

Abstract
Alzheimer disease (AD) is one of the most common neurodegenerative diseases characterized by memory loss and cognitive impairment. While the majority of AD cases are sporadic, some are caused by mutations in early-onset familial AD (FAD) genes. One FAD gene encodes presenilin 1 (PS1), and a PS1 mutation in methionine 146 impairs homeostatic synaptic plasticity (HSP). We have previously shown that Ca2+ and calcineurin activity are critical regulators of HSP. Here, we confirm that ER-mediated Ca2+ signals are increased in mutant PS1 neurons. We further show that calcineurin activity is abnormally elevated in the mutant, and that inhibition of increased calcineurin activity stabilizes GluA1 phosphorylation, promoting synaptic trafficking of Ca2+-permeable AMPA receptors, contributing to the recovery of impaired HSP found in the mutant. Because HSP is suggested to have roles during learning and memory formation, increased calcineurin activity-induced impairment of HSP can cause cognitive decline in FAD. Thus, reducing abnormally increased calcineurin activity in AD brain may be beneficial for improving AD-related cognitive decline.
Keywords: AMPA receptor, calcineurin, homeostatic synaptic plasticity, Alzheimer disease, presenilin1
1. Introduction
Alzheimer disease (AD) is a progressive, neurodegenerative disease characterized by memory loss, cognitive impairment, and neuron death (Selkoe, 2011). Amyloid beta (Aβ), a major component of amyloid plaque (the main characteristic pathology in the advanced stages of AD), is produced by sequential cleavages of the amyloid precursor protein (APP) by two proteases, β and γ-secretase (Selkoe, 2011). Aβ accumulation has been proposed to be the main cause of the disease, while Aβ-independent disease factors also contribute (Pimplikar, et al., 2010). While the majority of AD cases are sporadic, some are hereditary and caused by mutations that give rise to familial AD (FAD) (Selkoe, 2011). One FAD gene encodes presenilin 1 (PS1), a 9-pass transmembrane (TM) protein and the catalytic subunit of γ-secretase (LaFerla, 2002). The γ-secretase complex is important for producing amyloid plaques, and increased γ-secretase activity in the PS1 mutant is thought to be a basis for FAD (Selkoe, 2011). One group of FAD PS1 mutations involves methionine 146 (M146V, M146L and M146I), which lies in PS1 TM2, far from the catalytic site (TM6 and 7) (Li, et al., 2013), suggesting that its action may not be related to APP cleavage (Zhang, et al., 2013). Indeed, the role of PS1 mutations has recently been reinterpreted in terms of the AD Ca2+ hypothesis, which states that AD is a disease of Ca2+ deregulation (Berridge, 2011). Several studies confirm that PS1 mutants show alteration of the endoplasmic reticulum (ER) Ca2+ activity (Guo, et al., 1999, LaFerla, 2002). One of the mechanisms proposed to account for Ca2+ deregulation in PS1 mutants is that mutant PS1 activates the inositol 1,4,5-trisphosphate receptors (IP3Rs) and increases their Ca2+ conductance (Foskett, 2010, Muller, et al., 2011). This suggests that PS1 mutations increase intracellular Ca2+ levels by activating ER Ca2+ channels, which may contribute to brain dysfunction in AD.
Calcineurin is a Ca2+/calmodulin-activated phosphatase and a major regulator of several key proteins mediating synaptic transmission and neuronal excitability in both pre and postsynaptic areas (Baumgartel and Mansuy, 2012). It regulates AMPA receptor (AMPAR) trafficking by dephosphorylating one of the receptor subunits, GluA1, playing important roles in synaptic plasticity (Kim and Ziff, 2014, Lee, et al., 1998, Sanderson, et al., 2012). Phosphorylation of GluA1 within its intracellular carboxyl-terminal domain can regulate AMPAR membrane trafficking (Derkach, et al., 2007). Phosphorylation of serine 845 in GluA1 is important for activity-dependent trafficking of GluA1-containing AMPARs, and cAMP-dependent protein kinase A and cGMP-dependent protein kinase II can mediate this phosphorylation (Derkach, et al., 2007, Serulle, et al., 2007). Calcineurin dephosphorylates serine 845 in GluA1, which enables GluA1-containing AMPARs to be endocytosed from the plasma membrane during long-term depression (LTD) (Lee, et al., 1998, Sanderson, et al., 2012). Furthermore, mice expressing a dominant active calcineurin, as would be found in neurons with elevated Ca2+ activity, display impairment in both long-term potentiation (LTP) and long-term memory (Mansuy, et al., 1998, Winder, et al., 1998). The memory defects seen in these mice can be reversed by suppression of calcineurin (Dineley, et al., 2007, Dineley, et al., 2010). Therefore, calcineurin-mediated AMPAR trafficking is important for not only synaptic plasticity but also memory formation. Notably, overactivated calcineurin has been implicated in various aspects of AD-related pathology, including synaptic plasticity, spine morphology, and cognitive decline (Abdul, et al., 2009, Cavallucci, et al., 2013, Hudry, et al., 2012, Kuchibhotla, et al., 2008, Rozkalne, et al., 2011). Although these studies suggest that neuronal calcineurin can be activated in amyloid-based AD models, the cellular mechanisms underlying hyperactivation of calcineurin in PS1 FAD mutants and mutant PS1-mediated pathogenesis are not yet understood.
