomas with TP53 mutations.
Fig 2. CNAs frequently detected in IDH mutant gliomas.

A comparison of CNAs found in IDH mutant gliomas (A) with wild-type TP53 and (B) mutant TP53. The number of CNAs detected in both IDH mutant gliomas with wild-type and mutant TP53 is summarized in a table.
Correlation between TP53 mutation and CNAs in IDH mutant gliomas
We subsequently investigated the correlation between the TP53 mutation and CNA statuses. As expected, gliomas with TP53 mutation and −1p/19q were mutually exclusive. Most IDH mutant gliomas with TP53 mutations had at least one of the CNAs +7q, +8q, −9p, and −11p (Fig 3), and those CNAs overlapped with −1p/19q in tumors lacked TP53 mutations. IDH mutant gliomas with and without +7q, +8q, −9p, and −11p are summarized in Table 2.
Fig 3. The correlation between TP53 mutations and the CNAs −1p/19q, +7q, +8q, −9p, and −11p in IDH mutant gliomas.

A comparison between (A) primary (n = 47) and (B) recurrent disease (n = 24). The CNA −1p/19q represents a favorable prognostic marker; the CNAs +7q, +8q, −9p, and −11p represents unfavorable prognostic markers.
Table 2. A list of IDH mutant glioma patients with (A) and without (B) +7q, +8q, −9p, and/or −11p according to comparative genomic hybridization (CGH) analysis as well as their prognosis and TP53 mutation status.
| A | |||||||||
| Histology | Age, Sex | Surgery | CNAs | Genetic type | TP53 mut | Tumor regrowth | PFS, mo | Follow-up, mo | Outcome |
| A GII | 22M | 1 st | +8q21.3-ter, +10pter-q23, −10q25-ter, −X | +8q | NA | No | 85 | 85 | Alive |
| A GII* 1 | 25M | 1 st | −4q22-ter, +5pter-23.3, −5q31.2-ter, +7, +8q, +13, −19q, −22 | +8q | NA | Yes | 15 | 25 | Dead |
| A GII | 27M | 1 st | +7q31.3-ter, +10, +20 | +7q | p.K132fs | No | 30 | 30 | Alive |
| A GII | 28F | 1 st | −6q, +8q21.1-ter, −9p, −14q22-ter, −Xp | +8q, −9p | NA | Yes | 62 | 77 | Dead |
| A GII | 30F | 1 st | −6q, −9p, −14q22-ter, −19q | −9p | p.D281G | Yes | 41 | 67 | Alive |
| A GII* 2 | 33M | 1 st | +8q22.3-ter, −12q13-24.1 | +8q | p.R273H | Yes | 12 | 83 | Alive |
| A GII | 35F | 1 st | −4q, +6petr-q21, −8p21, +8q21.1–22, −13q22-ter, −14q21-ter | +8q | NA | Yes | 76 | 76 | Alive |
| A GII* 3 | 44F | 1 st | −3p22-21, +7q, +8q22-ter, +11q23.3-ter, +12p, −13q21-31, −19q | +7q, +8q | p.R273C | Yes | 27 | 65 | Dead |
| A GII | 45M | 1 st | +8q21.1–24.1, −11q23-ter, +19 | +8q | NA | No | 48 | 48 | Alive |
| A GII | 46F | 1 st | +7q, −19q | +7q | p.H179Y | No | 14 | 14 | Alive |
| A GII | 54F | 1 st | −4q, +8, +10pter-q22.3, −11p | −11p | p.N247S | No | 15 | 15 | Alive |
| A GII | 56F | 1 st | +7q, −10, +19q, +20 | +7q | NA | Yes | 5 | 9 | Dead |
| A GII | 57F | 1 st | +1q31-ter, −11p, +12p, −12q12-22, +12q23-ter, −18, −19q, −20p, −21 | −11p | NA | No | 2 | 2 | Alive |
| A GIII | 27F | 1 st | −3p21-13, +7p, +8q, +10p, −13q, +14q24.3-ter, −16q, −19q, −X | +8q | NA | Yes | 13 | 32 | Dead |
