

Abstract
Leishmania donovani, a protozoan parasite, causes the disease visceral leishmanisis (VL), characterized by inappropriate CD8+ T-cell activation. Therefore, we examined whether the Toll-like Receptor 2 (TLR2) ligand Ara-LAM, a cell wall glycolipid from non-pathogenic Mycobacterium smegmatis, would restore CD8+ T-cell function during VL. We observed that by efficient upregulation of TLR2 signaling-mediated NF-κB translocation and MAPK signaling in CD8+ T-cells (CD25+CD28+IL-12R+IFN-γR+), Ara-LAM triggered signaling resulted in the activation of T-bet, which in turn, induced transcription favourable histone modification at the IFN-γ, perforin, granzyme-B promoter regions in CD8+ T-cells. Thus, we conclude that Ara-LAM induced efficient activation of effector CD8+ T-cells by upregulating the expression of IFN-γ, perforin and granzyme-B in an NF-κB and MAPK induced T-bet dependent manner in VL.
Introduction
Visceral leishmaniasis caused by the protozoan parasite, Leishmania donovani, is fatal, if untreated. Dysfunctions of macrophages and T-cells during VL result in severe immunosuppression [1–3]. CD4+ T-cells activation is essential for IFN-γ-mediated protection against leishmaniais [4] whereas CD8+ T-cells confer protection via perforin, granzyme-B-mediated direct killing of Leishmania-infected host cells [5–6]. L. donovani induces functional exhaustion of CD8+ T-cells [7]. Activation of Leishmania-specific CD8+ T-cells is indispensable for clearance of the pathogen. For naive CD8+ T cells’ antigen-specific activation and differentiation, signals from T-cell receptor, CD28, IL-12-induced IFN-γ and TLR2 are reqiured [8–12]. The transcription factor T-bet promotes naive CD8+ T-cell differentiation to effector cytotoxic T lymphocytes (CTL) [13–14], expressing CD25 (IL-2R-α), IL-12 receptor (IL-12R) and IFN-γR.
TLR2, forming heterotypic associations with TLR1 or TLR6, recognizes triacetylated or diacetylated lipopeptides, respectively [15–17]. Ara-LAM, a cell wall glycolipid of Mycobacterium smegmatis, has been reported to confer protection against leishmanial pathogenesis via TLR2-dependent induction of the proinflammatory responses [18]. Ara-LAM–induced activation of p38 MAPK signalling in Leishmania infected macrophages shifts their Th2 phenotype towards Th1 via chromatin modification at various proinflammatory cytokine gene loci [19]. However, it was unclear if Ara-LAM would modulate TLR2 signalling in CD8+ T-cells, which might play a potential role in the regression of leishmanial pathogenesis.
In this study, we have demonstrated that Ara-LAM drives the activation of CD8+ T-cells, which are CD25+CD28+IL-12R+IFN-γR+ and specifically destroys the L. donovani-infected macrophages. Ara-LAM enhances T-bet expression in CD8+ T-cells via upregulation of NF-κB and p38MAPK in a TLR2-dependent pathway. T-bet ultimately enhances the expression of IFN-γ, perforin, and granzyme-B in CD8+ T-cells via histone modifications at their respective promoter regions to restore host-protective CD8+ T-cell responses.
Materials and Methods
Reagents and chemicals
RPMI-1640 medium, penicillin and streptomycin, SB203580 (p38MAP Kinase inhibitor), SN50 (NF-κB inhibitor) were from Sigma (St. Louis, MO, USA). dNTPs, Revert Aid M-MuLV Reverse Transcriptase, oligo dT, RNase inhibitor and others for cDNA synthesis were from Fermentas (Ontario, Canada). Phospho-H3 and acetyl-H3 Abs were from Abcam (Cambridge, UK) and chromatin immunoprecipitation (ChIP) assay kit was from Millipore (Bedford, MA). TLR2, MyD88, IRAK 1, NF-κB, p38, phospho-p38, ERK1/2, phospho-ERK1/2, T-bet, β-Actin antibodies were from Santa Cruz Biotechnology (Texas, USA). Ara-LAM was isolated as previously described [20]. Lipopolysaccharide contamination (<25 ng/mg) was checked by the Limulus test. All antibodies for FACS were from BD Biosciences (San Diego, USA).
Animals and parasites
L. donovani strain AG-83 (MHOM/IN/1983/AG-83) was maintained in vitro in Medium-199 (Sigma, St. Louis, MO) with 10% FCS (Gibco-BRL) and virulence was maintained by passage through BALB/c mice. Stationary-phase promastigotes obtained by suitable transformation were used for experiments [21]. BALB/c mice were infected with stationary phase L. donovani promastigotes (i.v., 2×107/mouse). BALB/c mice (6–8 weeks, NCLAS, Hydrabad, India) were divided into the following experimental groups: (1) control (receiving PBS); (2) infected (receiving L. donovani); (3) Infected and Ara-LAM–treated infected (Ara-LAM 30μg/kg body weight-injected 2 days prior to infection); (4) control shRNA and (5) TLR2 shRNA (TLR2 shRNA or control shRNA [bearing scrambled sequence] treatment 72h before Ara-LAM treatment). Mice were sacrificed on 14 and 28 days after infection by cervical dislocation method as mentioned by Institutional Animal Ethical Committee (Bose Institute), bearing a registration number: 1796/PO/ERe/S/14/CPCSEA. This study followed the Institutional Animal Ethical Committee approval. L. donovani infection was expressed in Leishman-Donovan units.
Isolation and purification of macrophages and CD8+ T-cells
Thioglycolate-elicited (i.p., 4% w/v, 1.0 ml/mouse) macrophages from different experimental groups of BALB/c mice were infected with stationary phase Leishmania promastigotes at a ratio of 1:10 [22]. Splenic CD8+ T-cells (purity >99% as ascertained by FACS) from the indicated mice were isolated by positive selection using CD8+ IMag beads, according to the manufacturer’s instructions (BD Biosciences). CD8+ T-cells were cultured in RPMI-1640 with plate-bound anti-CD3ε (5μg/mL) and CD28 (1μg/mL).