Homeostatic synaptic plasticity (HSP) also known as synaptic scaling is an important regulatory mechanism that compensates for altered neuronal activity (Turrigiano, 2012) and that recently has drawn attention as a means for counterbalancing synaptic alterations induced by LTP and thus stabilizing neuronal activity (Vitureira and Goda, 2013). Although LTP is strongly suggested to be a cellular basis of learning and memory, LTP can interact mechanistically with HSP, suggesting it has a role during learning and memory formation (Vitureira and Goda, 2013). PS1 M146V knockin (KI) mice display age-dependent deficits in hippocampal LTP and in learning and memory (Auffret, et al., 2010, Sun, et al., 2005). Significantly, PS1 M146V hippocampal neurons are deficient in synaptic scaling (Pratt, et al., 2011), suggesting that HSP may contribute cognitive impairment in FAD. However, the molecular mechanism underlying this defect is not yet determined. We have shown that calcineurin activity is important for activity deprivation-induced synaptic scaling in cultured cortical neurons (Kim and Ziff, 2014) and functional compensation in gene knockout in the hippocampus (Kim, et al., 2015). Inhibition of action potentials in cultured cortical neurons blocks Ca2+ entry, which decreases calcineurin activity (Kim and Ziff, 2014). This increases GluA1 phosphorylation, leading to elevation of synaptic trafficking of Ca2+-permeable AMPARs (CPARs) as a homeostatic response, which maintains not only synaptic strength but also Ca2+ signaling (Kim and Ziff, 2014). Therefore, reduction of calcineurin activity can be a trigger for synaptic homeostasis when neuronal activity is decreased. Because calcineurin activity is regulated by ER Ca2+ activity (Mehta and Zhang, 2015), the greatly elevated ER Ca2+ signals in PS M146V, both in the basal state and following IP3 release (Shilling, et al., 2014), could activate calcineurin, and in turn dephosphorylate synaptic substrates, diminish synaptic transmission, and block synaptic scaling.
Although many biochemical and genetic studies support the amyloid hypothesis, AD etiology is complex and amyloid alone is unable to explain all aspects of AD pathogenesis (Pimplikar, et al., 2010). Indeed, FAD-linked mutant PS1 enhances Ca2+ activity, which is implicated in AD pathogenesis (Cheung, et al., 2010, Cheung, et al., 2008, Muller, et al., 2011, Shilling, et al., 2014), however, downstream effects of such elevated Ca2+ signals on synaptic plasticity are unknown. Here, we describe for the first time that reduction of activated calcineurin rescues impaired synaptic scaling in PS1 M146V by promoting synaptic trafficking of CPARs. We confirm that ER-mediated Ca2+ activity is higher in the mutant, which accounts for an increase in calcineurin activity, leading to dephosphorylation of GluA1. Pharmacological inhibition of such enhanced calcineurin activity in cultured mutant hippocampal neurons recovers impaired synaptic scaling via synaptic CPAR expression. Therefore, reducing abnormally increased calcineurin activity in AD brain may be beneficial for overcoming memory loss.
2. Methods
2.1. Mouse hippocampal neuron cultures and neuronal transfection
Hippocampal primary neurons and transfection were carried out by the previously described method (Kim, et al., 2015, Kim and Ziff, 2014). Neurons were isolated from embryonic day 17-18 C57Bl6 or PS1 M146V knockin mouse embryonic brain tissues. All animal studies were performed with an approved protocol from New York University Langone Medical Center's Institutional Animal Care and Use Committee.
2.2. Ca2+ imaging
Ca2+ imaging was performed as the previous study (Kim, et al., 2015, Kim and Ziff, 2014). DIV4 Neurons were transfected with GCaMP5 (Addgene, 31788). Neurons were grown for 10-12 days after transfection in Neurobasal medium without phenol red and supplemented with B27 and 0.5mM Glutamax. Glass-bottom dishes were mounted on a temperature-controlled stage on Zeiss Axiovert 200M and maintained at 37°C and 5% CO2 using a Zeiss digital temperature and humidity controller. Images were captured for exposure time of 0.5 seconds depending on the intensity of the fluorescence signal using a 63× oil-immersion objective. Spontaneous Ca2+ signals in the cell body (excluding dendrites) were measured as changes of GCaMP5 fluorescence intensity (F). 100 images (Ft=1-100) were obtained with 1s interval, and analyzed using the ImageJ software. Relative fluorescence difference (ΔF/F0) was calculated by ΔF/F0 = (Ft-F0)/F0 in each image (F0 was determined as the minimum value during the imaging). Total Ca2+ signals were obtained by combining 100 values of ΔF/F0 in each image, and values of ΔF/F0 <0.3 were rejected due to bleaching.
2.3. Miniature EPSC (excitatory postsynaptic currents) recording
Miniature EPSCs were measured in cultured hippocampal neurons (Kim, et al., 2015, Kim and Ziff, 2014). Neurons were voltage clamped with the whole cell ruptured path technique during the recording. 1μM TTX (Tocris Biosciences) and 10μM bicuculline (Tocris Biosciences) were added to the bath. mEPSCs were recorded at -60mV with a Warner amplifier (PC-501A) and filtered at 1kHz. Recordings were digitized (Digidata 1440, Molecular Devices) and analyzed using the Mini Analysis software (Synaptosoft). The access resistance (Ra<25MΩ) was monitored during recording to eliminate artifacts. Events whose amplitude was less than 7.5pA were rejected. 20μM naspm (1-naphthylacetyl spermine trihydrochloride, Tocris Biosciences) was added to suppress CPAR-mediated transmission in the bath solution.
2.4. Synaptosome and PSD purification, surface biotinylation, and immunoblots
Synaptosomal fractions from DIV14 primary hippocampal neurons were prepared as described previously (Kim, et al., 2015, Kim and Ziff, 2014). PSD fractions from the heterozygotes and homozygotes of the PS1 M146V hippocampus were prepared as described previously (Kim, et al., 2015). Surface biotinylation was performed according to the previous study (Kim, et al., 2015, Kim and Ziff, 2014). Immunoblots were carried out as the previous reports (Kim, et al., 2015, Kim and Ziff, 2014).