| A GIII | 35F | 1 st | −11, −17p, −18q, −20p | −11p | p.R175H | Yes | 31 | 37 | Dead |
| A GIII | 35M | 1 st | −11, −12q | −11p | p.R273C | No | 9 | 9 | Alive |
| A GIII | 39F | 1 st | +1q21-31, −1q32.2-ter, +2p16-11.2, −5q, −6q, +7q, +8q23-ter, −9q22-33, −10p, +17q, −19q, −Xp | +7q, +8q | NA | No | 18 | 18 | Alive |
| A GIII | 40F | 1 st | +10q23-ter, −11, −13, −14, −17p, +17q, −18 | −11p | NA | Yes | 4 | 4 | Alive |
| A GIII | 43M | 1 st | +8q, +12p, -19q, +20, +X | +8q | NA | Yes | 50 | 50 | Alive |
| A GIII | 47F | 1 st | −8p, −9p, +9q, −10q24-ter, +12q14-15, −12q21-ter, −14q21-31 | −9p | p.R273C | Yes | 21 | 36 | Dead |
| A GIII* 4 | 48M | 1 st | −4q13-21, +7q, +13q31-ter, +X | +7q | p.R175H | Yes | 51 | 67 | Dead |
| A GIII* 4 | 52M | 2 nd | +2pter-22, +3pter-23, +4q21-24, +4q26-33, −6pter-22, −6q21-ter, +7q,−9pter-21, −12p13, +20q | +7q, −9p | p.R175H | Yes | 1 | 67 | Dead |
| A GIII | 55M | 1 st | −2pter-23, +2p21-13, +3pter-22, +3q24.1-ter, −4q28, +8p, −8q12-13, +8q21.3–22.3, −9p, −10q22.1-ter, +12q13.1–23, −13q12.1–22, −17p, +17q, +19q, −20p | +8q, −9p | NA | Yes | 3 | 26 | Dead |
| A GIII | 59F | 1 st | −4q, −5p, +7, +8q | +8q | NA | No | 10 | 10 | Alive |
| A GIII | 65F | 1 st | +1p35-33, +1q, +3p22-21, -6, +7p, +7q32-ter, +8q23-ter, +9pter-21, -9q, −10q, −13, −14, +15q, -22q | +7q, +8q | p.R244S, p.R245D | Yes | 10 | 30 | Dead |
| A GIII* 5 | 74F | 2 nd | +1p21, +1q, +7p15-cen, +8q21.3, +10p, +16p11.2, +16q | +8q | p.A138V | No | 13 | 19 | Dead |
| OA GII* 6 | 34M | 1 st | −1p, +2p, −9p, −19q | −1p/19q, −9p | none | Yes | 25 | 34 | Alive |
| OA GII* 7 | 39M | 1 st | +7q, +10q24-ter | +7q | p.R175H | Yes | 47 | 60 | Alive |
| OA GII* 7 | 43M | 2 nd | −5p, +7q, −18q | +7q | p.R175H | No | 11 | 60 | Alive |
| OA GII | 46F | 1 st | +3p, −5p, +8, −11p, −13q12.1–21.3, +13q22-ter | −11p | p.R306X | No | 19 | 19 | Alive |
| OA GI * 8 | 61F | 1 st | +7q31-ter, −X | +7q | p.Y163C | Yes | 20 | 74 | Alive |
| OA GII* 8 | 63F | 2 nd | +7q31.1-ter, +12q22-ter, −Xp | +7q | p.Y163C | No | 7 | 74 | Alive |
| OA GIII* 9 | 30M | 2 nd | −1p, −4, +7q21.3-ter, +8, +11, −14q22-23, −18, −19q | −1p/19q, +7q | none | Yes | 16 | 111 | Alive |
| OA GIII* 2 | 34M | 2 nd | +4p, −4q, −5qcen-13, −5q21-ter, +8q13-ter, −9pter-21.3, −11p, +12p, −12q22-23 | +8q, −9p, −11p | p.R273H | Yes | 48 | 83 | Alive |
| OA GIII | 36F | 1 st | −1q41-ter, −6q, +7q31-ter, −9p, −14q22-ter, −Xq21-ter | +7q, −9p | p.G245S | No | 36 | 36 | Alive |
| OA GIII | 40F | 1 st | −1p, +1q, +3, −9, +12q14, −15q, +17, +18, −19q, +20 | −1p/19q, −9p | none | Yes | 105 | 115 | Alive |
| OA GIII* 10 | 44F | 3 rd | +2p, −3p21.3–11.2, +7, +8q23-ter, +10pter-12.3, −19q13.2-ter | +8q | p.Y220C | Yes | 2 | 80 | Alive |
| OA GIII* 3 | 47F | 2 nd | −4q28-ter, +7q, +8q23-ter, +12p, −Xq | +7q, +8q | p.R273C | Yes | 29 | 65 | Dead |
