

Abstract
PKR is a member of the eIF2α family of protein kinases that inhibit translational initiation in response to stress stimuli and functions as a key mediator of the interferon-induced antiviral response. PKR contains a dsRNA binding domain that bind to duplex regions present in viral RNAs, resulting in kinase activation and autophosphorylation. An emerging theme in the regulation of protein kinases is the allosteric linkage of dimerization and activation. The PKR kinase domain forms a back-to-back parallel dimer which is implicated in activation. We have developed a sensitive homo-FRET assay for kinase domain dimerization to directly probe the relationship between RNA binding, activation, and dimerization. In the case of perfect duplex RNAs, dimerization is correlated with activation and dsRNAs containing 30 bp or more efficiently induce kinase domain dimerization and activation. However, more complex duplex RNAs containing a 10–15 bp 2'-O-methyl RNA barrier produce kinase dimers but do not activate. Similarly, inactivating mutations within the PKR dimer interface that disrupt key electrostatic and hydrogen binding interactions fail to abolish dimerization. Our data support a model where activating RNAs induce formation of a back-to-back parallel PKR kinase dimer whereas nonactivating RNAs either fail to induce dimerization or produce an alternative, inactive dimer configuration, providing an additional mechanism for distinguishing between host and pathogen RNA.
PKR is a key mediator of the Type I interferon-induced antiviral response.1 PKR belongs to the eIF2α family of protein kinases that inhibit translational initiation in response to different stress stimuli. PKR is activated by duplex regions present in viral RNAs2 to undergo autophosphorylation. Phosphorylation of eukaryotic initiation factor 2 (eIF2α) by activated PKR inhibits protein synthesis within infected cells, leading to apoptosis and the inhibition of viral replication. PKR also participates in stress response and inflammation pathways and regulates cellular growth and proliferation, nutrient signaling and metabolism.3–5
The N-terminal regulatory module of PKR consists of two dsRNA binding domains (dsRBDs.6 Although the PKR dsRBDs bind dsRNA without sequence specificity,7 there is evidence for shape recognition and sequence readout in the minor groove in some other dsRBDs.8 The catalytic domain has the typical bilobal architecture found in protein kinases, consisting of a smaller N terminal (N-) lobe and a larger C terminal (C-) lobe.9
A key mechanistic issue is how interaction of the PKR dsRBDs with dsRNA leads to activation of the kinase. Early studies supported an autoinhibition model in which the latent form of PKR is locked in a closed conformation where the second dsRBD interacts with the kinase and blocks substrate binding.10 In this scenario, RNA binding to the dsRBDs leads to an open, active state. This view is supported by NMR chemical shift perturbation measurements that reveal interaction between the second dsRBD and the kinase.11,12 However, this interaction is extremely weak (Kd ~ 250 μM).13 SAXS,14 AFM,15 and NMR16 analysis of latent PKR reveal that the long (~90 amino acid) linker between the dsRBDs and the kinase domain is flexible and unstructured, giving rise to a range of open and closed conformations that is inconsistent with the autoinhibition model.
An emerging mechanism in the regulation of protein kinases is the allosteric linkage of dimerization with a conformational transition to an active state.17 Structural and biophysical analyses support a pivotal role for dimerization in activation.10 The PKR kinase domain forms a dimer in both active, phosphorylated9 and inactive states.18 PKR dimerizes only weakly (Kd ~ 500 μM); however, this reaction is sufficient to activate autophosphorylation in the absence of dsRNA.19 In the context of duplex RNA, the minimum length of ~30bp of duplex required to activate PKR20,21 correlates with the minimum length capable of binding two PKR monomers with high affinity.22 PKR activation exhibits a characteristic bell-shaped curve where high dsRNA concentrations inhibit20, consistent with a model where PKR dimers are dissociated by dilution onto separate dsRNAs.23 These results support a PKR activation model where RNA binding functions to bring two kinase domains into close proximity and facilitate dimerization.
Dimerization plays a role in the activation of all eIF2α kinases. PERK dimerization is linked to activation24 whereas HRI25 and GCN226 exist as constitutive dimers. The structure of a phosphorylated PERK kinase domain reveals a back-to-back parallel dimer similar to PKR27 Although a similar N-lobe dimer interface is observed in a structure of an unphosphorylated GCN2 kinase domain, it adopts a novel, antiparallel orientation where one subunit is rotated approximately 180°.28 Interdimer hydrogen bonding (Y323-D289) and salt-bridge (R262-D266) residues are conserved within the eIF2α kinase family. Mutations designed to disrupt the salt-bridge in PKR, GCN2 and PERK block kinase activity and charge reversal restores biological function29–31 This contact is formed in the parallel PERK dimer, but the corresponding residues are too far apart in the antiparallel GCN2 dimer to form a salt bridge. The antiparallel GCN2 dimer likely corresponds to an inactive conformation17,28 and activation may be associated with rotation to a parallel structure that results in formation of the essential salt-bridge.29
Interestingly, simultaneous binding of two or more PKRs to an RNA does not necessarily activate. For example, at low salt two PKRs can bind RNAs as short as 20 bp without inducing autophosphorylation.22 Typical protein-RNA interaction measurements can count the number of protein monomers bound to an RNA but cannot determine whether the proteins directly interact on the RNA to form a dimer. Thus, we have developed a sensitive fluorescence assay to directly probe the role of PKR kinase domain dimerization in RNA-induced activation. We observe efficient dimerization upon binding to activating duplex RNAs. Surprisingly, several non-activating RNAs that bind multiple PKRs also induce dimerization. We propose that PKR can adopt an alternative, non-activating dimer configuration.
Materials and Methods
Reagents
RNAs were chemically synthesized (GE Life Sciences). Duplexes were annealed at 80°C and slowly cooled to room temperature.