Preparation of TLR2 and T-bet-specific siRNA
TLR2 and T-bet-specific siRNA were synthesized using the Silencer siRNA Construction kit (Ambion). Scrambled siRNA was synthesized with the similar GC content. Silencing primers are listed in the Table 1.
Table 1. Sequences of the PCR primers.
| Gene | Sequences |
|---|---|
| TLR2 | FP 5’- TCTGGGCAGTCTTGAACATTT -3’ |
| RP 5’- AGAGTCAGGTGATGGATGTCG -3’ | |
| IFN-γ | FP 5’- AGCTCTTCCTCATGGCTGTTTC -3’ |
| RP 5’- TGTTGCTGATGGCCTGATTGT -3’ | |
| Perforin | FP 5’- CTGAGCGCCTTTTTGAAGTC -3’ |
| RP 5’- AAGGTAGCCAATTTTGCAGC -3’ | |
| Granzyme-B | FP 5’- CTCTCGAATAAGGAAGCCCC -3' |
| RP 5’- CTGACCTTGTCTCTGGCCTC -3' | |
| T-bet | FP 5’- CCTCTTCTATCCAACCAGTATC -3’ |
| RP 5’- CTCCGCTTCATAACTGTGT -3’ | |
| IFN-γ promoter | FP 5’- GAGAAATTCACATTACAAGGGC -3’ |
| RP 5’- TTAAGATGGTGACAGATAGGTGG -3’ | |
| Perforin promoter | FP 5’- GTACTAGCCTGCTCAAACCT -3’ |
| RP 5’- CTAATCACAGTGTCCCATGAG -3’ | |
| Granzyme promoter | FP 5’- ATGCTCCTGATTACCCTCAC -3’ |
| RP 5’- CAGAGAACCACCACTTACAG -3’ | |
| TLR-2 siRNA | FP 5’-AAAGAGAAAGTACTTACTGCACCTGTCTC-3’ |
| RP 5’- AATGCAGTAAGTACTTTCTCTCCTGTCTC -3’ | |
| T-bet siRNA | FP 5’- AAACAAACATCCTGTAATGGCCCTGTCTC -3’ |
| RP 5’-AAGCCATTACAGGATGTTTGTCCTGTCTC-3’ | |
| GAPDH | FP 5’- GTTGTCTCCTGCGACTTCAACA -3’ |
| RP 5’- TCTCTTGCTCAGTGTCCTTGCT -3’ |
Flow cytometry
CD8+ T-cells from differently treated mice groups were stained with PE-labeled TLR2, IFN-γ, IFN-γR, IL-12R, CD28 or IL-10, APC-Cy7 labelled CD25, FITC-lebelled IFN-γ. For intracellular cytokine staining, brefeldin A (10μg/mL) was added 4h prior to harvest, fixed, and permeabilized (0.1% saponin) and stained with anti-IFN-γ-PE, anti-perforin-PE and anti-granzyme-B-PE antibodies. Cells were analyzed using a FACS Verse flow cytometer.
Isolation of RNA and Reverse Transcriptase polymerase chain reaction
Total RNA from purified CD8+ T-cells were extracted using TRI reagent using standard protocol [23]. The total RNA was reverse transcribed using Revert Aid M-MuLV reverse transcriptase (Fermentas). GAPDH was used as a loading control. Sequences of the PCR primers are given in the Table 1.
CD8+ T-cell proliferation assay
Splenic CD8+ T-cells were cultured with autologus infected macrophages (10:1) for 72h and labellled with [3H]-thymidine (1μCi/105 cells, JONAKI, DAE) for 18h before harvesting. [3H]-thymidine incorporation was determined using a liquid scintillation counter (Tri-Carb 2100TR; Packard Instrument) [24].
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were conducted using the ChIP Assay kit following the manufacturers protocol. Purified CD8+ T-cells (1×106) from the indicated mice were co-cultured with autologous L. donovani–infected macrophages (1×105) for 45 min, paraformaldehyde(1%)-fixed for 10 min at 37°C and washed with ice-cold PBS containing 1mM PMSF, harvested and lysed in SDS lysis buffer. DNA was sheared by ultrasonication using a High Intensity Ultrasonic Processor (Hielscher, Teltow, Germany) for 3 × 10s pulses at 20% amplitude. Lysates were cleared by centrifugation and diluted in ChIP dilution buffer. Lysates were pre-cleared using protein A-agarose and a sample of “input DNA” was collected at this point. Protein-DNA complexes were immunoprecipitated with 5μg of antibodies (phospho H3, acetyl H3, T-bet) overnight at 4°C. Antibody-protein-DNA complexes were then captured using protein A-agarose for 1 h at 4°C. After washing beads with low and high salt, LiCl, and TE buffers, the protein/DNA complexes were eluted in buffer (1%SDS, 0.1M NaHCO3). DNA was then extracted and precipitated. PCR was conducted using promoter specific primers (Table 1).
Preparation of nuclear and cytoplasmic extracts
The nuclear extracts were prepared from CD8+ T-cells as described previously [25]. Briefly, cells were resuspended in ice-cold hypotonic buffer [10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.2 mM PMSF, and 0.5 mM DTT] for 10 min, homogenized and the nuclei were precipitated (3300×g, 5 min, 4°C). The supernatant was used as the cytoplasmic extract. The nuclear pellet was extracted in ice-cold nuclear extraction buffer [20mM HEPES (pH 7.9), 0.4M NaCl, 1.5mM MgCl2, 0.2mM EDTA, 25% glycerol, 0.5mM PMSF and 0.5mM DTT] for 30min (12,000×g for 30 min, 4°C). The supernatant was used as nuclear extract.