| OA GIII* 5 | 74F | 1 st | +1p21, +1q, +2p16-ter, +2q, −3p21, +7p21, +7qcen-21, +8, +10p, +10qcen-24, +11, +12p, -12q14-23, +16q | +7q | p.A138V | Yes | 6 | 19 | Dead |
| O GII | 57F | 1 st | −1p, −9pter-21, −18, −19q, +21 | −1p/19q, −9p | none | No | 51 | 51 | Alive |
| O GII | 59M | 1 st | +7q, +8q21.1-ter, +10p, −13q14-32, −16q, −19q | +7q, +8q | p.R248W | No | 36 | 36 | Alive |
| O GII | 71M | 1 st | −6pter-16, +8q21.1–21.3 | +8q | NA | No | 40 | 40 | Alive |
| O GIII* 11 | 37F | 2 nd | −1p, +1q, −2, +6, +7, +8, −9, +11, −16, +17, −18, +19p, −19q, +21, −22 | −1p/19q, −9p | none | No | 8 | 26 | Alive |
| O GIII | 41M | 5 th | −1p, +7q, +8, +18p, −18q, −19q, +22q | −1p/19q, +7q | none | Yes | 5 | 76 | Dead |
| O GIII | 62M | 1 st | −1p, +7q31.1-ter, −19q | −1p/19q, +7q | none | No | 0 | 0 | Dead |
| O GIII* 12 | 64M | 2 nd | −1p, +2, +7, −9p, +9q, −15q, −19q | −1p/19q, −9p | none | Yes | 27 | 134 | Alive |
| O GIII | 72M | 1 st | +1q, −2q37, −4p, +4q, −6pter-21.3, −6q16-ter, −8, −9pter-23, −11p, −14q, −17p, +17q22-ter | −9p, −11p | p.R248W | Yes | 4 | 15 | Dead |
| GBM* 1 | 26M | 2 nd | −3q11.2–24, +3q24.1-ter, +4p, −4q, +5pter-5q23.3, −5q31.2-ter, −6pter-22.1, +6p22.2–18.3, −6q21-26, +7, +8q, −9p, +13, −14q, +16q, +17q, −18, −19q, +21, −22 | +8q, −9p | none | Yes | 1 | 25 | Dead |
| GBM* 13 | 28F | 2 nd | −3pter-3q24, −5p, +7, −11p, −11q22-23.1, −13q, −19q, −22, −X | −11p | p.Y220C, p.R248W | No | 24 | 102 | Alive |
| GBM* 14 | 28M | 3 rd | −4q28-ter, +5pter-q23.3, +7q, +8q, −9p, −9q, −11pter-15.1, −11q23.1-ter, +12p, −13q21.1–22, +13q31-ter | +7q, +8q, −9p | p.Y236D | Yes | 4 | 67 | Dead |
| GBM | 62M | 1 st | +2, −6p, −7p, +7q, +8q22-ter, −9p, +9q, −11, −13q, −14q, +15q, +18q21 | +7q, +8q, −9p, −11p | NA | Yes | 3 | 4 | Dead |
| ND* 14 | 26M | 1 st | +7q, +8q22.1-ter, +11q23.3-ter, +12p, +19 | +7q, +8q | NA | Yes | 27 | 67 | Dead |
| ND* 14 | 30M | 4 th | +7q, +8q, −9p, −X | +7q, +8q, −9p | p.Y236D | Yes | 4 | 67 | Dead |
| ND* 2 | 38M | 3 rd | −5q31.1-ter, +8q22.3-ter, +10p, −10q, +12p | +8q | p.R273H | No | 20 | 83 | Alive |
| B | |||||||||
| A GII* 13 | 22F | 1 st | −11q22-23.1 | another | p.Y220C, p.R248W | Yes | 78 | 102 | Alive |
| A GII* 15 | 22F | 1 st | none | none | p.H193Y | Yes | 72 | 85 | Alive |
| A GII | 37M | 1 st | +2q24-33, −3p22-q25, −4q28-ter, +7 | another | NA | No | 7 | 7 | Alive |
| A GII | 38F | 1 st | −1p, −19q | −1p/19q | none | No | 14 | 14 | Alive |
| A GII | 41M | 1 st | −4 | another | p.H179R | No | 9 | 9 | Alive |
| A GIII | 55M | 1 st | −1p, +7, −10p, −19q | −1p/19q | NA | No | 12 | 12 | Alive |
| A GIII | 60M | 1 st | −1p, −4, −9q22-ter, −14q21.3-ter, −19q | −1p/19q | NA | No | 118 | 118 | Alive |
| OA GII* 9 | 24M | 1 st | −1p, −19q | −1p/19q | none | Yes | 69 | 111 | Alive |
| OA GII | 31F | 1 st | −12q, −13q14.3–22 | another | none | No | 5 | 5 | Alive |
| OA GII | 34F | 1 st | −1p, −19q | −1p/19q | none | No | 55 | 55 | Alive |