The pPET-PKR/PPase PKR expression vector19 was modified by substituting an amber stop codon (TAG) at K261, K371 or N301 and replacing the endogenous amber codon with ochre (TAA). PKR mutants were co-expressed with tRNA synthetase/suppressor tRNA pair for pAzF encoded by the pEVOL-pAzF vector32 in ArcticExpress (DE3) cells (Agilent Technologies). The cells were grown in LB media at 37°C. At an A600nm ~ 0.3, expression of the pAzF tRNA synthetase/suppressor tRNA pair was induced with 0.02% arabinose (Acros Organics) and 1 mM pAzF (Bachem) was added to the medium. PKR expression was induced with 1 mM IPTG at A600nm ~ 1.2 for 4 hours at room temperature. Subsequent purification steps were performed in the dark to avoid pAzF-mediated photocrosslinking. PKR constructs were purified as previously described22 and equilibrated into AU200 buffer (20 mM HEPES, 200 mM NaCl, 0.1 mM EDTA, pH 7.5) by gel filtration on a Superdex 200 column. Incorporation of pAzF was confirmed by electrospray ionization mass spectroscopy (predicted mass: 62154.3 Da, observed mass: 62023.3 Da).
Fluorescence labeling was performed by reacting 35 μM of pAzF-containing PKR with 80 μM dibenzocyclooctyne (DIBO) conjugate of Alexa Fluor 488 (Molecular Probes) or Cy3 (Click Chemistry Tools) for 1 hour at room temperature in AU200. Free dye was removed by buffer exchange (two times) into either AU200 or AU75 (20 mM HEPES, 75 mM NaCl, 0.1 mM EDTA, pH 7.5) containing 0.1 mM TCEP using spin columns packed with Bio-gel P-6 (Biorad). Specific labeling was confirmed by mass spectroscopy (predicted mass: 63032.16Da, observed mass: 62858.9Da). The efficiency of labeling with Alexa Fluor 488 determined by absorbance is approximately 80%.
Fluorescence Measurements
Steady state fluorescence anisotropy and emission spectra were recorded with a FluoroMax-3 fluorimeter equipped with Glan-Thompson polarizers (Jobin Yvon Inc., New Jersey). All measurements were performed at 20°C using a 2 ×10 mm quartz cuvette (Precision Cells). Fluorescence anisotropy measurements were performed with Alexa 488-labeled PKR at excitation and emission wavelengths of 495 nm and 518 nm and bandwidths of 2 and 3 nm, respectively. Intensities were integrated at each polarization for 5 s with three repetitions. The data were corrected for buffer background and the anisotropy (r) was calculated as
| (1) |
where I∥ and I⊥ correspond to the fluorescence intensity with the excitation polarizer oriented in the vertical position and the emission polarizer in the vertical and horizontal positions, respectively. The G factor corrects for instrument polarization bias and was measured to be ~0.64.
Analytical ultracentrifugation
Sedimentation velocity analysis of PKR-dsRNA interactions was performed in AU200 buffer at 20°C using absorbance optics at 260 nm as previously described.22,33 Sedimentation equilibrium analysis of PKR dimerization was performed in AU200 buffer at 20°C using interference optics as previously described.19
Activity Assays
Quantitative PKR autophosphorylation assays were carried out using [γ-32P] ATP as previously described.19,22
Results
PKR Dimerization on activating RNAs
A homo-Förster Resonance Energy Transfer (homo-FRET) fluorescence assay was developed to directly monitor dimerization of PKR kinase domains induced by RNA binding. Homo-FRET occurs between fluorophores of the same kind provided the Stokes shift is small, and is readily detected by depolarization of the emission using steady-state anisotropy measurements. Based on the dsRNA dissociation constants for PKR,22 the equilibrium populations of PKR dimers are expected to be low and we have used homo-FRET to assay PKR kinase domain dimerization due to the high precision of the measurement and convenience of labeling. Because PKR contains 8 cysteines, 7 of which are highly conserved, unnatural amino acid mutagenesis32 was employed to introduce a single p-azido-l-phenylalanine (pAzF) residue into wild-type PKR for labeling with extrinsic fluorophores (Fig. 1A). Labeling sites were selected based on their proximity, solvent accessibility, and lack of interference with the dimer interface. Highly conserved residues were excluded. The K261pAzF mutant (Fig. 1B) was chosen for detailed analysis based on high expression, ease of labeling, and strong homo-FRET (see below).
Figure 1. Synthesis of pAzF-261-A488.

(A) Conjugation of fluorophores with pAzF-substituted PKR. (B) Structure of the PKR kinase domain parallel dimer (PDB accession 2A1A) rendered in PyMol (Schrödinger). Alexa Fluor 488 DIBO conjugated at pAzF-261 was modeling onto the structure, with the ligand indicated in stick representation and the chromophore in green.
K261pAzF PKR reacts readily with Alexa Fluor 488-dibenzocyclooctyne to produce a stable conjugate, denoted pAzF-261-A488. The integrity of the labeled protein was monitored by autophosphorylation assays, dsRNA binding, and solution dimerization analysis. pAzF-261-A488 is an active kinase and undergoes autophosphorylation upon addition of a 40 bp dsRNA at a level about 2.5 -fold higher than unlabeled PKR (Fig. 2A). As previously observed for unlabeled PKR,21,22 two monomers of pAz-F261-A488 bind to the activating 40 bp dsRNA with comparable dissociation constants (Fig. 2B). As assayed by sedimentation equilibrium experiments in the absence of RNA, pAzF-261-A488 PKR undergoes reversible dimerization with Kd = 94 (85, 103) μM (Fig. 2C) which is ~5 fold lower than the Kd for dimerization of unlabeled PKR.19 The enhancement in activation of pAzF-261-A488 relative to unlabeled PKR may be due to stronger dimerization. This enhancement in dimerization is not observed for PKR labeled at position 371, which is more distant from the dimer interface (Kd = 1,260 μM). However, the energy transfer efficiency is too low for useful analysis. For the pAzF-261-A488 dimer, the predicted inter-chromophore distance is R = 25 Å (Fig. 1B) and R0 for Alexa Fluor 488 is 48 Å,34 with a corresponding energy transfer efficiency of 98% and a steady-state anisotropy about half the monomer value (Supporting Information).