Preparation of cell lysate and immunoblot analysis
CD8+ T-cells were lysed using lysis buffer for isolation of protein [26]. Equal amounts of protein (30μg) were subjected to 10% SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was blocked overnight with 3%BSA in Tris-saline buffer (pH 7.5), and immunoblotting was performed to detect MyD88, IRAK 1, NF-κB, T-bet, β-Actin, GAPDH, and phosphorylated or total p38MAPK and ERK1/2, as described previously [27].
Coimmunoprecipitation
In coimmunoprecipitation studies, the lysates of differently treated CD8+ T-cells were incubated with specific antibody (TLR2, MyD88). The complexes were captured with immobilized Protein A-agarose beads, washed, resolved by 10% SDS-PAGE and developed with antibodies to MyD88 and IRAK 1 to detect TLR2-MyD88, MyD88-IRAK 1 interactions [28].
Statistical analysis
All experiments were performed in triplicate and a minimum of 4 mice was used per group. The data, represented as mean values ± SD, are from one experiment that was performed at least 3 times. One-way ANOVA test (using a statistical package, Instat3) was employed to assess the significance of the differences. p≤0.001 was considered significant.
Results
Ara-LAM upregulates IL-12R and IFN-γR in CD8+ T-Cells in L. donovani infection
We studied the effect of Ara-LAM on BALB/c mice-derived CD8+ T-cells in indicated groups. Naïve CD8+ T cells proliferate in response to TCR and CD28 signals, but reqiure IFN-γ and IL-12 to develop effector functions [29–30]. We investigated the status of CD28 on CD8+ T cells expressing CD25, receptor for IL-12 (IL-12R) and IFN-γ (IFN-γR) [31–32]. 28 days after infection, compared to the splenic CD8+ T cells of untreated infected mice, Ara-LAM strongly induced the expression of IL-12R and a moderate induction of IFN-γR on splenic CD8+ T cells, co-expresseing CD25 (Fig 1A). Activation of TLR2 in CD8+ T-cells is associated with their enhanced effecter functions [18–19]. Therefore, we tested whether Ara-LAM, being a TLR2 ligand, could activate the CD8+ T-cells by upregulating the transcription of perforin and granzyme-B. We observed a significant enhancement in both perforin and granzyme-B expression in CD8+ T-cells isolated from Ara-LAM treated L. donovani infected mice compared to that of untreated infected mice (Fig 1B).
Fig 1. Characterization of CD8+ T cells at 28 days postinfection upon Ara-LAM treatment in Leishmania donovani infected BALB/c mice.
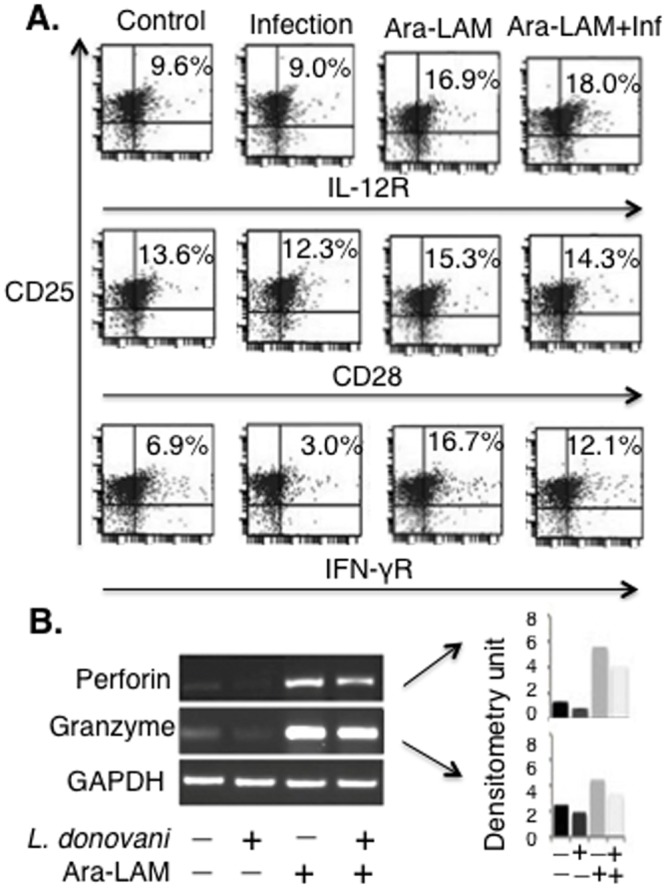
(A) CD8+ T from differently treated BALB/c mice 28 days postinfection were subjected to FACS analyis to check the expression of CD25+IL12R+, CD25+CD28+, CD25+IFN-γR+ cells. Data are from one of three representative experiments. (B) In separate set of experiment, CD8+ T cells from differently treated mice group were isolated and cultured in presence of plate-bound anti-CD3ε mAbs (5μg/mL) and CD28 (1μg/mL) and expresion of perforin and granzyme-B was done by conventional RT PCR. Data are from one of three representative experiments.
Ara-LAM-induced CD8+ T-cells activation in L. donovani infection is TLR2-dependent
We examined the effect of Ara-LAM treatment on TLR2 surface expression in CD8+ T-cells from different groups of BALB/c mice. Ara-LAM treatment significantly augmented the expression of TLR2 in splenic CD8+ T-cells on 14 and 28days post infection (Fig 2A). Because we observed significantly enhanced expressions of IFN-γ, perforin and granzyme-B in CD8+ T-cells isolated from Ara-LAM treated L. donovani infected mice compared to that of untreated infected mice (Fig 2A), we tested if TLR2 silencing could abrogate these effector functions. TLR2 silencing abrogated the Ara-LAM induced generation of IFN-γ, perforin, granzyme-B molecules in CD8+ T-cells isolated from the infected mice (Fig 2A and 2B).