2.5. FRET-based calcineurin activity assay
Neurons were transfected with the calcineurin activity biosensor, and FRET activity was measured at DIV14 according to the previously described method (Kim, et al., 2015, Kim and Ziff, 2014). Images were captured by using Applied Precision PersonalDV live-cell imaging system in the Microscopy Core of New York University Langone Medical Center.
2.6. Statistics
Most statistical comparisons were analyzed with the GraphPad Prism6 software. Unpaired two-tailed Student's t-tests were used in single comparisons. For multiple comparisons, we used one-way analysis of variance (ANOVA) followed by Fisher's Least Significant Difference (LSD) test to determine statistical significance. The Kolmogorov-Smirnov (K-S) test (http://www.physics.csbsju.edu/stats/KS-test.html) was used for comparisons of cumulative probabilities. Results were represented as a mean ± s.e.m. and p value<0.05 was considered statistically significant.
3. Results
3.1 Enhanced ER-mediated Ca2+ signaling in PS1 M146V hippocampal neurons
Abnormally elevated Ca2+ signaling is found in non-neuronal cells from presymptomatic FAD patients (Etcheberrigaray, et al., 1998) and in neurons in FAD mouse models (Stutzmann, et al., 2004), indicating that increased intracellular Ca2+ caused by FAD PS1 is an early phenotype and could contribute to disease pathogenesis. We thus investigated Ca2+ activity in DIV14 cultured mutant hippocampal neurons transfected with GCaMP5, a genetically encoded Ca2+ indicator (Akerboom, et al., 2012, Kim, et al., 2015, Kim and Ziff, 2014). We found active spontaneous Ca2+ transients in both wild-type (WT) and the PS1 M146V mutant, however, total Ca2+ activity in the mutant was significantly higher than in WT neurons (p<0.0001) (Fig. 1), consistent with the previous finding in lymphoblasts carrying PS1 M146L mutant (Cheung, et al., 2010). It has been shown that PS1 can interact with IP3Rs and that FAD mutations in PS1 provide enhancement of IP3R channel functions (Cheung, et al., 2008). To confirm this, we treated neurons with 50μM 2APB, an IP3R blocker, and found that both WT and PS1 neurons displayed a significant decrease in GCaMP5 activity (p<0.0001) (Fig. 1). Ryanodine receptors (RyRs) are another Ca2+ channel located in the ER, and are also involved in intracellular Ca2+ release (Bading, 2013). Notably, PS1 regulates neurotransmitter release, mediated by RyRs (Zhang, et al., 2009). It also regulates RyR protein expression in neurons (D'Adamio and Castillo, 2013). To test whether increased Ca2+ activity found in the PS1 mutant is mediated by RyRs, we added 25μM dantrolene, a RyR blocker, and found that dantrolene treatment significantly lowered GCaMP5 activity in mutant neurons (p<0.0001) (Fig. 1) while it had no effect on Ca2+ currents in WT cells (Fig. 1). This suggests that RyRs have minor roles in Ca2+ release under the basal conditions, but are activated in the mutant and contribute to elevation of Ca2+ activity in the mutant. Taken together, these results indicate that PS1 M146V hippocampal neurons have abnormally high Ca2+ activity, which is induced by both IP3R and RyR-mediated ER Ca2+ release.
Figure 1. Enhanced ER-mediated Ca2+ signaling in PS1 M146V hippocampal neurons.
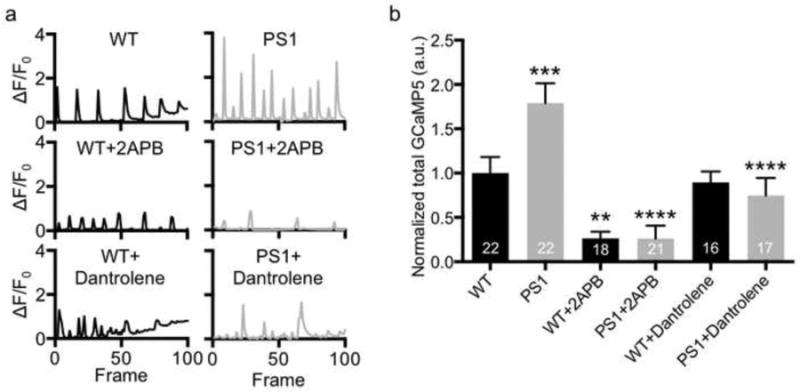
a) Example bar graphs of Ca2+ signals in each condition. Each bar represents the GCaMP5 fluorescence intensity detected in a single exposure frame. b) Normalized average of total Ca2+ activity in each condition showing that PS1 mutant neurons have higher ER-mediated Ca2+ signals (n=number of neurons, **p<0.01, ***p<0.001, and ****p<0.0001, one-way ANOVA, uncorrected Fisher's LSD).