| OA GII | 34M | 1 st | −1p, −14q, −19q | −1p/19q | none | No | 161 | 161 | Alive |
| OA GII* 16 | 35F | 1 st | −1p, −19q | −1p/19q | none | Yes | 52 | 102 | Alive |
| OA GII | 37M | 1 st | −1p, −14, −19q | −1p/19q | none | Yes | 35 | 35 | Alive |
| OA GII* 10 | 40F | 2 nd | +7 | another | p.Y220C | Yes | 22 | 80 | Alive |
| OA GII | 41M | 1 st | −4q26-ter, −5q21-ter, +7, −11q, −12q | another | none | No | 79 | 79 | Alive |
| OA GII | 41F | 1 st | −1p, −19q | −1p/19q | none | No | 43 | 43 | Alive |
| OA GII | 44F | 1 st | −1p, −14q, −19q | −1p/19q | none | No | 81 | 81 | Alive |
| OA GII | 45F | 1 st | −1p, +7, −19q | −1p/19q | none | No | 5 | 5 | Alive |
| OA GII | 48M | 1 st | −1p, −19q | −1p/19q | none | No | 39 | 39 | Alive |
| OA GIII* 15 | 28F | 2 nd | −13q22, +18p, +19, −X | another | p.H193Y | No | 4 | 85 | Alive |
| OA GIII* 17 | 29M | 1 st | −1p, −19q | −1p/19q | none | Yes | 49 | 68 | Alive |
| OA GIII* 9 | 32M | 3 rd | −1p, +3, −4, +5, +7, +9q, +10p, −10q, −13q, −15q11.2–22.3, +15q22.2-ter, −19q | −1p/19q | none | Yes | 11 | 111 | Alive |
| OA GIII* 17 | 34M | 2 nd | −1p, −19q | −1p/19q | none | No | 9 | 68 | Alive |
| OA GIII | 35M | 1 st | −1p, −4, −13, −18, −19q | −1p/19q | none | No | 40 | 40 | Alive |
| OA GIII | 36M | 1 st | −1p, −4, +11, −19q | −1p/19q | none | No | 116 | 116 | Alive |
| OA GIII | 37F | 1 st | −1p, −14q13-24, −19q | −1p/19q | none | No | 151 | 151 | Alive |
| OA GIII | 43M | 1 st | −1p, +1q12-32.1, −1q32.2-ter, +11, +17, +19p, −19q | −1p/19q | none | Yes | 44 | 64 | Dead |
| OA GIII | 44M | 1 st | −1p, −19q | −1p/19q | none | No | 11 | 11 | Alive |
| OA GIII | 74M | 1 st | +1q, +3, +4q12-24, −5q13.1–14, +9p, −14q, −18p | another | NA | No | 3 | 3 | Alive |
| O GII | 33M | 1 st | −1p, −14q22-24.3, −19q | −1p/19q | NA | No | 15 | 15 | Alive |
| O GII* 18 | 34M | 1 st | −1p, −14, −19q | −1p/19q | none | Yes | 63 | 69 | Alive |
| O GII | 52F | 1 st | −1p, −4, +7, +11q, −15q21-ter, −18q, −19q | −1p/19q | NA | No | 12 | 12 | Alive |
| O GII* 12 | 53M | 1 st | −1p, +7, −15q, −19q, +22q | −1p/19q | none | Yes | 104 | 134 | Alive |
| O GIII* 9 | 34M | 4 th | −1p, −4, −10q, −13q, −14q, −15qcen-21, +15q24-ter, −19q | −1p/19q | none | Yes | 9 | 111 | Alive |
| O GIII* 11 | 36F | 1 st | −1p, −19q | −1p/19q | none | Yes | 17 | 26 | Alive |
| O GIII* 6 | 36M | 2 nd | −1p, −17p, −18q, −19q | −1p/19q | none | No | 7 | 34 | Alive |
| O GIII* 18 | 39M | 2 nd | −1p, −14q, −19q, +21q | −1p/19q | none | No | 6 | 69 | Alive |
| O GIII* 16 | 40F | 2 nd | −1p, +11, −14, −19q | −1p/19q | none | No | 48 | 102 | Alive |
| O GIII* 19 | 57M | 2 nd | −1p, −14q13-24, −19q | −1p/19q | none | Yes | 12 | 85 | Alive |
| O GIII* 19 | 58M | 3 rd | −1p, +7, −14q21-24.3, −15q15-22.1, −19q | −1p/19q | none | Yes | 6 | 85 | Alive |
| O GIII | 68F | 1 st | −1p, +2, −3p, +3q, −4, +5, +7, +8, +9q, 13q, +14q, +17, −19q | −1p/19q | none | No | 21 | 21 | Alive |
