

Abstract
Glioblastoma can originate from terminally differentiated astrocytes and neurons, which can dedifferentiate to a stem cell-like state upon transformation. In this study, we confirmed that transformed dedifferentiated astrocytes and neurons acquired a stem/ progenitor cell state, although they still retained gene expression memory from their parental cell. Transcriptional network analysis on these cells identified upregulated genes in three main pathways: Wnt signaling, cell cycle and focal adhesion with the gene Spp1, also known as osteopontin (OPN) serving as a key common node connecting these three pathways. Inhibition of OPN blocked the formation of neurospheres, affected the proliferative capacity of transformed neurons and reduced the expression levels of neural stem cell markers. Specific inhibition of OPN in both murine and human glioma tumors prolonged mice survival. We conclude that OPN is an important player in dedifferentiation of cells during tumor formation, hence its inhibition can be a therapeutic target for glioblastoma.
Introduction
Glioblastoma multiforme (GBM) is the most common and most aggressive form of primary brain tumors in adults. It accounts for almost 20% of all primary brain tumors diagnosed annually and about half of all cases of malignant brain cancer.
Despite some advances in therapy over the past two decades, treatment outcomes remain poor, with typical median survival times after diagnosis ranging from 1 to 2 years. GBM has a very high recurrence rate, and transplantation of as few as 10–100 malignant tumor cells can lead to tumorigenesis. Thus, it appears that GBM tumor cells have the potential to function as stem cells. Cancer stem cells were initially described in hematopoietic cell malignances1 and glioma cancer stem cells were among the first to be identified and isolated from solid tumors.2 The isolation of tumor cells with stem cell features from human gliomas implied neural stem cells (NSCs) as possible cells of origin. However, these NSCs-like properties of glioma cancer stem cells could be acquired during transformation and as our group and others have recently shown,3–6 differentiated non-stem cancer cells can undergo dedifferentiation to form tumor stem-like cells. In this study, we investigated the mechanisms of dedifferentiation/reprogramming achieved by cortical mature neurons and astrocytes upon transduction with lentiviral vector containing HRasV12 and shp53.4 We performed whole genome transcriptome analysis of the dedifferentiated neurons and astrocytes along with the enriched populations of mouse embryonic stem cells (mESCs), NSCs, neurons and astrocytes to characterize the point of regression of these dedifferentiated cells on the differentiation axis. Our transcriptome data revealed that the dedifferentiated cells have significantly lower expression of known markers of their parental cell types. They also exhibited increased expression of progenitor NSC markers. Enrichment analysis of the differentially regulated genes in the dedifferentiated cell types revealed upregulation of the Wnt signaling, cell cycle and the focal adhesion pathways as compared with the mature parental cell types. Furthermore, we identified a functional network that was conserved in the dedifferentiated neurons and astrocytes, thus revealing significant interactions between the genes responsible for the phenotype observed in the dedifferentiated cell types. We identified Spp1 (secreted phosphoprotein 1), which encodes for the protein osteopontin (OPN), as an important node in this network and validated the role of OPN in the dedifferentiation of transformed neurons.
Results
Experimental design
To understand the molecular mechanism involved in the dedifferentiation of mature neurons and astrocytes upon onco-genic insult, we followed the in vitro system that we described previously.4 Briefly, cortical neurons and astrocytes were derived from 11-day-old SynapsinI-Cre and GFAP-Cre mice, respectively. The cells were cultured in their respective media to maintain their identity (see materials and methods section). These cells were then transduced with HRas-shp53 lentivirus with a transduction efficiency of >90%.4 The transduced neurons and astrocytes were later switched to NSC media devoid of serum and supplemented with FGF-2 (NSC media). Within 1 week, these cells became proliferative and aggregated to form free-floating neurospheres. These cells, hereinafter referred to as NSynR53 and AGR53, respectively, were later harvested and mRNA collected for sequencing library generation using DP-seq.7 To assess the regression of these cells to an undifferentiated state along the differentiation axis, enriched populations of mESC and NSC were also grown in vitro and mRNA obtained from these cells were subjected to library preparation (Figure 1a).
Figure 1.