3.2. Elevated calcineurin activity in PS1 M146V can decrease GluA1 S845 phosphorylation
Although an increase in Ca2+ activity is evident in the PS1 mutant, the consequence of this exaggerated Ca2+ release in vivo is not clear, nor is the contribution of exaggerated Ca2+ release to PS1-associated FAD pathogenesis known. Because ER Ca2+ activity can regulate major neuronal phosphatase, calcineurin (Mehta and Zhang, 2015), the abnormally increased ER Ca2+ signals in the mutant could activate calcineurin. To measure in vivo calcineurin activity directly, we used a FRET (fluorescence resonance energy transfer)-based calcineurin activity sensor that utilizes a calcineurin activity-dependent molecular switch based on the N-terminal regulatory domain of NFAT (nuclear factor of activated T-cells) as a specific substrate, which was inserted between CFP and YFP (Kim, et al., 2015, Kim and Ziff, 2014, Newman and Zhang, 2008). Using this biosensor, we found that in vivo calcineurin activity in PS1 M146V hippocampal neurons was significantly higher than in WT neurons (WT; 1.40±0.01 and PS1; 1.56±0.02, p<0.0001) (Fig. 2). This suggests that abnormally increased Ca2+ in the mutant elevates calcineurin activity. We have shown that activated calcineurin decreases GluA1 S845 phosphorylation (Kim and Ziff, 2014), thus PS1 M146V could exhibit lower phosphorylation of GluA1. To test this, we first isolated synaptososomes from WT and mutant cultured hippocampal neurons and measured AMPAR levels (Fig. 3a). As expected, GluA1 S845 phosphorylation was significantly reduced (p=0.0003) while total GluA1 and GluA2/3 levels were not altered (Fig. 3a). Moreover, we purified postsynaptic density (PSD) from the hippocampus of 18-month old PS1 M146V homozygous and heterozygous animals to determine synaptic AMPAR levels (Fig. 3b). Hippocampal PSD from PS1 homozygotes contained significantly less GluA1 S845 phosphorylation (p<0.001) and synaptic GluA1 (p=0.034) compared with PS1 heterozygotes (Fig. 3b). However, GluA2/3 levels were not altered (Fig. 3b), suggesting that the effect of enhanced calcineurin activity in the mutant on GluA1 is selective. This suggests that abnormally elevated calcineurin activity in PS1 M146V selectively dephosphorylates GluA1, which is also found in the amyloid-based AD mouse model (Cavallucci, et al., 2013).
Figure 2. Elevated calcineurin activity in PS1 M146V can be reduced by FK506 treatment.

Representative images of CFP channel, FRET channel, and pseudocolored emission ratio (FRET/CFP) in each condition (Blue (L): low emission ratio and red (H): high emission ratio). A scale bar indicates 10μm. A summary graph showing an increase in calcineurin activity in PS1 M146V, which can be reduced by FK506 treatment (n=number of cells, *p<0.05, **p<0.01, and ****p<0.0001, one-way ANOVA, uncorrected Fisher's LSD).
Figure 3. Elevated calcineurin activity in PS1 M146V can decrease GluA1 S845 phosphorylation.
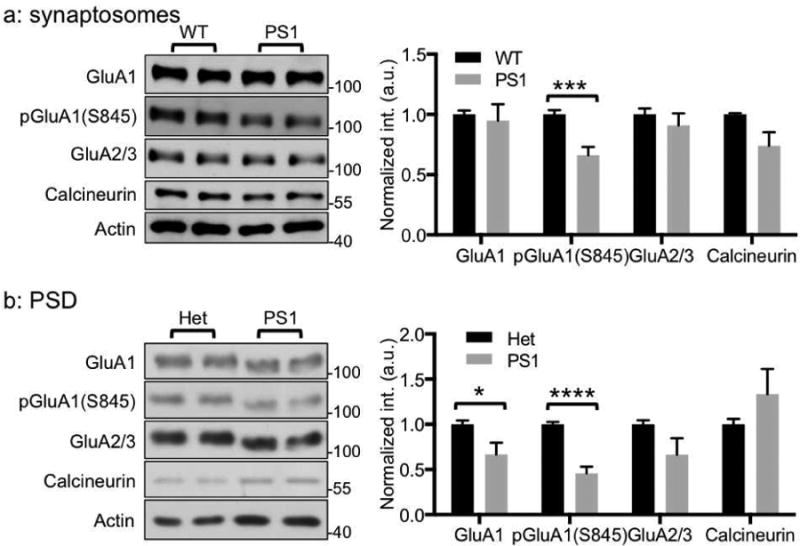
a) Representative immunoblots and quantitative analysis of synaptosomes from cultured hippocampal neurons of WT and PS1 M146V showing that selective reduction of GluA1 S845 phosphorylation [pGluA1(S845)] (n=6 experiments, ***p<0.001, unpaired two-tailed student's t-test). b) Representative immunoblots and quantitative analysis of PSD from the hippocampus of 18-month old heterozygotes (het) and homozygotes (PS1) of PS1 M146V showing GluA1 and GluA1 S845 phosphorylation are decreased (n=5 WT and 5 KO animals, *p<0.05 and ****p<0.0001, unpaired two-tailed student's t-test).
3.3 Elevated calcineurin activity in PS1 M146V can be reduced by FK506 treatment
We have shown that reduction of calcineurin activity is required for activity deprivation-induced synaptic scaling in cultured cortical neurons, and constitutively active calcineurin blocks TTX-induced synaptic scaling (Kim and Ziff, 2014). These findings suggest that synaptic scaling can fail to take place in PS1 mutant neurons due to their higher calcineurin activity. Indeed, TTX treatment is unable to induce synaptic scaling in PS1 M146V hippocampal neurons (Pratt, et al., 2011). Thus, we tested whether TTX was sufficient to reduce calcineurin activity in the mutant. We treated mutant neurons with 2μM TTX for 48hr and found that it was not capable of reducing calcineurin activity (PS1; 1.56±0.02 and PS1+TTX; 1.53±0.03) (Fig. 2). This could explain why TTX is not able to induce synaptic scaling in the mutant. To reduce elevated calcineurin activity in the mutant, we treated neurons with FK506, a drug that forms a drug-immunophilin complex that is a highly specific inhibitor for calcineurin (Liu, et al., 1991), and found that 12hr treatment with 5μM FK506 significantly decreased calcineurin activity as compared with that under the basal condition (PS1; 1.56±0.02 and PS1+FK506; 1.29±0.01, p<0.0001) (Fig. 2). Therefore, exacerbated Ca2+ activity in PS1 M146V increases calcineurin activity, which inhibits TTX-induced synaptic scaling, and the decreased calcineurin activity can be recovered by FK506 treatment.