The genetic type indicates detected the CNAs which are regarded as a favorable CNA (–1p/19q) or unfavorable CNAs (+7q, +8q, −9p, and/or −11p). A repeated number denoted by an asterisk indicates ta single patient who underwent multiple surgeries. Abbreviations: NA, not available.
Because TP53 mutations in IDH mutant gliomas were indicative of a poor prognosis and as +7q, +8q, −9p, and −11p were frequently observed in IDH mutant gliomas with TP53 mutations, we hypothesized that these CNAs were associated with a poor prognosis in patients with IDH mutant gliomas. Accordingly, patients with IDH mutant gliomas who harbored at least one of the abovementioned CNAs had a significantly worse prognosis than did patients with IDH mutant gliomas without these CNAs (p < 0.0001; Fig 1E and 1F). The median PFS was 31 months for patients with IDH mutant gliomas harboring +7q, +8q, −9p, and/or −11p compared with 78 months for all other IDH mutant gliomas (hazard ratio = 0.254; 95% CI: 0.128–0.506; p < 0.0001). The median OS for patients with IDH mutant gliomas who harbored these CNAs was 65 months, whereas the median OS for all others could not be defined (hazard ratio = 0.255; 95% CI: 0.111–0.586; p < 0.0001). In addition, a multivariate logistic regression analysis revealed that the 3-year recurrence rate was higher for patients with gliomas who harbored these CNAs than for patients with other types of gliomas (S1 Table). Therefore, +7q, +8q, −9p, and −11p should be considered negative prognostic factors in IDH mutant gliomas.
The CNAs +8q, −9p, −11p, and +12p are candidate markers for tumor progression in IDH mutant gliomas
In the present copy number analyses, gliomas with +7q were mainly detected in cases involving first surgeries. However, tumors harboring +8q, −9p, −11p, and +12p were frequently found after subsequent surgeries (Fig 3); +7q, +8q, −9p, −11p, and +12p emerged between the initial surgery and recurrent surgical interventions in 0, 2, 4, 3, and 4 cases of IDH mutant gliomas with TP53 mutations, respectively. Moreover, +7q was frequently detected in grade II gliomas, whereas +8q, −9p, −11p, and +12p were observed in high grade cases. These observations suggest that +7q is an early event, whereas +8q, −9p, −11p, and +12p may reflect tumor progression in IDH mutant gliomas with TP53 mutations. Among patients with IDH mutant gliomas with TP53 mutations, the median PFS was 31 months for gliomas harboring any one of +8q, −9p, −11p, or +12p compared with 47 months for patients harboring all other CNAs (n = 24; p = 0.067). On the other hands, the average MIB-1 indexes was 25.4% among cases harboring +8q, −9p, −11p, and/or +12p and 8.04% in gliomas without these CNAs (n = 31; p = 0.011). These results suggest that malignancy-related genes are present in these regions.