Figure 2. Characterization of pAzF-261-A488.
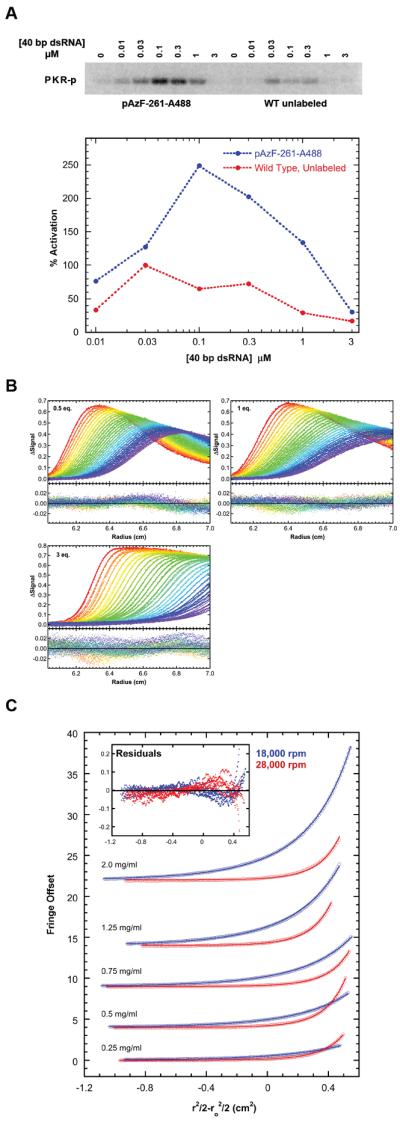
(A) Activation of pAzF-261-A488 by 40 bp dsRNA. Top: Phosphorimager analysis of wild type, unlabeled PKR (red) and pAzF-261-A488 (blue) activation. Bottom: quantitation. Data were normalized to wild type, unlabeled PKR at 30 nM 40 bp. (B) Sedimentation velocity analysis of pAzF-261-A488 binding to 0.75 μM 40 bp dsRNA. Global analysis of difference curves for at three concentrations of pAzF-261-A488 fit to a 2:1 binding model provides best-fit dissociation constants of Kd1= 0.324 (0.280, 0.376) μM and Kd2 = 0.517 (0.448, 0.579) μM with RMSD = 0.0060 OD. The top panels contain the data (points) and fits (lines) and the bottom panels contain the residuals. (C) Sedimentation equilibrium analysis of pAzF-261-A488 self-association. Global analysis of 5 concentrations and two rotor speeds (18000 rpm – blue, 28000 rpm – red) to a monomer-dimer model results in Kd = 94 (85, 103) μM with RMSD = 0.0327 fringes.
Significant anisotropy changes are induced by binding of pAz-F261-A488 to duplex RNAs (Fig. 3A). The anisotropy decreases upon addition of dsRNAs ≥ 30 bp capable of activating PKR and binding two or more monomers, but non-activating, 20 and 25 bp duplexes that bind a single PKR22 produce negligible anisotropy changes. Thus, RNA binding alone does not significantly affect the steady state anisotropy of pAzF-261-A488, and the depolarization induced upon addition of the longer duplexes can be ascribed to homo-FRET within kinase domain dimers. Note that the dsRBD is separated from the kinase domain by a long (~ 90 residue) unstructured linker, so that the changes in rotational correlation time of the dsRBD due to RNA binding are not expected to affect the kinase.
Figure 3. Analysis of PKR dimerization induced by activating dsRNAs.

(A) Anisotropy titrations of pAzF-261-A488 with dsRNAs. Samples contained 200 nM pAzF-261-A488. (B) Species distribution for pAz-F261-A488 binding to the 40 bp dsRNA. The fractional concentrations of free protein (P, blue), the the 1:1 complex (RP, red) and the 1:2 complex (RP2, green) are plotted. The fractional concentrations were determined using the experimentally determined dissociation constants Kd1= 0.324 μM and Kd2 = 0.517 μM (Fig. 2) and numerically solving the equilibria with IGOR Pro (Wavemetrics, Lake Oswego OR). The width of the distributions corresponds to the two standard deviation confidence intervals.
Quantitative analysis of the anisotropy changes produced by the activating dsRNAs provides useful mechanistic insights. The amplitude of the anisotropy change increases with the length of the dsRNA, but even for the longest (40 bp) RNA the maximum decrease of 12% does not approach the ~50% effect predicted to occur upon formation of a dimer of pAzF-261-A488 (Supporting Information). Also, the anisotropy returns to the monomer value at higher RNA concentrations. These observations can be understood in the context of the binding equilibria for pAzF-261-A488 titrated with the 40 bp RNA depicted in Figure 3B. RNA-induced formation of the kinase domain dimer requires that two PKR monomers bind to the same RNA (Fig. 4). For an RNA that binds two protein ligands, the concentration of the species containing two PKRs bound to the same RNA (RP2) is expected to rise and then fall as PKR monomers are diluted onto separate RNAs, as is observed in the anisotropy titrations (Fig 3A). The maximum fraction of PKR present in the RP2 species is dependent on the binding affinities (Fig 2B). In a simulation of a titration of 200 nM pAz-F261-A488 with the 40 bp dsRNA, the peak value of RP2 is predicted to correspond to only 14% of the total protein (Fig. 3B, Table 1). We can compare this value with the maximum amount of kinase domain dimer to determine the efficiency of dimerization upon binding of two PKRs to a single dsRNA (Fig. 4). The amplitude of the anisotropy changes for the 40 bp dsRNA indicates that a maximum of ~30% of the kinase domains exist as dimers (Table 1). If the kinase domains present in the RP2 species quantitatively formed dimers, the ratio of the maximum % dimer to % RP2 would equal one. For the 40 bp dsRNA this value is about two, implying that the maximum dimer population must be overestimated due to additional sources of depolarization. Regardless of the origin of this discrepancy, the shape of the anisotropy titration is consistent with the RNA concentration dependence of the RP2 species and the amplitude implies that RNA-induced dimerization of the kinase domain occurs efficiently upon sequential binding of two PKRs to the same RNA.