Fig 2. Ara-LAM facilitates TLR2 dependent activation and expansion of CD8+ T-cells in Leishmania donovani infected BALB/c mice.

(A) Purified CD8+ T-cells were subjected to FACS analysis for TLR2 expression. Separately, purified CD8+ T-cells from differently treated mice were co-cultured with autologous infected macrophages (10:1) for 48hrs and IFN-γ, perforin, granzyme-B expression were determined by intracellular FACS. (B) CD8+ T-cells from differently treated mice groups were stimulated as described previously and conventional RT PCR was done after RNA extraction. (C) Purified CD8+ T-cells from differently treated mice and autologous L. donovani–infected macrophages were co-cultured for 72 hours. Proliferation was determined by an 18 h [3H] thymidine incorporation assay. Data were presented as count/million (×103). Results were mean value ±SD. from triplicate wells. The asterisk indicated a statistically significant induction (**P<0.001) of T-cell proliferation, compared with that in infected mice.
It has been noted earlier that Leishmania infection of the susceptible host results in apoptosis of T-cells, leading to impairment of cell-mediated immunity [33]. Therefore, we investigated whether Ara-LAM could restore the impaired CD8+ T-cell proliferation in Leishmania-infected mice. Ara-LAM treatment resulted in significant enhancement of splenic CD8+ T-cell proliferation compared to the splenic CD8+ T-cells from untreated infected mice. The Ara–LAM–induced CD8+ T-cell proliferation was significantly attenuated during TLR2 silencing condition (Fig 2C). These results suggested that Ara-LAM stimulation leads to the enhanced effector function as well as the proliferation of CD8+ T-cells via TLR2 dependent pathway.
Ara-LAM triggers T-bet recruitment and histone modification at the IFN-γ, Perforin and Granzyme-B promoter regions in CD8+ T-cells
The expressions of IFN-γ, perforin and granzyme-B in CD8+ T cells is principally regulated at the level of transcription, which, in turn, is strictly dependent upon the favourable histone modifications at their respective promoter regions [19]. Therefore, we performed ChIP assays to investigate whether the Ara-LAM-mediated enhancement of IFN-γ, perforin and granzyme-B expression in CD8+ T-cells was due to chromatin modification at their respective promoter regions. We observed a significantly higher level of phosphorylated (Fig 3A) and acetylated (Fig 3B) histones at the promoter regions of IFN-γ, perforin, granzyme-B in the splenic CD8+ T-cells of Ara-LAM treated L. donovani infected BALB/c mice relative to the splenic CD8+ T-cell from untreated infected mice. These Ara-LAM mediated histone modifications at the IFN-γ, perforin and granzyme-B promoter regions in CD8+ T-cells were significantly attenuated in TLR2 silenced condition (Fig 3A and 3B). Therefore, Ara-LAM induced transcription favourable histone modifications at the IFN-γ, perforin, and granzyme-B loci of CD8+ T-cells in a TLR2 dependent pathway.
Fig 3. Histone H3 modifications at the IFN-γ, Perforin, Granzyme-B promoter of CD8+ T-cells in different groups of BALB/c mice.

(A-B) CD8+ T cells from differently treated mice groups were co-cultured with autologous infected macrophages for 45 min and chromatin immunoprecipitation (ChIP) assays were conducted. Immunoprecipitations were performed using Abs specific to phosphorylated H3 histone (IP: phospho-H3), acetylated H3 histone (IP: acetyl-H3) and conventional RT PCR was performed using primers specific to the IFN-γ, perforin and granzyme-B promoter region. (C) CD8+ T-cells were co-cultured with infected macrophages, lysed and the nuclear protein extracts were analyzed for the activation of T-bet by Western blot. (D) CD8+ T-cells from differently treated mice group were co-cultured with autologous L. donovani–infected macrophages, Immunoprecipitations were conducted using T-bet (IP: T-bet) specific Abs. Conventional RT-PCR was performed for amplifying the putative T-bet binding sites of the IFN-γ, perforin, granzyme-B promoter. Data represented were one of the three indepenedent experiments with similar results performed in the same way.
The effector functions of CD8+ T-cells are predominantly regulated by the transcription factor T-bet [34]. Therefore, we investigated the accumulaion of T-bet in the splenic CD8+ T-cells of different experimental mice groups. Immunoblot analysis showed that accmulation of T-bet was significantly higher in the splenic CD8+ T-cells of Ara-LAM treated L. donovani infected mice compared to that of the untreated infected mice (Fig 3C).
T-bet binding at the promoter regions of IFN-γ, perforin, granzyme-B is a crucial event for the optimal induction of these effectors molecules in CD8+ T-cells [13]. Therefore, we performed ChIP-on-ChIP assay to investigate the interaction of T-bet with the promoter regions of IFN-γ, perforin, and granzyme-B in CD8+ T-cells from Ara-LAM treated and untreated L. donovani infected BALB/c mice. Ara-LAM treatment induced a strong association of T-bet with the IFN-γ, perforin, and granzyme-B promoter regions in CD8+ T-cells compared with that of the untreated L. donovani infected mice; TLR2 silencing decreased Ara-LAM induced T-bet recruitment to the promoter regions of IFN-γ, perforin, granzyme-B in CD8+ T-cells (Fig 3D). Collectively, Ara-LAM-induced TLR2 dependent activation of T-bet augmented the transcription of target effector genes in CD8+ T-cells of infected mice.