3.4. FK506 treatment is sufficient for inducing synaptic scaling in PS1 M146V
Because inhibition of calcineurin activity in cultured cortical neurons is sufficient to induce CPAR-mediated synaptic scaling (Kim and Ziff, 2014), and FK506 treatment is able to reduce elevated calcineurin activity in PS1 M146V hippocampal neurons, we asked whether FK506 treatment can induce synaptic scaling in mutant neurons. To test this, we studied spontaneous synaptic transmission by measuring mEPSCs in DIV14-17 mutant hippocampal neurons (Fig. 4a). We first treated mutant neurons with 2μM TTX for 48hr and found that there were no changes in mEPSC amplitude and frequency (Fig. 4b-c), consistent with the previous report (Pratt, et al., 2011). However, treatment for 12hr with 5μM FK506 significantly increased average mEPSC amplitude (DMSO; 14.99±0.69pA and FK506; 19.16±0.51pA, p<0.0001) (Fig. 4b). Consistent with the previous finding (Kim and Ziff, 2014), the average mEPSC frequency was also increased when neurons were treated with FK506 (DMSO; 3.37±0.57Hz and FK506; 5.87±0.29Hz, p<0.0004) (Fig. 4c). There was a significant decrease in mEPSC decay time (peak to 10%) with FK506 treatment (DMSO; 3.65±0.10ms and FK506; 3.08±0.08ms, p=0.005) (Fig. 4d), indicating that CPARs mediate mEPSC changes in FK506-treated neurons. We next used 20μM naspm (1-naphthyl acetyl spermine), a blocker of CPARs, to determine if CPARs were responsible for the FK506-mediated increase of the mEPSC amplitude (Fig. 4a). Consistent with the previous findings (Kim, et al., 2015, Kim and Ziff, 2014), naspm treatment significantly reduced the FK506-induced increase in amplitude (FK506; 19.16±0.51pA and FK506+naspm; 15.06±0.84pA, p<0.0001) (Fig. 4b) and frequency (FK506; 5.87±0.29Hz and FK506+naspm; 3.10±0.56Hz, p=0.0002) (Fig. 4c). Moreover, the FK506-induced reduction of decay time was recovered by naspm treatment (FK506; 3.08±0.08ms and FK506+naspm; 3.53±0.15ms, p=0.03) (Fig. 4d). This confirms that FK506-induced synaptic scaling in the mutant is mediated by CPARs. Synaptic scaling can be global and multiplicative, properties that are important for preserving the relative strength differences between synapses. Because each synapse strength is multiplied or divided by the same factor, each synaptic strength is increased or decreased in proportion to its initial strength (Turrigiano and Nelson, 2004). In addition, synaptic scaling is induced by synapse-specific processes, providing local control of synaptic strength (Beique, et al., 2011). To determine whether FK506-induced synaptic scaling in the mutant occurs in a multiplicative manner, we analyzed the mEPSC amplitude distribution (Fig. 4e). The cumulative probability of mEPSC amplitude in FK506-treated neurons shifted towards larger values compared with that in DMSO-treated neurons (Kolmogrov-Smirnov test, p<0.0001) (Fig. 4e). However, when mEPSC amplitude of FK506-treated neurons was divided by a factor (1.27) to match the average mEPSC amplitude of DMSO-treated cells, the cumulative probability curve was not completely superimposed (Kolmogrov-Smirnov test, p<0.05) (Fig. 4e). Taken together, we are able to rescue impaired homeostatic synaptic plasticity found in PS1 M146V hippocampal neurons by pharmacological inhibition of calcineurin activity, and FK506 treatment induces CPAR-mediated non-multiplicative scaling in the mutant.
Figure 4. FK506 treatment is sufficient for inducing synaptic scaling in PS1 M146V.

a) Representative traces of mEPSC recordings in each condition (n=number of cells). b) An average mEPSC amplitude graph showing FK506 treatment increases the CPAR-dependent amplitude (****p<0.0001, one-way ANOVA, uncorrected Fisher's LSD). c) An average mEPSC frequency graph showing FK506 treatment increases the CPAR-mediated frequency (***p<0.001, one-way ANOVA, uncorrected Fisher's LSD). d) An average decay time (peak to 10%) graph showing that FK506 treatment induces synaptic expression of CPARs (*p<0.05, one-way ANOVA, uncorrected Fisher's LSD). e) Average cumulative probability of mEPSC amplitude. FK506-induced distribution is significantly different from the DMSO-treated control (p<0.0001, K-S test). Distribution of FK506 scaled down by a factor of 1.27 does not fitted to the DMSO-treated control (p<0.05, K-S test).
3.5. FK506 treatment-induced selective synaptic trafficking of GluA1
Because GluA1 S845 phosphorylation is required not only for homeostatic scaling (Diering, et al., 2014, Goel, et al., 2011, Kim and Ziff, 2014) but also for maintaining CPARs on the synaptic membrane (He, et al., 2009), we measured the effects of FK506 on GluA1 S845 phosphorylation levels by purifying synaptosomes from mutant neurons treated with 5μM FK506 for 12hr and measuring protein and phosphorylation levels of AMPAR subunits. FK506 treatment significantly increased both GluA1 S845 phosphorylation (p=0.001) and total GluA1 (p=0.016) while GluA2/3 and calcineurin levels were not changed compared with DMSO-treated neurons (Fig. 5a), indicating synaptic trafficking of GluA1 homomeric AMPARs. We further determined that surface GluA1 levels were elevated after FK506 treatment (p=0.03) (Fig. 5b). In contrast, surface GluA2/3 was not altered (Fig. 5b), confirming that GluA1 homoermic CPARs are trafficked after FK506 treatment in PS1 M146V neurons.