Discussion
From this study, we report two major findings. First, we have shown that copy number-independent genetic subgroups determined using IDH1/2 and TP53 as markers for Sanger sequencing could sufficiently substitute for genetic classification with 1p/19 co-deletions. Second, via a whole-chromosome CNA analysis of IDH mutant gliomas with TP53 mutations, we have clarified the CNAs that contribute to poor prognosis in patients with IDH mutant gliomas.
Previous studies have confirmed that specific genetic features including IDH mutation and 1p/19p co-deletions are excellent prognostic markers for gliomas [17, 18]. In the present study, we aimed to identify copy number-independent methods that would allow a widespread clinical application of genetic classification of gliomas. Several previous studies reported various prognostic genes identified via mutation analyses, and ATRX and TERT promoters have recently been recognized as prognostic markers of gliomas [6, 19, 20]. In the present study, we selected TP53 as a prognostic marker because gliomas with 1p/19q co-deletions and TP53 mutations were previously shown to be mutually exclusive [8]; accordingly, we hypothesized that IDH mutant gliomas with wild-type TP53 would predominantly harbor 1p/19q co-deletions. Our results support the previous finding that 1p/19q co-deletions and TP53 mutation are mutually exclusive. The survival curves for patients with gliomas carrying 1p/19q co-deletions were almost identical to those of patients with wild-type TP53, suggesting that wild-type TP53 is sufficiently indicative of 1p/19q co-deletions. In addition to the convenience of PCR-based TP53 mutation analysis, we are now investigating the relevance of prognosis, CNA, and the mutated TP53 exon for demonstrating the advantage of subgrouping according to TP53 mutation versus subgrouping according to 1p/19q co-deletions. IDH and TP53 mutant gliomas that carry +7q also tend to carry mutations in TP53 exon 5, suggesting that an exon 5 mutation is associated with a better prognosis in IDH and TP53 mutant gliomas comparing with other types of IDH and TP53 mutant gliomas. However, the sample size is extremely low, and we would need to increase the number of analyzed samples to support this conclusion.
As shown in Fig 1E and 1F, IDH mutant gliomas harboring any one of the CNAs +7q, +8q, −9p, or −11p were associated with a significantly worse survival when compared with other IDH mutant gliomas, indicating that these CNAs are negative prognostic factors for IDH mutant gliomas. Several studies previously reported that specific CNAs were candidate negative prognostic markers in gliomas. Our previous studies suggested that gliomas carrying +7q were more likely to be associated with a shorter PFS than were gliomas carrying −1p/19q; −9p was found to be a negative prognostic factor in grade II and III gliomas [1]. Moreover, Kitange et al. and Trost et al. indicated that +8q was associated with short survival durations in patients with oligodendrogliomas [21, 22], and recent studies have reported that −10q, −11p, and −19q were negative prognostic factors for low grade gliomas [16, 23]. Via a whole-chromosome analysis of CNAs for IDH mutant gliomas with TP53 mutations, we clarified that +7q, +8q, −9p, and −11p are unfavorable prognostic factors for IDH mutant gliomas. In addition, because +12p was unique to IDH mutant gliomas with TP53 mutations, we suspected that this CNA will be associated with poor survival in patients with IDH mutant gliomas. Accordingly, this CNA tended to emerge in cases involving recurrent surgical interventions or high grade gliomas (Figs 2 and 3). However, correlations between gliomas and +12p remain elusive. The chromosomal regions 7q, 8q, 9p, 11p, and 12p contain various oncogenes or tumor suppressor genes, including MET (7q31), MYC (8q24.21), CDKN2A (9p21), CDKN1C (11p15.5), and KRAS (12p12.1), and these genes might be associated with tumor progression in IDH mutant gliomas with TP53 mutations. The mechanisms underlying the associations of TP53 mutations with CNAs in the abovementioned specific regions remain unclear. p53 is a transcription factor that regulates target genes in response to DNA damage and is best known as a tumor suppressor gene [24]. Recent studies have correlated the absence of TP53 with chromosome segregation errors and chromosomal instability [25, 26], suggesting that TP53 mutations occur during the early phase of tumorigenesis in IDH mutant gliomas and cause chromosomal instability and gene dysregulation in specific regions such as 7q, 8q, 9p, 11p, or 12p. Further studies of these chromosomal changes may facilitate interpretations of tumor growth processes in IDH mutant gliomas with TP53 mutations.