Ara-LAM promotes NF-κB activation and MAPK signaling in CD8+ T-cells
Ara-LAM triggers the TLR2 mediated downstream signaling in host macrophages [18–19]. However, Ara-LAM induced modulation of TLR2 signaling in CD8+ T-cells has not been explored till date. The association between TLR2 and MyD88 is a crucial event for the initiation of TLR2 downstream signaling [18]. Therefore, we carried out co-immunoprecipitation studies to investigate the TLR2-MyD88 interaction in Ara-LAM–treated CD8+ T-cell. We observed a strong association between TLR2 and MyD88 in Ara-LAM–treated CD8+ T-cell compared with that of the untreated CD8+ T-cells. IRAK-1, crucial for activation of TLR2 downstream signaling [18], was found to be intricately associated with MyD88 in Ara-LAM–treated CD8+ T-cells compared to the untreated CD8+ T-cells (Fig 4A). Furthermore, nuclear translocation of NF-κB were significantly augmented in Ara-LAM treated CD8+ T-cell compared to the untreated CD8+ T-cells. Ara-LAM induced TLR2 downstream signaling mediated NF-κB translocation was completely abrogated under TLR2 silenced condition (Fig 4B).
Fig 4. Ara-LAM activates TLR2 signalling via activation of NF-κB and p38 MAPK in naive CD8+ T-cells.

(A) Purified CD8+ T-cells (1×106/mL) were stimulated with plate-bound anti-CD3ε mAbs (5μg/mL) and CD28 (1μg/mL) for 24 hrs and transfected with control siRNA or TLR2 siRNA, followed by Ara-LAM (3μg/mL) treatment for 24 hr. The cells were then lysed and subjected to immunoprecipitation with anti-TLR2 antibody, and the blots were probed with anti-MyD88 antibody. (B) Cells were lysed and immunoprecipitated with anti-MyD88 antibody; the blots were probed with anti-IRAK1 antibody. Cytosolic and nuclear protein extracts were analyzed for nuclear translocation of NF-κB. (C-D) In yet separate experiments, CD8+ T cells were treated by Ara-LAM for 5, 15, 30, and 60 min, and lysed. The lysate was subjected to Western blot analysis for the expression of p38MAPK, phospho-p38MAPK and ERK1/2, phospho-ERK1/2. Data represented were one of the three indepenedent experiments with similar results performed in the same way.
In addition to the NF-κB activation, TLR2 signaling can also modulate p38 and ERK-1/2 MAPK signaling cascades [19]. Therefore, we investigated whether Ara-LAM treatment could induce MAPK phosphorylation in CD8+ T-cells. Ara-LAM treatment resulted in significantly higher level of p38MAPK phosphorylation (Fig 4C), however; it failed to induce significant ERK1/2 phosphorylation in CD8+ T-cells (Fig 4C). TLR2 silencing abrogated the Ara-LAM induced p38 phosphorylation (Fig 4D). Taken together, Ara-LAM activated p38MAPK and NF-κB activation in infected CD8+ T-cells.
Ara-LAM enhances T-bet expression in CD8+ T-cells via upregulation of NF-κB and p38MAPK signaling
Since, Ara-LAM induced efficient activation of T-bet in CD8+ T-cells (Fig 3C) and enhanced the activation of NF-κB and p38MAPK in CD8+ T-cell (Fig 4), we hypothesized that T-bet activation in CD8+ T-cells might be regulated either by the NF-κB or MAPK signalling or by both of these signalling molecules. Ara-LAM mediated activation as well as mRNA expression of T-bet was attenuated in the presence of SB203580 and SN50, the pharmacological inhibitors of p38MAPK and NF-κB, respectively (Fig 5A and 5B), suggesting that the activation of T-bet in Ara-LAM treated CD8+ T-cells was regulated by TLR2 triggered NF-κB and p38MAPK signalling cascade. Besides, the enhanced IFN-γ, perforin and granzyme-B expression in Ara-LAM stimulated CD8+ T-cells were significantly decreased during T-bet silencing, as well as, NF-κB and the p38MAPK inhibition (Fig 5B). Therefore, the upregulation of IFN-γ, perforin and granzyme-B expression in Ara-LAM stimulated CD8+ T-cells were due to the NF-κB and the p38MAPK dependent activation of T-bet.
Fig 5. Ara-LAM promptly regulates effector functions in CD8+ T-cells through NF-κB and p38MAPK mediated T-bet signalling.

(A) CD8+ T-cells were isolated by MACS from the spleen BALB/c mice. Purified CD8+ T cells were stimulated as described previously and allowed to transfect with control siRNA or T-bet siRNA, or treated with SB203580 (SB) (5μg/ml), or SN50 (SN) (20μg/ml), subsequently followed by Ara-LAM (3μg/mL) treatment for 24 hr. The cells were then lysed and nuclear protein extracts were prepared, followed by subjected to Western blot with anti-T-bet. (B) The blot shown is representative of triplicate experiments that yield similar type of results. In a separate set of experiments, after the treatment schedule, the cells were collected in Trizol for RNA extraction, and conventional RT PCR analysis was performed to determine the expression of T-bet, IFN-γ, perforin, granzyme-B. Data represented were one of the three indepenedent experiments with similar results performed in the same way.
Ara-LAM reduces hepatic and splenic parasitic burden in BALB/c mice
We have observed that Ara-LAM activates the CD8+ T cells via upregulation of IFN-γ and the cytotoxic molecules perforin and granzyme-B. These activated T cells have the ability to kill the infected macrophages to reduce the parasite burden [5–6]. In accordance with this phenomenon, we observed a significant reduction of the hepatic as well as splenic parasite burden in the Ara-LAM treated infected mice groups compared to the untreated infected mice groups (Fig 6) on 14 and 28days postinfection. However, Ara-LAM mediated clearance of parasites was significantly attenuated in TLR2 silenced condition. Collectively, the data clearly suggest that Ara-LAM plays a very important role to clear the L. donovani infection via a TLR2 dependent mechanism.
Fig 6. Ara-LAM treatment in L. donovani infected BALB/c mice showed reduced parasite burden.