Figure 4. Schematic of formation of the kinase domain dimer.

The kinase domain is depicted in green, dsRBD1 in blue and dsRBD2 in red. The fluorophore is depicted as an orange star. Upon binding of two pAz-F261-A488 monomers to a single dsRNA the kinase domains may dimerize in the back-to-back parallel configuration, leading to homo-FRET (top). Alternatively, the protein may bind to the dsRNA in a configuration that does not support kinase domain dimerization that leads to homo-FRET (bottom). The term RP2 encompasses all forms of the RNA containing two bound PKR monomers.
Table 1.
Correlation of PKR dimerization and RNA binding.
| Sample | Max. % Dimera | Max. % RP2b | Dimer / RP2c |
|---|---|---|---|
| WT + 40 bp | 29.5 ± 0.6 | 14.2 ± 1.4d | 2.08 ± 0.21 |
| R262D + 40 bp | 11.4 ± 1.5 | 14.2 ± 1.4d | 0.81 ± 0.13 |
| Y323A + 40 bp | 11.1 ± 2.2 | 14.2 ± 1.4d | 0.78 ± 0.17 |
| WT + 20 bp (75 mM NaCl) | 9.2 ± 0.3 | 48.7 ± 4.5e | 0.19 ± 0.02 |
| WT + 5-Me | 20.9 ± 2.1 | 17.9 ± 2.0f | 1.17 ± 0.18 |
| WT + 10-Me | 11.6 ± 2.3 | 10.2 ± 2.2f | 1.14 ± 0.34 |
| WT + 15-Me | 9.5 ± 1.3 | 4.7 ± 0.5f | 2.02 ± 0.35 |
Maximum % dimer calculated from the largest anisotropy change and the anisotropy of the dimer (Eq. S3).
Maximum population of the RP2 species based on Kd1 and Kd2 for sequential binding of two PKR monomers under the conditions indicated.
The ratio of the maximal % dimer to the maximal % RP2.
Data from Fig. S1.
Data previously reported.22
Data from Table 2.
Control experiments confirm that the dsRNA-induced anisotropy changes are due to FRET within kinase domain dimers. Addition of unlabeled PKR to pAzF-261-A488 is expected to reduce the extent of homo-FRET by formation of mixed labeled/unlabeled heterodimers. Assuming random formation of heterodimers, the anisotropy change should be linear in the fraction of labeled PKR (see Supporting Information). For higher order oligomers a nonlinear dependence is predicted. Figure 5A demonstrates that mixing increasing amounts of unlabeled PKR with pAzF-261-A488 linearly reduces the anisotropy decrease induced by binding to a 40 bp dsRNA (R = 0.991 for a linear fit).
Figure 5. Depolarization is due to FRET-induced dimerization.

(A) Dependence of anisotropy on the fraction of labeled PKR. Samples contained 100 nM 40 bp dsRNA, 200 nM pAz-F261-A488, and variable amounts of unlabeled PKR. A linear fit to the data gives a linear correlation coefficient of R=0.991 and a y-intercept of 0.2846. (B) Analysis of PKR dimerization by hetero-FRET. Donor: pAz-F261-A488, acceptor: pAz-F261 PKR labeled with DIBO-Cy3. Spectra: 1:1 mixture (200 nM total) of donor and acceptor (blue), 1:1 mixture (200 nM total) of donor and acceptor with 100 nM 40 bp dsRNA (red), algebraic sum of 100 nM donor and 100 nM acceptor spectra (grey). (C) Fluorescence anisotropy analysis of pAzF-261-A488 dimerization in the absence of dsRNA. Top: raw anisotropy as a function of concentration. Bottom: experimental % dimer calculated from the anisotropy data compared to the % dimer predicted using the Kd determined by sedimentation equilibrium of 94 μM. The slits were adjusted during the titration to maintain the count rate in the linear range.
In principle, the increase in rotational correlation time of pAzF-261-A488 upon dimerization could result an increase in anisotropy. However, if this were the case, the y-intercept of Fig. 5A, corresponding to the anisotropy at infinite dilution, would be greater than the anisotropy of free, monomeric pAzF-261-A488. The y-intercept of 0.2846 is very close to the anisotropy of the free protein (0.2869), indicating that changes in rotational mobility upon dimerization do not contribute substantially to the observed signal.
Alexa Fluor 488 can serve as an efficient FRET donor to Cy3 (R0 ~ 67 Å)35. The fluorescence spectrum of a 1:1 mixture of PKR labeled at pAzF261 with Alexa Fluor 488 and Cy3 overlays with the sum of the spectra of the two components, indicating the absence of FRET (Fig. 5B). Binding of the mixture to the 40 bp dsRNA results in significant donor quenching and enhanced Cy3 emission due to hetero-FRET.
Finally, the fluorescence anisotropy of pAz-F261-A488 in the absence of RNA decreases at high protein concentrations consistent with formation of a dimer (Fig. 5C). The extent of dimerization is on the order predicted by the weak Kd measured by sedimentation equilibrium (Fig. 5C). Thus, depolarization is due to dimerization rather than RNA binding per se.