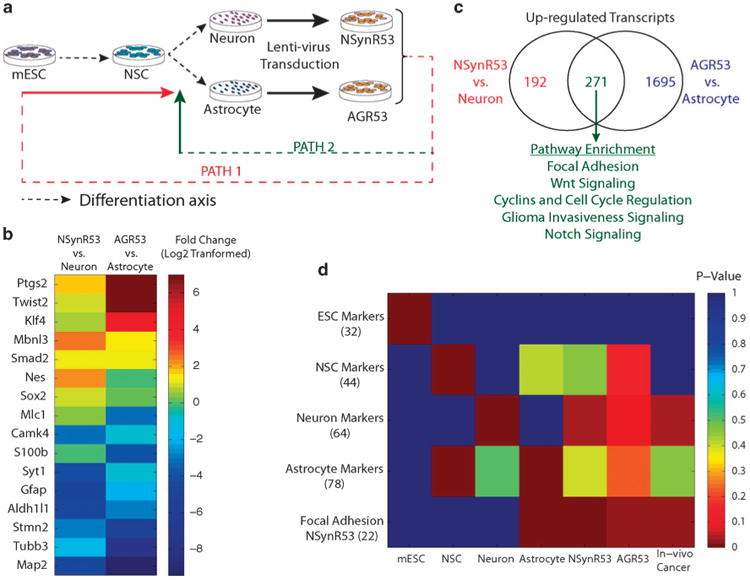
Scheme of experimental design. (a) mRNA collected from enriched populations of mESCs, NSCs, primary cultures of cortical neurons and astrocytes, and dedifferentiated neurons and astrocytes were subjected to sequencing library generation using DP-seq. Dedifferentiation of neurons and astrocytes was achieved by transducing the primary cultures of neuron and astrocytes by lentiviral vector comprising of HRas and shp53. The transduced neurons and astrocytes were switched to stem cell media devoid of serum and supplemented with FGF-2 for 3 weeks. (b) Differential expression of NSC markers and differentiation specific markers in dedifferentiated cell types in comparison with their mature parental cell types. (c) Pathway enrichment. The genes commonly upregulated in the dedifferentiated cell types showed enrichment for Wnt signaling, cell cycle and focal adhesion pathways. (d) Single sample gene set enrichment analysis. Gene lists comprising of the known markers (number of genes in the parentheses) showed significant enrichment in the respective populations. The dedifferentiated cell types exhibited high enrichment scores for NSC markers, neuron markers and a distinct set of focal adhesion genes.
Sequencing libraries prepared from these samples exhibited high transcriptome coverage with a vast majority of the reads mapping to the NCBI Refseq database (Supplementary Table 1). To validate our sequencing libraries, we investigated the expression of known markers of different cell types. MESC markers,8 which were significantly enriched in mESC libraries, showed low expression in other cell types (Supplementary Figure S1). The enriched populations of other cell types also showed upregulation of their respective markers.9 In case of dedifferentiated neurons and astrocytes, majority of the mESC markers had low expression. Additionally, these cells exhibited diminished expression of their parental cell type markers whereas the expression of known NSC markers were significantly high in these cells (Figure 1b). This demonstrated that the dedifferentiated cells acquired an undifferentiated progenitor/ stem cell state.
Differential gene expression analysis
The biological cell types considered in this study were highly divergent with many housekeeping genes exhibiting differential expression. Therefore, we normalized the sequencing libraries using quantile normalization. Differential expression analysis identified 463 genes upregulated in NSynR53 cells in comparison with the parental mature neurons (Supplementary Figure S2). AGR53 biological samples showed higher differential expression (1966 genes upregulated in comparison with the parental astrocytes) owing to high biological variations in the neuron samples (Supplementary Figure S3). Majority of the 463 genes upregulated in NSynR53 were also upregulated in AGR53 (Figure 1c) highlighting that the genetic alterations introduced by the oncogenic lentivirus affected the same set of genes in the two parental cell types. Similar observations were made for the downregulated genes in the dedifferentiated NSynR53 and AGR53 cells.