Figure 5. FK506 treatment-induced selective synaptic trafficking of GluA1.

a) Representative immunoblots and quantitative analysis of synaptosomes from cultured mutant hippocampal neurons in the presence and absence of FK506 treatment showing a selective increase in total GluA1 and GluA1 S845 phosphorylation [pGluA1(S845)] (n=10 experiments, *p<0.05 and ***p<0.001, unpaired two-tailed student's t-test). b) Representative immunoblots of surface biotinylation and a summary graph in DMSO (D) or FK506 (F)-treated neurons showing that FK506 treatment selectively increases surface GluA1 levels (n=10 experiments, *p<0.05, unpaired two-tailed student's t-test).
4. Discussion
Although many reports have suggested that neuronal calcineurin can be hyperactivated in AD (Abdul, et al., 2009, Cavallucci, et al., 2013, Hudry, et al., 2012, Kuchibhotla, et al., 2008, Rozkalne, et al., 2011), these studies are mainly carried out in the amyloid-based models, and the PS1 mutant-mediated alteration of calcineurin activity has not been investigated. Furthermore, only in vitro calcineurin activity has been measured in most reports, thus accurate in vivo calcineurin activity has not been determined. Finally, downstream effects of hyperactivated calcineurin on synapses in AD are largely unknown. We describe a novel mechanism, in which mutant PS1-mediated impairment of synaptic scaling is dependent on overactivtaion of calcineurin, which can be recovered by suppression of calcineurin activity. We find that PS1 M146V hippocampal neurons display enhanced ER-linked Ca2+ activity (Fig. 1), which activates calcineurin, a major brain phosphatase (Fig. 2). Such elevated calcineurin activity dephosphorylates GluA1 (Fig. 3), indicating synaptic impairment in the PS1 mutant hippocampus, possibly providing the cellular basis for memory loss in FAD. We have previously shown that increased calcineurin activity blocks synaptic scaling while inhibition of calcineurin activity is sufficient to induce HSP (Kim and Ziff, 2014). In the current study, we show for the first time that the impaired synaptic scaling found in the mutant (Pratt, et al., 2011), which is mediated by CPARs, is rescued by pharmacological reduction of elevated calcineurin activity (Fig. 4-5). We note that although TTX reduced GCaMP5 activity in both WT and PS1 mutant neurons (Data not shown), it only reduced calcineurin activity in the WT (Fig. 2). A computational model shows that calcineurin can remain active at moderate Ca2+ levels (Graupner and Brunel, 2010). A recent study suggests that the presenilin FAD mutant elevates lysosomal pH to increase Ca2+ efflux from the lysosomes (McBrayer and Nixon, 2013), an increase that would be voltage independent. Although TTX blocks the voltage-dependent changes in Ca2+ activity to which GCaMP5 is responsive, the actual Ca2+ concentration may be higher in the PS1 mutant than the WT and thus higher calcineurin activity can be maintained in TTX-treated mutant neurons. Taken together, calcineurin activity is pathologically elevated in PS1 M146V hippocampal neurons, impairing HSP, and pharmacological suppression of calcineurin activity recovers such defects in the mutant, suggesting a novel therapeutic strategy for AD.
Expression of a dominant active calcineurin mutant in animals impairs both synaptic plasticity and learning and memory, which impairment can be reversed by suppression of the mutant calcineurin (Dineley, et al., 2007, Dineley, et al., 2010, Mansuy, et al., 1998, Winder, et al., 1998). This suggests that abnormal activation of calcineurin can cause pathology in neurons. Indeed, calcineurin hyperactivation impairs long-term depression and spine integrity, possibly contributing to synaptic loss in amyloid-based AD models (Abdul, et al., 2009, Cavallucci, et al., 2013, Kuchibhotla, et al., 2008, Rozkalne, et al., 2011). It is also strongly correlated with cognitive decline in AD (Hudry, et al., 2012). Moreover, a decrease in calcineurin activity is beneficial for such pathological changes (Abdul, et al., 2009, Cavallucci, et al., 2013, Hudry, et al., 2012, Kuchibhotla, et al., 2008, Rozkalne, et al., 2011). Although these studies clearly suggest that hyperactivation of calcineurin is pathological in AD, how amyloid affects Ca2+ and calcineurin activity is largely unknown. Recently, inhibition of calcineurin rescues structural plasticity in PS1 M146V neurons (Zhang, et al., 2015). However, it is not understood how the direct downstream effectors of calcineurin affect plasticity. Therefore, we suggest that our finding is novel because we: 1) directly measure an increase in in vivo Ca2+ and calcineurin activity in PS1 M146V neurons, 2) confirm that GluA1 is the target of calcineurin, contributing synaptic impairment, and 3) can rescue loss of HSP by inhibition of calcineurin activity in the PS1 M146V.
The M146V mutation may also show pathological changes by a second pathway. This pathway involves phosphatidylinositol 3-kinase (PI3K) and Akt (also known as protein kinase B)-mTOR-dependent signaling, which controls local GluA1 synthesis(Schratt, et al., 2004, Troca-Marin, et al., 2011), which is also known to contribute to synaptic plasticity by providing newly synthesized GluA1 to synapses (Aoto, et al., 2008). Indeed, PS1 mutants suppress PI3K-Akt signaling, which inactivates mTOR-dependent downstream effects (Baki, et al., 2008), suggesting that mTOR-mediated GluA1 local translation is impaired in the mutant, which could account for HSP failure. Indeed, activation of Akt is able to recover impaired synaptic scaling in the mutant although the molecular mechanism is not known (Pratt, et al., 2011).