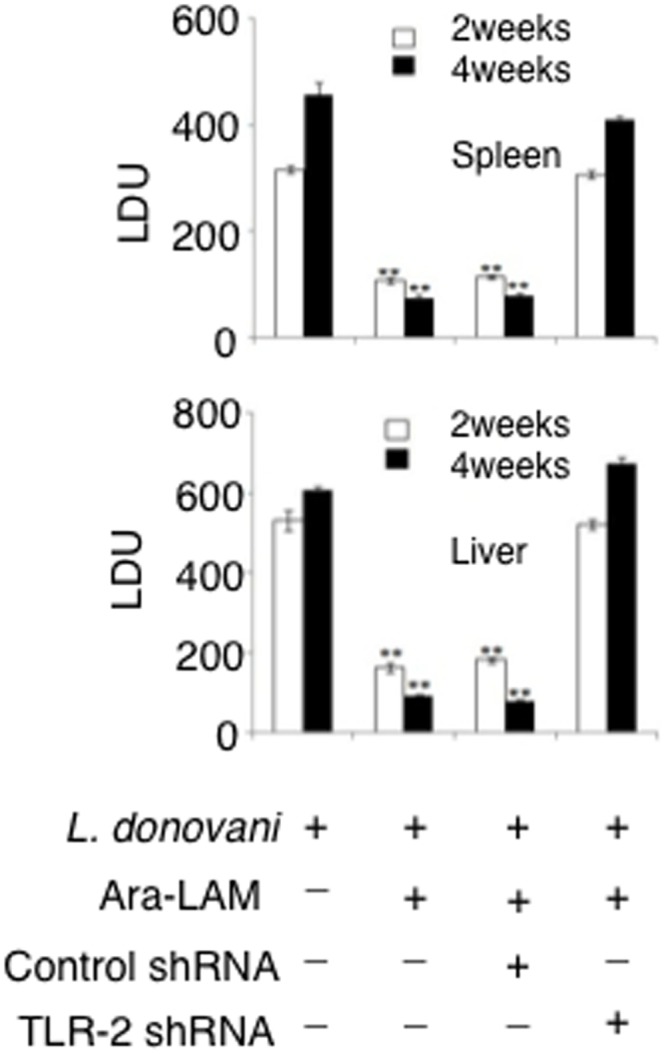
Mice were treated as described in Fig 1 legend. Mice from different groups were sacrificed on day 14 and 28 post-infection. The parasite burden in liver and spleen were expressed in Leishman-Donovan units (LDUs). Results were for 3 independent experiments and represented the mean values ± SD for four animals per group. **P<0.001 for the comparison with infected mice.
Reversion of the Th subset expansion in L. donovani infected mice
We checked the effect of Ara-LAM on IFN-γ and IL-10 producing-CD8+ T-cells in both spleen and liver by flow cytometry. In both uninfected and infected mice, Ara-LAM up-regulated the IFN-γ secreting CD8+ T-cells in spleen as well as liver. IL-10 producing CD8+ T-cells were suppressed by Ara-LAM pretreatment in both spleen and liver of infected as well as uninfected mice. Although in case of hepatic CD8+ T-cells, the effect of Ara-LAM on both IFN-γ and IL-10 secretion was not very significant (Fig 7).
Fig 7. Ara-LAM up-regulates IFN-γ secreting CD8+ T-cells in an organ-dependent manner during L. donovani infection.

28days post-infection, different groups of mice were sacrificed. Splenocytes and hepatocytes were stimulated with soluble leishmanial antigen (SLA, 10 μg/mL) for 48hrs. Before harvesting, cells were incubated with brefeldin A (10 μg/mL) for 4 hrs, CD8+ T cells were MACS sorted (see Materials and Methods), permeabilized (0.1% saponin) and stained with anti-mouse IFN-γ-FITC and anti-mouse IL-10-PE antibodies and were analyzed by flow cytometry. Data are from 1 of 3 experiments conducted in the same way with similar results.
Discussion
Leishmanial pathogenesis is associated with abrogated pro-inflammatory responses, resulting in immune suppression of the host [9–11]. The persistence of the infection during VL was due to the impaired cell-mediated immunity which in turn was intricately associated with the severe dysfunction of cytotoxic CD8+ T-cells [7]. Therefore, activation of CD8+ T cells is very much important to eradicate L. donovani mediated infection. Recently, it has been reported that activation of TLR signalling could restore the impaired effector function of CD8+ T-cells in various models of infectious diseases [35]. Therefore, we intended to study whether Ara-LAM, so far known to be a TLR2 ligand [18], could restore the functional capacity of the effector CD8+ T-cells during VL.
We confirmed that Ara-LAM pre-treatment in L. donovani infected BALB/c mice significantly augmented the expression of TLR2 along with the concomitant increase in IFN-γ, perforin, and granzyme-B in the splenic CD8+ T-cells (Figs 1 and 2). Because knowing phenotype is important for its functional significance, we examined the phenotype of these T cells, which are IL-12R+IFN-γ+CD28+ co-expressing CD25 (Fig 1A). Moreover, Ara-LAM pre-treatment was found to be associated with significant enhancement of CD8+ T-cell proliferation (Fig 2C), a prerequisite for its effector function. TLR2 silencing significantly attenuated such Ara-LAM mediated enhanced expression of these effector molecules in CD8+ T-cells (Fig 2).
The optimal transcriptional induction of the IFN-γ, perforin, granzyme-B genes in CD8+ T-cells requires histone modification at their respective promoter regions [19]. Our study revealed that Ara-LAM pretreatment led to transcription favourable histone H3 phosphorylation and acetylation specifically at the promoter region of IFN-γ, perforin, granzyme-B in the splenic CD8+ T-cells of L. donovani-infected BALB/c mice (Fig 3A and 3B). In line with the fact that the activation of the transcription factor T-bet is a crucial event for the expression of IFN-γ, perforin, granzyme-B in CD8+ T-cells [13, 34], we observed an increased T-bet accumulaion (Fig 3C) along with enhanced T-bet binding to the promoter regions of IFN-γ, perforin, granzyme-B in the Ara-LAM treated splenic CD8+ T-cells of L. donovani infected BALB/c mice (Fig 3D). Hence, our study revealed that Ara-LAM induces T-bet dependent activation of effector molecules in CD8+ T-cells to hinder leishmanial pathogenesis.