PKR dimerization without kinase activation
In the PKR dimer interface, R262 forms a salt bridge with D266 (Fig. 6A) and disruption of this interaction in a R262D PKR mutant blocks kinase activity.31 Similarly, disruption of a hydrogen-bonding interaction between Y323 and D289 in PKR Y323A reduces activation. We expected that these mutations block PKR activation by preventing kinase dimerization and would serve as negative controls for the homo-FRET assay. Surprisingly, fluorescence anisotropy titrations indicate that pAzF-261-A488 containing either interface mutation undergoes dimerization upon binding to the 40 bp dsRNA (Fig. 6B). The maximal amplitudes of the anisotropy changes are somewhat smaller than observed for wild-type PKR with a corresponding dimer/RP2 ~ 0.8 (Table 1). PKR autophosphorylation assays confirm that these constructs are inactive (Fig. S1). Consistent with the anisotropy data, sedimentation equilibrium analysis of R262D PKR demonstrates reversible dimerization in free solution with Kd of 923 (714, 1267) μM (Figure 6C), or about 2-fold weaker than WT. Similarly, Y323A PKR also dimerizes with a Kd of 689 (590, 820) μM. Thus, the disruption of interdimer salt-bridge or hydrogen bonding interactions abolishes PKR activation while only slightly reducing dimerization affinity.
Figure 6. Dimerization without activation of PKR.

(A) Critical intermolecular salt bridge (R262 – D266) and hydrogen bond (Y323-D289) interactions in the PKR kinase dimer interface (PDB accession 2A1A) rendered in PyMol (Schrödinger). (B) Anisotropy titrations of PKR dimer interface mutants with 40 bp dsRNA. The anisotropy of the free protein was subtracted from each data set. (C) Sedimentation equilibrium analysis of R262D PKR. The points are the data and the solid lines are the best fit. For clarity, only every fourth point is shown. The inset shows the residuals. Global analysis reveals that R262D dimerizes, with a best-fit Kd = 923 (714, 1267) μM (the values in parentheses correspond to the one standard deviation confidence intervals) and an RMS deviation of 0.041 fringes. The Kd for WT PKR dimerization under the same conditions is 450 μM.19 (D) Anisotropy titration of pAzF-261-A488 PKR with a 20 bp dsRNA in AU200 and AU75 buffers.
The reduction in the dimer/RP2 ratio for R262D suggests that this mutant dimerizes less efficiently. Alternatively, the dimers could form with the same frequency but adopt an altered geometry that places the fluorophores further apart, resulting in reduced energy transfer and an apparent decrease in the % dimer. Formation of an antiparallel dimer modeled on the GCN2 structure would increase the distance between pAzF-261-A488 fluorophores from 25 to 49 Å (Fig. S2) resulting in diminished energy transfer. To test this model, wild-type and R262D PKR were labeled at position 301 where the energy transfer efficiency should be enhanced in a GCN2-type antiparallel, inactive dimer relative to the active parallel structure, with interfluorophore distances of 24 and 60 Å, respectively. Titration of either pAzF-301-A488 or pAzF-301-A488/R262D PKR with 40 bp dsRNA results in a similar low amplitude anisotropy change (Fig. S3), indicating that the inactive R262D dimers do not adopt a GCN2-type structure.
The 20 bp dsRNA binds a second PKR upon reducing the concentration of NaCl from 200 to 75 mM.22 Although this short RNA does not activate PKR at either NaCl concentration,21,22 it does induce an anisotropy change in 75 mM NaCl (Fig. 6D) corresponding to a maximum dimer population of about 9%. However, PKR binds very strongly to the 20 bp RNA at low salt22 so that the maximum population of RP2 is high (49%), resulting in dimer/RP2 of only 0.19 (Table 1). Thus, only a small fraction of the bound PKRs form dimers upon binding to this short dsRNA.
Dimerization on barrier containing RNAs
The introduction of helical imperfections into dsRNAs typically reduces PKR binding and activation.36,37 However, PKR is also activated by certain RNAs that contain bulges, loops, pseudoknots and single-stranded tails 2. PKR can be activated by dimers of TAR38 and the HDV ribozyme39 that contain short duplex regions separated by a distorted central region that serves as a barrier. To systematically probe the effect of structural distortions on PKR dimerization and activation by dsRNA, we introduced structurally defined, inert, rigid barriers into a model duplex. The dsRBD interacts with the RNA 2'-OH8 and the barriers consist of 2'-O-methyl dsRNA. The RNAs contain two dsRNA regions of 15 bp, the minimal length required to each bind one PKR with high affinity, separated by variable length, 2'-O-methyl dsRNA barriers and are denoted as X-Me, where X is the length of 2'-O-methyl dsRNA (Fig. 7A). PKR is not activated by 2'-O-methyl dsRNA40 and does not bind to a 15 bp 2'-O-methyl duplex RNA (Fig. S4).
Figure 7. Interaction of PKR with dsRNAs containing 2'-O-methyl barriers.

(A) Schematic of 2'-O-methyl (2'-O-Me) barrier-containing dsRNAs. Chimeric dsRNAs were designed to contain 5, 10 and 15 bp 2'-O-Me barriers inserted between two 15 bp dsRNA regions. (B) Activation of WT PKR by 2'-O-Me barrier-containing dsRNAs. The percent activation is normalized to the signal from the 40 bp dsRNA at 100 nM. (C) Anisotropy titration of pAzF-261-A488 PKR with 2'-O-Me barrier-containing dsRNAs. For reference, the maximal anisotropy changes associated with PKR binding to regular 30 and 40 bp dsRNAs are indicated by arrows. (D) Fractional concentrations of the 1:2 RNA:PKR complex (RP2) for PKR binding to the 2'-O-Me barrier-containing dsRNAs. The fractional concentrations were determined using the experimentally determined dissociation constants (Table 1). The maximal fraction of RP2 for PKR binding to a 40 bp dsRNA is indicated with an arrow.
PKR binding to the barrier-containing RNAs was characterized using sedimentation velocity analytical ultracentrifugation. As expected, each of the RNAs binds two PKR monomers (Table 2). The dissociation constants for 5-Me are similar to a 30 bp dsRNA (zero-length barrier).21 The dissociation constants for the binding of the first PKR are not very sensitive to barrier length but Kd2 increases systematically from 5-Me to 15-Me.