Hebbian plasticity, including LTP and LTD, have been extensively studied as cellular mechanisms of learning and memory and considered as input specific, in which changes of synaptic strength are limited to active synapses (Bi and Poo, 2001). Conversely, HSP has been generally referred to as employing global changes, such that all synaptic inputs received by a given neuron are scaled equally (Turrigiano, et al., 1998). Thus, HSP can preserve the relative differences in synaptic strengths resulting from input-specific Hebbian changes, providing a basis for the possible interaction of these forms of synaptic plasticity for memory storage (Vitureira and Goda, 2013). However, increasing evidence supports the idea that both global and local homeostatic mechanisms might operate in parallel (Pozo and Goda, 2010). Indeed, we found that FK506-induced rescue of impaired synaptic scaling in PS1 mutant neurons is not multiplicative (Fig. 4). Thus, such recovery occurs in local active synapses, suggesting that effects of PS1 mutant-induced pathogenesis on synaptic plasticity can be confined to subset of synapses rather than in global synapses.
Both intracellular Ca2+ levels and calcineurin activity are elevated in aged and AD brains (Reese and Taglialatela, 2011), suggesting that the Ca2+-calcineurin pathway that we describe in the PS1 FAD model may also apply to sporadic forms of the disease. FK506 is a clinically approved inhibitor of calcineurin that has been reported to improve memory in an amyloid-based AD mouse model (Dineley, et al., 2007, Dineley, et al., 2010). Thus, reducing abnormally increased calcineurin activity by using FK506 in AD brain may be beneficial for improving AD and aging related cognitive decline.
Highlights.
PS1 M146V neurons have higher Ca2+ and calcineurin activity.
GluA1 phosphorylation is decreased in mutant synapses.
A calcineurin inhibitor rescues GluA1 phosphorylation and loss of synaptic scaling.
Decreasing calcineurin activity can be a novel therapeutic target.
Acknowledgments
We appreciate all members of the Ziff lab. We think Drs. Mark P. Mattson (National Institute on Aging) and Jin Zhang (Johns Hopkins University) for providing PS1 M146V KI mice and FRET-based calcineurin biosensor, respectively. FRET assay was done in the Microscopy Core of NYU Langone Medical Center. We thank Dr. Loren Looger (Janelia farm) for providing the GCaMP5 construct. This work was supported by a Blas Frangione postdoctoral fellowship (SK), and NIH grant, 5R01NS061920 (EBZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29(41):12957–69. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ, Tian L, Marvin JS, Mutlu S, Calderon NC, Esposti F, Borghuis BG, Sun XR, Gordus A, Orger MB, Portugues R, Engert F, Macklin JJ, Filosa A, Aggarwal A, Kerr RA, Takagi R, Kracun S, Shigetomi E, Khakh BS, Baier H, Lagnado L, Wang SS, Bargmann CI, Kimmel BE, Jayaraman V, Svoboda K, Kim DS, Schreiter ER, Looger LL. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. J Neurosci. 2012;32(40):13819–40. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, Chen L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron. 2008;60(2):308–20. doi: 10.1016/j.neuron.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffret A, Gautheron V, Mattson MP, Mariani J, Rovira C. Progressive age-related impairment of the late long-term potentiation in Alzheimer's disease presenilin-1 mutant knock-in mice. Journal of Alzheimer's disease: JAD. 2010;19(3):1021–33. doi: 10.3233/JAD-2010-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H. Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci. 2013;14(9):593–608. doi: 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- Baki L, Neve RL, Shao Z, Shioi J, Georgakopoulos A, Robakis NK. Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J Neurosci. 2008;28(2):483–90. doi: 10.1523/JNEUROSCI.4067-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartel K, Mansuy IM. Neural functions of calcineurin in synaptic plasticity and memory. Learning & memory. 2012;19(9):375–84. doi: 10.1101/lm.027201.112. [DOI] [PubMed] [Google Scholar]
- Beique JC, Na Y, Kuhl D, Worley PF, Huganir RL. Arc-dependent synapse-specific homeostatic plasticity. Proc Natl Acad Sci U S A. 2011;108(2):816–21. doi: 10.1073/pnas.1017914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Calcium signalling and Alzheimer's disease. Neurochemical research. 2011;36(7):1149–56. doi: 10.1007/s11064-010-0371-4. [DOI] [PubMed] [Google Scholar]
- Bi G, Poo M. Synaptic modification by correlated activity: Hebb's postulate revisited. Annual review of neuroscience. 2001;24:139–66. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- Cavallucci V, Berretta N, Nobili A, Nistico R, Mercuri NB, D'Amelio M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer's disease. Neuromolecular medicine. 2013;15(3):541–8. doi: 10.1007/s12017-013-8241-2. [DOI] [PubMed] [Google Scholar]
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Science signaling. 2010;3(114):ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58(6):871–83. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Adamio L, Castillo PE. Presenilin-ryanodine receptor connection. Proc Natl Acad Sci U S A. 2013;110(37):14825–6. doi: 10.1073/pnas.1313996110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8(2):101–13. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Diering GH, Gustina AS, Huganir RL. PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron. 2014;84(4):790–805. doi: 10.1016/j.neuron.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiology of learning and memory. 2007;88(2):217–24. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. Journal of neuroscience research. 2010;88(13):2923–32. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etcheberrigaray R, Hirashima N, Nee L, Prince J, Govoni S, Racchi M, Tanzi RE, Alkon DL. Calcium responses in fibroblasts from asymptomatic members of Alzheimer's disease families. Neurobiology of disease. 1998;5(1):37–45. doi: 10.1006/nbdi.1998.0176. [DOI] [PubMed] [Google Scholar]