Our results confirmed that most IDH mutant gliomas with TP53 mutations involve at least one of the CNAs +7q, +8q, −9p, and −11p, and that most IDH mutant gliomas with wild-type TP53 carry 1p/19q co-deletions. On the other hands, +7 and −10q are frequently detected in IDH wild-type gliomas [5]. These results suggest that gliomas can be separated into different lineage depending on IDH mutation, and IDH mutant gliomas are further separated into two distinct linages according to the TP53 mutation developing the specific CNAs in each lineage. As mentioned above, TP53 mutation did not affect the prognosis of patients with IDH wild-type gliomas. In a comparison of prognosis between IDH wild-type gliomas and primary glioblastomas, the median PFS (6 and 6 months, respectively) and median OS (17 and 15 months, respectively) were almost identical, suggesting that histological diagnosis can sufficiently predict prognosis in cases of primary glioblastomas.
Given the high recurrence rate among IDH mutant gliomas with TP53 mutations, efforts are required to prevent progression to high grade gliomas or secondary glioblastomas, which are difficult to control with multidisciplinary treatments. To this end, studies are in progress now using OncoScan arrays (Affymetrix) for this type of glioma to identify specific regions with common losses, gains, or high copy number gains, and consequent changes in gene expression. In addition, some patients with 1p/19q co-deleted gliomas developed recurrence within a few years and these gliomas lacked TP53 mutations, suggesting the presence of other genes that contribute to a poor prognoses in patients with IDH mutant gliomas.
In this study, we showed that PCR-based mutation analyses using IDH1/2 and TP53 as markers could rapidly and simply classify glioma with prognostic relevance. Although pathological diagnoses facilitate evaluations of malignancy at the time of surgery, genetic classifications provide better prognostic predictions, particularly in cases of WHO grade II and III gliomas. Specifically, IDH mutant gliomas carrying at least one of the CNAs +7q, +8q, −9p, or −11p were associated with a shorter survival and were predominantly associated with TP53 mutations. In conclusion, both pathological and genetic classifications are essential for glioma diagnosis and the present observations could be used to facilitate genetic classification.
Supporting Information
(TIF)
(DOCX)
Acknowledgments
The authors thank all the participating patients. We also thank Mrs. Fujiko Sueishi and Mrs. Misa Matsudo for technical support as well as Dr. Hideo Hagihara and Dr. Takema Kato for technical advice.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The authors received no specific funding for this work.
References
- 1. Hirose Y, Sasaki H, Miwa T, Ohba S, Ikeda E, Abe M, et al. Whole genome analysis from microdissected tissue revealed adult supratentorial grade II-III gliomas are divided into clinically relevant subgroups by genetic profile. Neurosurgery. 2011; 69: 376–90. 10.1227/NEU.0b013e318212bcd8 [DOI] [PubMed] [Google Scholar]
- 2. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321: 1807–12. 10.1126/science.1164382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360: 765–73. 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hartmann C, Hentschel B, Simon M, Westphal M, Schackert G, Tonn JC, et al. Long-term survival in primary glioblastoma with versus without isocitrate dehydrogenase mutations. Clin Cancer Res. 2013; 19: 5146–57. 10.1158/1078-0432.CCR-13-0017 [DOI] [PubMed] [Google Scholar]
- 5. Hirose Y, Sasaki H, Abe M, Hattori N, Adachi K, Nishiyama Y, et al. Subgrouping of gliomas on the basis of genetic profiles. Brain Tumor Pathol. 2013; 30: 203–8. 10.1007/s10014-013-0148-y [DOI] [PubMed] [Google Scholar]
- 6. Killela PJ, Pirozzi CJ, Healy P, Reitman ZJ, Lipp E, Rasheed BA, et al. Mutation in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget. 2014; 5: 1515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huse JT, Aldape KD. The evolving role of molecular markers in the diagnosis and management of diffuse glioma. Clin Cancer Res. 2014; 20: 5601–11. 10.1158/1078-0432.CCR-14-0831 [DOI] [PubMed] [Google Scholar]