Table 2.
Analysis of PKR binding to barrier-containing dsRNAs.
| RNA | Kd1 (nM) | Kd2 (nM) | RMSb |
|---|---|---|---|
| 5-Me | 173 (127, 235) | 375 (319, 440) | 0.0067 |
| 10-Me | 360 (263, 492) | 784 (606, 1036) | 0.0082 |
| 15-Me | 266 (231, 306) | 1,930 (1709, 2194) | 0.0091 |
Parameters were obtained by global nonlinear least square analysis of sedimentation velocity measurements. The values in parentheses represent the 95% joint confidence intervals obtained using the F-statistic.
Uncorrected sedimentation coefficient (Svedbergs).
Root mean square deviation of the fit in absorbance units.
PKR activation is strongly affected by the introduction of longer barriers (Fig. 7B). 5-Me activates PKR but 10-Me and 15-Me elicits little to no autophosphorylation. These results are supported by previous work where introduction of a 11 bp DNA-RNA chimeric duplex between two 20 bp dsRNAs greatly attenuated PKR activation.36 Binding of pAzF-261-A488 to all three barrier-containing dsRNAs results in a significant anisotropy decrease due to kinase domain dimerization (Fig. 7C). The maximal anisotropy change is slightly attenuated with increasing barrier length, but even for the inactive 10-Me and 15-Me constructs it is comparable to that induced by the activating 30 bp dsRNA. All three RNAs have a high dimer/RP2 ratio, indicating efficient kinase domain dimerization.
Discussion
The homo-FRET assay directly probes PKR kinase domain dimerization on RNA and provides new insights into the factors that distinguish RNA activators and inhibitors. As previously inferred10 dsRNA activators promote kinase domain dimerization and the extent of dimerization correlates with activation potency. These data support a dimerization model 10,23 where activating duplex RNAs function to bring PKR monomers into close proximity, leading to dimerization of the kinase domains.
PKR binds to dsRNA nonspecifically in multiple configurations41 so that PKR monomers may be quite far apart along the nucleic acid lattice. Thus, it is noteworthy that duplex RNA activators ≥30 bp induce efficient dimerization following assembly of two PKRs onto a single RNA. Other proteins harboring dsRBDs can travel along dsRNA by passive diffusion42 and efficient assembly of kinase domain dimers may occur via this mechanism.
In contrast to longer dsRNAs, kinase domain dimerization is inefficient on short, nonactivating dsRNA that bind two PKRs in low salt. The impaired dimerization for short duplexes is correlated with a difference in RNA binding mode. Affinity cleavage,43 NMR shift perturbation,41 and RNA binding22,41 analyses reveal that short dsRNAs that fail to activate predominantly interact with dsRBD1 whereas longer activators interact extensively with both dsRBDs. Thus, interaction of dsRBD2 with RNA appears to regulate kinase domain dimerization despite the fact that these regions are separated by a long linker. Although this linker is unstructured in the free enzyme,14–16 it may become at least partially ordered in the presence of RNA and serve to sterically regulate kinase domain interactions.
In contrast to perfect duplexes, the correlation between PKR activation and kinase domain dimerization breaks down in the context of more complex RNAs, indicating that dimerization is required but not sufficient to activate the kinase. Abrogation of PKR activation by insertion of 2'-O-Me dsRNA barriers ≥ 10 bp does not appreciably affect the efficiency of dimerization. A barrier of 10 bp of A-form RNA duplex corresponds to a distance along the helical axis of only ~ 28 Å. It is reasonable that the barrier does not disrupt dimerization given that the linker between the dsRBD and the kinase is long and would be capable of spanning this short distance. However, barriers ≥ 10 bp induce a change in RNA binding mode that modulates activation, indicating long-range communication between the dsRBDs and kinase domain. The structural basis for the dramatic dependence of activation on barrier length is not clear. The effect of barrier length on binding affinity for the second PKR (Table 1) implies that weak cooperative interactions that stabilize binding of the second PKR become attenuated as the length of the barrier increases.
The relationship between PKR dimerization and activation can be interpreted within the broader context of the other members of the eIF2α family of protein kinases. PKR9,18 and PERK27 share a similar back-to-back parallel dimer and conservation of interface residues suggests that this structure is a common element among the other eIF2α kinases. We propose that PKR activation requires formation of a back-to-back parallel interface. Although R262D and Y323A PKR mutations that disrupt interdimer salt bridge and hydrogen-bonding interactions block PKR activation (Fig. S1)30,31 they do not prevent dimerization at high concentration in the absence of RNA or upon binding to an activating dsRNA. Although the energy transfer efficiency of pAzF-261-A488 is reduced by these mutations, implying a rearrangement in the dimer interface, it is not enhanced in pAzF-301-A488/R262D, indicating that the inactive dimer does not correspond to the model based on the antiparallel GCN2 interface. Some nonactivating RNAs, such as the 20 bp duplex at low salt, bind multiple PKRs but fail to induce kinase domain dimerization. Other nonactivating RNAs, such as the barrier containing molecules, induce formation of an inactive dimer configuration analogous to that found in the R262D and Y323A PKR mutants. The allosteric pathway linking the PKR dimer interface to the kinase active site9,17 is likely disrupted in this inactive dimer configuration.