Further, we intended to investigate the underlying molecular mechanism of Ara-LAM induced CD8+ T-cells activation during VL. Since, Ara-LAM confers its protective functions against VL via activation of the TLR2 downstream signaling in host macrophages [18–19], we investigated Ara-LAM mediated modulation of TLR2 signaling in CD8+ T-cells. Ara-LAM treatment led to successful initiation of TLR2 signalling in naive CD8+ T-cells via the TLR2-MyD88 association (Fig 4A), resulting in the selective activation of intermediate signalling molecules, IRAK1 and ultimately leading to nuclear translocation of NF-κB (Fig 4B). Ara-LAM treatment also led to increased p38MAPK phosphorylation along with concomitant attenuation of ERK1/2 phosphorylation in splenic CD8+ T-cells (Fig 4C and 4D). These observations indicate that Ara-LAM induced activation of TLR2 downstream signalling molecules leads to enhanced effector function of CD8+ T-cells during VL.
We observed that inhibition of NF-κB and p38MAPK, by their respective pharmacological inhibitors, significantly abrogated the Ara-LAM–induced T-bet expression and activation in CD8+ T-cells (Fig 5A and 5B). Our results indicated the active involvement of NF-κB and p38MAPK signalling in the regulation of Ara-LAM driven T-bet activation. Further, Ara-LAM treatment significantly reduced the parasite burden in the liver and spleen 14 and 28 days after the infection (Fig 6) accompanied with the expansion of IFN-γ+CD8+T-cells (Fig 7). It is therefore feasible to conclude that Ara-LAM mediated reduced parasitic burden may be due to the CD8+ T-cell driven killing of the infected macrophages. It is noteworthy to mention that Ara-LAM works well when utilized as an immunotherapiutic agent administered 2days prior to L. donovani infection [18–20, 28].
In summary, Ara-LAM confers significant protection through activation of CD8+ T-cells in L. donovani infected BALB/c mice. The novelty of our work lies in the fact that Ara-LAM-induced TLR2 signaling leads to the activation of the transcription factor T-bet which plays a pivotal role in restoring the effector functions of CD8+ T-cells in L. donovani infected susceptible host. Thus, we are one the way to devise a strategy in near future so that Ara-LAM can be used as a suitable vaccine during L. donovani infection.
Acknowledgments
The work was supported by the Department of Biotechnology (DBT), India (Grant No. BT/PR14435/MED/29/201/2010). We are grateful to the Director, Bose Institute (Kolkata, India) for his continous encouragement. We thank Mr. Prabal Gupta for his technical assistance and the Central Instrument Facility for their assistance in FACS analysis and Gel Documentation system.
Data Availability
All relevant data are within the paper itself.
Funding Statement
Funding was provided by the Department of Biotechnology, Ministry of Science and technology (IN), http://www.dbtindia.nic.in/, BT/PR14435/MED/29/201/2010. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ahmed S, Colmenares M, Soong L, Goldsmith-Pestana K, Munstermann L, Molina R, et al. (2003) Intradermal infection model for pathogenesis and vaccine studies of murine visceral leishmaniasis. Infect Immun 71: 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander J, Bryson K (2005) T helper Th1/Th2 and Leishmania: paradox rather than paradigm. Immunol Lett 99: 17–23. [DOI] [PubMed] [Google Scholar]
- 3. Kumar R, Nylén S (2012) Immunobiology of visceral leishmaniasis. Front Immunol 3:251/ 10.3389/fimmu.2012.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wakil AE, Wang ZE, Ryan JC, Fowell DJ, Locksley RM (1998) Interferon-γ derived from CD4 (+) T cells is sufficient to mediate T helper cell type 1 development. J Exp Med 188: 1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kaushal H, Bras-Gonçalves R, Negi NS, Lemesre JL, Papierok G, Salotra P. (2014) Role of CD8+T cells in protection against Leishmania donovani infection in healed Visceral Leishmaniasis individuals. BMC Inf Dis 14: 653–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colmenares M, Kima PE, Samoff E, Soong L, McMahon-Pratt D (2003) Perforin and gamma interferon are critical CD8+ T-cell-mediated responses in vaccine-induced immunity against Leishmania amazonensis infection. Infect Immun 71: 3172–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gigley JP, Bhadra R, Moretto MM, Khan IA (2012) T cell exhaustion in protozoan disease. Trend in Parasitology. 28: 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tian S, Maile R, Collins EJ, Frelinger JA (2007) CD8+ T Cell Activation Is Governed by TCR-Peptide/MHC Affinity, Not Dissociation Rate. J Immuonl 179: 2952–2960. [DOI] [PubMed] [Google Scholar]
- 9. Bodas M, Jain N, Awasthi A, Martin S, Penke Loka RK, Dandekar D, et al. (2006) Inhibition of IL-2 Induced IL-10 Production as a Principle of Phase-Specific Immunotherapy. J Immunol 177: 4636–4643. [DOI] [PubMed] [Google Scholar]
- 10. Gozalbo D, Maneu V, Gil ML (2014) Role of IFN-gamma in immune responses to Candida albicans infections. Front Biosci (Landmark Ed). 19: 1279–1290. [DOI] [PubMed] [Google Scholar]
- 11. Kared H, Camous X, Larbi A (2014) T cells and their cytokines in persistent stimulation of the immune system. Curr Opin Immunol 29: 79–85. 10.1016/j.coi.2014.05.003 [DOI] [PubMed] [Google Scholar]
- 12. Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH (2003) Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Nat Acad Sci 100: 15818–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sutherland AP, Joller N, Michaud M, Liu SM, Kuchroo VK, Grusby MJ (2013) IL-21 promotes CD8+ CTL activity via the transcription factor T-bet. J Immunol 190: 3977–3984. 10.4049/jimmunol.1201730 [DOI] [PubMed] [Google Scholar]