- 8. Ueki K, Nishikawa R, Nakazato Y, Hirose T, Hirato J, Funada N, et al. Correlation of histology and molecular genetic analysis of 1p, 19q, 10q, TP53, EGFR, CDK4, and CDKN2A in 91 astrocytic and oligodendroglial tumors. Clin Cancer Res. 2002; 8: 196–201. [PubMed] [Google Scholar]
- 9. Walker DR, Bond JP, Tarone RE, Harris CC, Makalowski W, Boguski MS, et al. Evolutionary conservation and somatic mutation hotspot maps of p53: correlation with p53 protein structural and functional features. Oncogene. 1999; 19: 211–8. [DOI] [PubMed] [Google Scholar]
- 10. Holstege H, Joosse SA, van Oostrom CT, Nederlof PM, de Vries A, Jonkers J. High incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Cancer Res. 2009; 69: 3625–33. 10.1158/0008-5472.CAN-08-3426 [DOI] [PubMed] [Google Scholar]
- 11. Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum Mutat. 2014; 35: 672–88. 10.1002/humu.22552 [DOI] [PubMed] [Google Scholar]
- 12. Vegran F, Rebucci M, Chevrier S, Cadouot M, Boidot R, Lizard-Nacol S. Only missense mutations affecting the DNA binding domain of P53 influence outcomes in patients with breast carcinoma. PLOS one. 2013; 8: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Watanebe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009; 174: 1149–53. 10.2353/ajpath.2009.080958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koga Y, Yasunaga M, Moriya Y, Akasu T, Fujita S, Yamamoto S, et al. Detection of the DNA point mutation of colorectal cancer cells isolated from feces stored under different conditions. Jpn J Clin Oncol. 2009; 39: 62–9. 10.1093/jjco/hyn129 [DOI] [PubMed] [Google Scholar]
- 15. Solis OE, Mehta RI, Lai A, Mehta RI, Farchoukh LO, Green RM, et al. Rosette-forming glioneuronal tumor: a pineal region case with IDH1 and IDH2 mutation analyses and literature review of 43 cases. J Neuro Oncol. 2011; 102: 477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alentorn A, van Thuijl HF, Marie Y, Alshehhi H, Carpentier C, Boisselier B, et al. Clinical value of chromosome arms 19q and 11p losses in low grade gliomas. Neuro Oncol. 2014; 16: 400–8. 10.1093/neuonc/not227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998; 90: 1473–9. [DOI] [PubMed] [Google Scholar]
- 18. McLendon RE, Herndon JE 2nd, West B, Reardon D, Wiltshire R, Rasheed BK, et al. Survival analysis of presumptive prognostic markers among oligodendrogliomas. Cancer. 2005; 104: 1693–9. [DOI] [PubMed] [Google Scholar]
- 19. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012; 3: 709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J pathol. 2012; 226: 7–16. 10.1002/path.2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kitange G, Misra A, Law M, Passe S, Kollmeyer TM, Maurer M, et al. Chromosomal imbalances detected by array comparative genomic hybridization in human oligodendrogliomas and mixed oligoastrocytomas. Genes Chromosomes Cancer. 2005; 42: 68–77. [DOI] [PubMed] [Google Scholar]
- 22. Trost D, Ehrler M, Fimmers R, Felsberg J, Sabel MC, Kirsch L, et al. Identification of genomic aberrations associated with shorter overall survival in patients with oligodendroglial tumors. Int. J. Cancer. 2007; 120: 2368–76. [DOI] [PubMed] [Google Scholar]
- 23. van Thuijl HF, Scheinin I, Sie D, Alentorn A, van Essen HF, Cordes M, et al. Spatial and temporal evolution of distal 10q deletion, a prognostically unfavorable event in diffuse low-grade gliomas. Genome Biol. 2014; 15: 471 10.1186/s13059-014-0471-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shatz M, Menendez D, Resnick MA. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 2012; 72: 3948–57. 10.1158/0008-5472.CAN-11-4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Carcer G, Malumbres M. A centrosomal route for cancer genome instability. Nat. Cell Biol. 2014; 16: 504–6. 10.1038/ncb2978 [DOI] [PubMed] [Google Scholar]
- 26. Nam HJ, van Deursen JM. Cyclin B2 and p53 control proper timing of centrosome separation. Nat. Cell Biol. 2014; 16: 535–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.