- Foskett JK. Inositol trisphosphate receptor Ca2+ release channels in neurological diseases. Pflugers Archiv: European journal of physiology. 2010;460(2):481–94. doi: 10.1007/s00424-010-0826-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Xu LW, Snyder KP, Song L, Goenaga-Vazquez Y, Megill A, Takamiya K, Huganir RL, Lee HK. Phosphorylation of AMPA receptors is required for sensory deprivation-induced homeostatic synaptic plasticity. PLoS One. 2011;6(3):e18264. doi: 10.1371/journal.pone.0018264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M, Brunel N. Mechanisms of induction and maintenance of spike-timing dependent plasticity in biophysical synapse models. Front Comput Neurosci. 2010;4 doi: 10.3389/fncom.2010.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nature medicine. 1999;5(1):101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc Natl Acad Sci U S A. 2009;106(47):20033–8. doi: 10.1073/pnas.0910338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Wu HY, Arbel-Ornath M, Hashimoto T, Matsouaka R, Fan Z, Spires-Jones TL, Betensky RA, Bacskai BJ, Hyman BT. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer's disease. J Neurosci. 2012;32(9):3176–92. doi: 10.1523/JNEUROSCI.6439-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Titcombe RF, Zhang H, Khatri L, Girma HK, Hofmann F, Arancio O, Ziff EB. Network compensation of cyclic GMP-dependent protein kinase II knockout in the hippocampus by Ca2+-permeable AMPA receptors. Proc Natl Acad Sci U S A. 2015;112(10):3122–7. doi: 10.1073/pnas.1417498112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ziff EB. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS biology. 2014;12(7):e1001900. doi: 10.1371/journal.pbio.1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59(2):214–25. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3(11):862–72. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21(5):1151–62. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493(7430):56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66(4):807–15. doi: 10.1016/0092-8674(91)90124-h. doi:0092-8674(91)90124-H [pii] [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Winder DG, Moallem TM, Osman M, Mayford M, Hawkins RD, Kandel ER. Inducible and reversible gene expression with the rtTA system for the study of memory. Neuron. 1998;21(2):257–65. doi: 10.1016/s0896-6273(00)80533-4. [DOI] [PubMed] [Google Scholar]
- McBrayer M, Nixon RA. Lysosome and calcium dysregulation in Alzheimer's disease: partners in crime. Biochemical Society transactions. 2013;41(6):1495–502. doi: 10.1042/BST20130201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S, Zhang J. Dynamic visualization of calcium-dependent signaling in cellular microdomains. Cell calcium. 2015 doi: 10.1016/j.ceca.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Cardenas C, Mei L, Cheung KH, Foskett JK. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer's disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci U S A. 2011;108(32):13293–8. doi: 10.1073/pnas.1109297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RH, Zhang J. Visualization of phosphatase activity in living cells with a FRET-based calcineurin activity sensor. Molecular bioSystems. 2008;4(6):496–501. doi: 10.1039/b720034j. [DOI] [PubMed] [Google Scholar]
- Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. J Neurosci. 2010;30(45):14946–54. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Goda Y. Unraveling mechanisms of homeostatic synaptic plasticity. Neuron. 2010;66(3):337–51. doi: 10.1016/j.neuron.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Zimmerman EC, Cook DG, Sullivan JM. Presenilin 1 regulates homeostatic synaptic scaling through Akt signaling. Nat Neurosci. 2011;14(9):1112–4. doi: 10.1038/nn.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese LC, Taglialatela G. A role for calcineurin in Alzheimer's disease. Current neuropharmacology. 2011;9(4):685–92. doi: 10.2174/157015911798376316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozkalne A, Hyman BT, Spires-Jones TL. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiology of disease. 2011;41(3):650–4. doi: 10.1016/j.nbd.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, Dell'Acqua ML. AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J Neurosci. 2012;32(43):15036–52. doi: 10.1523/JNEUROSCI.3326-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24(33):7366–77. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease. Cold Spring Harb Perspect Biol. 2011;3(7) doi: 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56(4):670–88. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilling D, Muller M, Takano H, Mak DO, Abel T, Coulter DA, Foskett JK. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer's disease pathogenesis. J Neurosci. 2014;34(20):6910–23. doi: 10.1523/JNEUROSCI.5441-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24(2):508–13. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Beglopoulos V, Mattson MP, Shen J. Hippocampal spatial memory impairments caused by the familial Alzheimer's disease-linked presenilin 1 M146V mutation. Neuro-degenerative diseases. 2005;2(1):6–15. doi: 10.1159/000086426. [DOI] [PubMed] [Google Scholar]
- Troca-Marin JA, Alves-Sampaio A, Montesinos ML. An increase in basal BDNF provokes hyperactivation of the Akt-mammalian target of rapamycin pathway and deregulation of local dendritic translation in a mouse model of Down's syndrome. J Neurosci. 2011;31(26):9445–55. doi: 10.1523/JNEUROSCI.0011-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4(1):a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391(6670):892–6. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5(2):97–107. doi: 10.1038/nrn1327. doi:10.1038/nrn1327nrn1327 [pii] [DOI] [PubMed] [Google Scholar]
- Vitureira N, Goda Y. Cell biology in neuroscience: the interplay between Hebbian and homeostatic synaptic plasticity. The Journal of cell biology. 2013;203(2):175–86. doi: 10.1083/jcb.201306030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92(1):25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Sudhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460(7255):632–6. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu J, Sun S, Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling, excitability, and synaptic plasticity defects in a mouse model of Alzheimer's disease. Journal of Alzheimer's disease: JAD. 2015;45(2):561–80. doi: 10.3233/JAD-142427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhang M, Cai F, Song W. Biological function of Presenilin and its role in AD pathogenesis. Translational neurodegeneration. 2013;2(1):15. doi: 10.1186/2047-9158-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]