The inactive PKR dimer configuration may provide an additional mechanism for distinguishing between host and pathogen RNA. About half of the mammalian genome is comprised of noncoding retrotransposons such as SINEs and Alus, which typically form dsRNA duplexes44 and high throughput transcriptional analysis has revealed the presence of hundreds of natural dsRNAs in human cells.45 PKR must be able to distinguish host RNAs from pathogen-derived molecules to avoid inappropriate activation of the innate immunity response. One level of recognition could be achieved based on dsRNA length, where a minimum of ~30 bp of duplex are required for activation.7,20–22 Replication of positive strand RNA viruses and DNA viruses induces formation of dsRNAs longer than 40 bp.46 Here, regulation of PKR is controlled by dimerization; short duplexes do not induce dimerization (Fig. 6D). Alternative regulation may be controlled by the dimer orientation. Our analysis of 2'-O-methyl barrier-containing duplexes suggests that some secondary structure defects may regulate PKR activation by induction of inactive dimers and this may serve to prevent aberrant response to host transcripts. In this regard, adenosine deaminases acting on RNA (ADARs) catalyze the conversion of adenosine to inosine, inducing I-U mismatches. ADAR1 carries out hyperediting of long dsRNAs and this activity suppresses activation of the dsRNA innate immunity pathway and PKR.47,48 A knock-in mutation to inactivate ADAR1 results in activation of the interferon and dsRNA sensing pathways.49 Further investigation is required to determine how I-U mismatches regulate PKR activity.
Supplementary Material
Acknowledgements
We thank Michael Bruno for assistance with unnatural amino acid mutagenesis.
Funding Source: Grant number AI-53615 from the NIH to J.L.C
Abbreviations
- dsRBD
double-stranded RNA binding domain
- dsRNA
double-stranded RNA
- eIF2α
eukaryotic initiation factor 2α
- PKR
RNA activated protein kinase
- RMSD
root mean square deviation.
Footnotes
Supporting Information Supporting Information Available. Supporting Text: Theory of homo-FRET within a dimeric cluster. Supporting Text: Effect of dilution of pAzF-261-A488 with unlabeled PKR. Fig. S1: Activation assays of PKR dimer interface mutants. Fig. S2: Structural model for an antiparallel PKR dimer. Fig. S3: Anisotropy titrations of PKR labeled at pAzF301. Fig. S4: Sedimentation velocity analysis of PKR binding to 2'-OH and 2'-O-Me dsRNAs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Toth AM, Zhang P, Das S, George CX, Samuel CE. Interferon action and the double-stranded RNA-dependent enzymes ADAR1 adenosine deaminase and PKR protein kinase. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:369–434. doi: 10.1016/S0079-6603(06)81010-X. [DOI] [PubMed] [Google Scholar]
- (2).Nallagatla SR, Toroney R, Bevilacqua PC. Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr Opin Struct Biol. 2011;21:119–127. doi: 10.1016/j.sbi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).García MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiology and Molecular Biology Reviews. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pindel A, Sadler A. The role of protein kinase R in the interferon response. J Interferon Cytokine Res. 2011;31:59–70. doi: 10.1089/jir.2010.0099. [DOI] [PubMed] [Google Scholar]
- (5).Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JPY, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bevilacqua PC, Cech TR. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–9994. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- (8).Masliah G, Barraud P, Allain FH-T. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell. Mol. Life Sci. 2013;70:1875–1895. doi: 10.1007/s00018-012-1119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- (10).Cole JL. Activation of PKR: an open and shut case? Trends Biochem Sci. 2007;32:57–62. doi: 10.1016/j.tibs.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gelev V, Aktas H, Marintchev A, Ito T, Frueh D, Hemond M, Rovnyak D, Debus M, Hyberts S, Usheva A, Halperin J, Wagner G. Mapping of the auto-inhibitory interactions of protein kinase R by nuclear magnetic resonance. J. Mol. Biol. 2006;364:352–363. doi: 10.1016/j.jmb.2006.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nanduri S, Rahman F, Williams BR, Qin J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000;19:5567–5574. doi: 10.1093/emboj/19.20.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Anderson E, Cole JL. Domain stabilities in protein kinase R (PKR): evidence for weak interdomain interactions. Biochemistry. 2008;47:4887–4897. doi: 10.1021/bi702211j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vanoudenhove J, Anderson E, Krueger S, Cole JL. Analysis of PKR structure by small-angle scattering. J Mol Biol. 2009;387:910–920. doi: 10.1016/j.jmb.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lemaire PA, Tessmer I, Craig R, Erie DA, Cole JL. Unactivated PKR exists in an open conformation capable of binding nucleotides. Biochemistry. 2006;45:9074–9084. doi: 10.1021/bi060567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).McKenna SA, Lindhout DA, Kim I, Liu CW, Gelev VM, Wagner G, Puglisi JD. Molecular Framework for the Activation of RNA-dependent Protein Kinase. Journal of Biological Chemistry. 2007;282:11474–11486. doi: 10.1074/jbc.M700301200. [DOI] [PubMed] [Google Scholar]
- (17).Lavoie H, Li JJ, Thevakumaran N, Therrien M, Sicheri F. Dimerization-induced allostery in protein kinase regulation. Trends Biochem Sci. 2014;39:475–486. doi: 10.1016/j.tibs.2014.08.004. [DOI] [PubMed] [Google Scholar]
- (18).Li F, Li S, Wang Z, Shen Y, Zhang T, Yang X. Structure of the kinase domain of human RNA-dependent protein kinase with K296R mutation reveals a face-to-face dimer. Chin. Sci. Bull. 2013;58:998–1002. [Google Scholar]
- (19).Lemaire PA, Lary J, Cole JL. Mechanism of PKR activation: dimerization and kinase activation in the absence of double-stranded RNA. J Mol Biol. 2005;345:81–90. doi: 10.1016/j.jmb.2004.10.031. [DOI] [PubMed] [Google Scholar]