- 14. Russ BE, Denton AE, Hatton L, Croom H, Olson MR, Turner SJ (2012) Defining the molecular blueprint that drives CD8+T cell differentiation in response to infection. Front Immunol 10.3389/fimmu.2012.00371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, et al. (2009) Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31: 873–884. 10.1016/j.immuni.2009.09.018 [DOI] [PubMed] [Google Scholar]
- 16. Pandey SP, Chandel HS, Srivastava S, Selvaraj S, Jha MK, Shukla D, et al. (2014) Pegylated Bisacycloxypropylcysteine, a Diacylated Lipopeptide Ligand of TLR6, Plays a Host-Protective Role against Experimental Leishmania major Infection. J Immunol 193: 3632–3643. 10.4049/jimmunol.1400672 [DOI] [PubMed] [Google Scholar]
- 17. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, et al. (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130: 1071–1082. [DOI] [PubMed] [Google Scholar]
- 18. Bhattacharya P, Bhattacharjee S, Gupta G, Majumder S, Adhikari A, Mukherjes A, et al. (2010) Arabinosylated Lipoarabinomannan–Mediated Protection in Visceral Leishmaniasisthrough Up-Regulation of Toll-Like Receptor 2 Signaling: An Immunoprophylactic Approach. J Inf Dis 202: 145–155. [DOI] [PubMed] [Google Scholar]
- 19. Bhattacharya P, Gupta G, Majumder S, Adhikari A, Banerjee S, Halder K, et al. (2011) Arabinosylated Lipoarabinomannan Skews Th2 Phenotype towards Th1 during Leishmania Infection by Chromatin Modification: Involvement of MAPK Signaling.PlosOne 6:e24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Majumder N, Dey R, Mathur RK, Datta S, Maitra M, Ghosh S, et al. (2006) An unusual pro-inflammatory role of interleukin-10 induced by arabinosylated lipoarabinomannan in murine peritoneal macrophages. Glycoconj J 23: 675–686. [DOI] [PubMed] [Google Scholar]
- 21. Bhattacharjee S, Bhattacharjee A, Majumder S, Majumdar SB, Majumdar S (2012) Glycyrrhizic acid suppresses Cox-2-mediated anti-inflammatory responses during Leishmania donovani infection. J Antimicrob Chemother 67: 1905–1914. 10.1093/jac/dks159 [DOI] [PubMed] [Google Scholar]
- 22. Rub A, Dey R, Jadhav M, Kamat R, Chakkaramakkil S, Majumdar S, et al. (2009) Cholesterol depletion associated with Leishmania major infection alters macrophage CD40 signalosome composition and effector function. Nat Immunol 10: 273–281. 10.1038/ni.1705 [DOI] [PubMed] [Google Scholar]
- 23. Das S, Banerjee S, Majumder S, Chowdhury BP, Goswami A, Halder K, et al. (2014) Immune Subversion by Mycobacterium tuberculosis through CCR5 Mediated Signaling: Involvement of IL-10. 9:e92477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Halder K, Banerjee S, Bose A, Majumder S, Majumdar S (2014) Overexpressed PKCd downregulates the expression of PKCa in B16F10 melanoma: induction of apoptosis by PKCd via ceramide generation. PlosOne 9:e91656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Majumdar S, Kane LH, Rossi MW, Volpp BD, Nauseef WM, Korchak HM (1993) Protein kinase C isotypes and signal transduction in human neutrophils: selective substrate specificity of calcium dependent b-PKC and novel calcium independent n-PKC. Biochim Biophys Acta. 1176: 276–286. [DOI] [PubMed] [Google Scholar]
- 26. Ghosh S, Bhattacharyya S, Sirkar M, Sa GS, Das T, Majumdar D, et al. (2002) Leishmania donovani suppresses activator protein-1 and NF-kB in host macrophages via ceramide generation: involvement of extracellular signal regulated kinase. Infect Immun. 70: 6828–6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76: 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das S, Bhattacharjee O, Goswami A, Pal NK, Majumdar S (2014) Arabinosylated lipoarabinomannan (Ara-LAM) mediated intracellular mechanisms against tuberculosis infection: Involvement of protein kinase C (PKC) mediated signaling. Tuberculosis (Edinb). 10.1016/j.tube.2014.11.007 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 29. Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-γ: an overview of signals, mechanisms and functions. Hum J of Leu Biol 75: 163–189. [DOI] [PubMed] [Google Scholar]
- 30. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, et al. (2009) Gene regulation and chromatin remodeling by IL-12 and Type I interferon in programming for CD8 T cell effector function and memory. J Immunol 183: 1695–1704. 10.4049/jimmunol.0900592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Elloso MM, Scott P (2001) Differential requirement of CD28 for IL-12 receptor expression and function in CD4+ and CD8+ T cells. Eur J Immunol 31: 384–395. [DOI] [PubMed] [Google Scholar]
- 32. Whitmire JK, Tan JT, Whitton JL (2005) Interferon-γ acts directly on CD8 T cells to increase their abundance during virus infection. J Exp Med 201: 1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Das G, Vohra H, Rao K, Saha B, Mishra GC (1999) Leishmania donovani infection of a susceptible host results in CD4+ T-cell apoptosis and decreased Th1 cytokine production. Scand J Immunol 49: 307–310. [DOI] [PubMed] [Google Scholar]
- 34. Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH (2003) Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Nat Aca Sci 100: 15818–15823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quigley M, Martinez J, Huang X, Yang Y (2009) A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood 113: 2256–64. 10.1182/blood-2008-03-148809 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper itself.