- (20).Manche L, Green SR, Schmedt C, Mathews MB. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol Cell Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lemaire PA, Anderson E, Lary J, Cole JL. Mechanism of PKR Activation by dsRNA. J Mol Biol. 2008;381:351–360. doi: 10.1016/j.jmb.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Husain B, Mukerji I, Cole JL. Analysis of high-affinity binding of protein kinase R to double-stranded RNA. Biochemistry. 2012;51:8764–8770. doi: 10.1021/bi301226h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kostura M, Mathews MB. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol Cell Biol. 1989;9:1576–1586. doi: 10.1128/mcb.9.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- (25).Yang JM, London IM, Chen JJ. Effects of hemin and porphyrin compounds on intersubunit disulfide formation of heme-regulated eIF-2 alpha kinase and the regulation of protein synthesis in reticulocyte lysates. J Biol Chem. 1992;267:20519–20524. [PubMed] [Google Scholar]
- (26).Qiu H, Garcia-Barrio MT, Hinnebusch AG. Dimerization by translation initiation factor 2 kinase GCN2 is mediated by interactions in the C-terminal ribosome-binding region and the protein kinase domain. Mol Cell Biol. 1998;18:2697–2711. doi: 10.1128/mcb.18.5.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cui W, Li J, Ron D, Sha B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr D Biol Crystallogr. 2011;67:423–428. doi: 10.1107/S0907444911006445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Padyana AK, Qiu H, Roll-Mecak A, Hinnebusch AG, Burley SK. Structural basis for autoinhibition and mutational activation of eukaryotic initiation factor 2alpha protein kinase GCN2. J Biol Chem. 2005;280:29289–29299. doi: 10.1074/jbc.M504096200. [DOI] [PubMed] [Google Scholar]
- (29).Dey M, Cao C, Sicheri F, Dever TE. Conserved intermolecular salt bridge required for activation of protein kinases PKR, GCN2, and PERK. J Biol Chem. 2007;282:6653–6660. doi: 10.1074/jbc.M607897200. [DOI] [PubMed] [Google Scholar]
- (30).Dey M, Mann BR, Anshu A, Mannan MA-U. Activation of protein kinase PKR requires dimerization-induced cis-phosphorylation within the activation loop. J Biol Chem. 2014;289:5747–5757. doi: 10.1074/jbc.M113.527796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, Dever TE. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- (32).Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- (33).Wong CJ, Launer-Felty K, Cole JL. Analysis of PKR-RNA interactions by sedimentation velocity. Meth Enzymol. 2011;488:59–79. doi: 10.1016/B978-0-12-381268-1.00003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Szabó A, Horvath G, Szöllosi J, Nagy P. Quantitative characterization of the large-scale association of ErbB1 and ErbB2 by flow cytometric homo-FRET measurements. Biophys J. 2008;95:2086–2096. doi: 10.1529/biophysj.108.133371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M, Periasamy A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods. 2003;29:58–73. doi: 10.1016/s1046-2023(02)00283-9. [DOI] [PubMed] [Google Scholar]
- (36).Heinicke LA, Nallagatla SR, Hull CM, Bevilacqua PC. RNA helical imperfections regulate activation of the protein kinase PKR: effects of bulge position, size, and geometry. RNA. 2011;17:957–966. doi: 10.1261/rna.2636911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Patel S, Blose JM, Sokoloski JE, Pollack L, Bevilacqua PC. Specificity of the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR for double-stranded RNA: insights from thermodynamics and small-angle X-ray scattering. Biochemistry. 2012;51:9312–9322. doi: 10.1021/bi300935p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Heinicke LA, Wong CJ, Lary J, Nallagatla SR, Diegelman-Parente A, Zheng X, Cole JL, Bevilacqua PC. RNA dimerization promotes PKR dimerization and activation. J Mol Biol. 2009;390:319–338. doi: 10.1016/j.jmb.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Heinicke LA, Bevilacqua PC. Activation of PKR by RNA misfolding: HDV ribozyme dimers activate PKR. RNA. 2012;18:2157–2165. doi: 10.1261/rna.034744.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Minks MA, West DK, Benvin S, Greene JJ, Ts'o POP, Baglioni C. Activation of 2'-5`-oligo-(A) polymerase and protein kinase of interferon-treated HeLa cells by 2'-O-methylated poly(inosinic acid)-poly(cytidylic acid) J Biol Chem. 1980;255:6403–6407. [PubMed] [Google Scholar]
- (41).Ucci JW, Kobayashi Y, Choi G, Alexandrescu AT, Cole JL. Mechanism of interaction of the double-stranded RNA (dsRNA) binding domain of protein kinase R with short dsRNA sequences. Biochemistry. 2007;46:55–65. doi: 10.1021/bi061531o. [DOI] [PubMed] [Google Scholar]
- (42).Koh HR, Kidwell MA, Ragunathan K, Doudna JA, Myong S. ATP-independent diffusion of double-stranded RNA binding proteins. Proc Natl Acad Sci USA. 2013;110:151–156. doi: 10.1073/pnas.1212917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Spanggord RJ, Vuyisich M, Beal PA. Identification of binding sites for both dsRBMs of PKR on kinase-activating and kinase-inhibiting RNA ligands. Biochemistry. 2002;41:4511–4520. doi: 10.1021/bi0120594. [DOI] [PubMed] [Google Scholar]
- (44).Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, la Bastide, de M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Szustakowki J. International Human Genome Sequencing Consortium. (2001) Initial sequencing and analysis of the human genome. Nature. 409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- (45).Portal MM, Pavet V, Erb C, Gronemeyer H. Human cells contain natural double-stranded RNAs with potential regulatory functions. Nat Struct Mol Biol. 2015;22:89–97. doi: 10.1038/nsmb.2934. [DOI] [PubMed] [Google Scholar]
- (46).Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. Journal of Virology. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).George CX, John L, Samuel CE. An RNA Editor, Adenosine Deaminase Acting on Double-Stranded RNA (ADAR1) J Interferon Cytokine Res. 2014;34:437–446. doi: 10.1089/jir.2014.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Vitali P, Scadden ADJ. Double-stranded RNAs containing multiple IU pairs are sufficient to suppress interferon induction and apoptosis. Nat Struct Mol Biol. 2010;17:1043–1050. doi: 10.1038/nsmb.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science (New York, NY) 2015 doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.