We next performed pathway enrichment analysis on the differentially regulated genes identified in the dedifferentiated cell types (Supplementary Table 2 and 3). In both cell types, canonical Wnt signaling, cell cycle and the focal adhesion pathways were significantly represented (Figure 1c). Aberrant regulation of Wnt signaling has been implicated in progression of various cancers10 and many of its components have been associated with maintenance of cancer stem cells.11 Expectedly, cell cycle-related genes were upregulated in dedifferentiated cell types as these cells were highly proliferative in contrast to their parental cell types. The dedifferentiated cell types underwent drastic transformation losing their flattened morphology and acquired an aggregated free-floating neurosphere-like structure. This transformation resulted in the differential expression of many focal adhesion genes. Interestingly, focal adhesion genes exhibited a bifurcated expression pattern where a unique set of genes were enriched in the dedifferentiated cell types while some genes lost their expression (Supplementary Figure. S4). Both, neurons and astrocytes, displayed conserved regulation of many of the focal adhesion-associated genes. Pathways downregulated in dedifferentiated cell types were necessary for maintenance of terminally differentiated cell types (neurons and astrocytes). This further highlights that the dedifferentiated cell types have distanced themselves from the differentiated state and acquired a progenitor stem cell-like state.
Gene set enrichment analysis
We performed single sample gene set enrichment analysis4,12 to assess the path adopted by mature neurons and astrocytes to dedifferentiate upon oncogenic insult to a progenitor stem celllike state. We compiled a list of known markers of the enriched populations: mESC, NSC, neurons and astrocytes and assessed their distribution in the sorted list of transcripts whose expression were normalized across these populations.
Expectedly, the known lineage markers showed significantly high enrichment scores in their respective populations (Figure 1d). The NSC population showed significant enrichment for astrocyte markers. Similarly, cortical astrocytes showed positive but not statistically significant enrichment of the NSC markers. This could be attributed to the plasticity observed in astrocytes with some astrocyte-specific markers sharing the expression with the NSCs.13 In the case of dedifferentiated neurons (NSynR53 cells), positive enrichment was observed for neuron markers. The astrocyte markers also displayed positive enrichment in the dedifferentiated astrocytes; however, the enrichment was not statistically significant. Surprisingly, dedifferentiated astrocytes (AGR53) also showed upregulation of a number of neuron markers. GO term enrichment analysis of the neuron markers that showed upregulation in both dedifferentiated neurons and astrocytes revealed biological processes associated with neural function (Supplementary Table 4). On the other hand, the neuron-specific markers that exhibited low expression in dedifferentiated neurons and astrocytes were mostly associated with ion transport and ligand interaction (Supplementary Table 5). This analysis suggests that at the whole transcriptome level, the dedifferentiated cell types have acquired the expression of NSC markers while retaining the memory of their parental mature cells. The low expression of the mESC-related markers and their negative enrichment scores in the dedifferentiated cells indicate that these cells adopted PATH 2 (Figure 1a) while regressing to a progenitor stem cell-like state.
We performed similar analysis on glioma tumors obtained from the stereotaxic injection of the lentiviral vector in the cortex of SynapsinI-Cre and GFAP-Cre mice. Positive enrichments for both neuronal markers and focal adhesion molecules upregulated in the dedifferentiated neurons were observed. This implies that even the cancer formed by dedifferentiation of neurons and astrocytes share neuronal traits and exhibit similar expression of focal adhesion molecules observed in the dedifferentiated cell types.
Identification of the functional network involved in dedifferentiation
We next sought to identify functional connectivity between genes that were upregulated in the dedifferentiated cell types. We compiled a database of known as well as predicted functional gene and protein interactions from four different sources including TRANFAC,14 STRINGS (Search Tool for the Retrieval of Interacting Genes/Proteins 8.3; http://string-db.org/), KEGG and BioGRID ver. 3.2.15 The resulting network consisted of more than 8000 nodes/genes that were connected by >40 000 edges/ interactions. We projected the upregulated genes in dedifferentiated neurons (in comparison with neurons) on this network using Cytoscape16 and identified a functional connectivity between 38 nodes that were connected by more than 53 edges (Figure 2). The network further demonstrated sub-networks representing the genes associated with the three signaling pathways: Wnt signaling, cell cycle and the focal adhesion pathway. These pathways were also identified as significantly enriched in dedifferentiated astrocytes.
Figure 2.
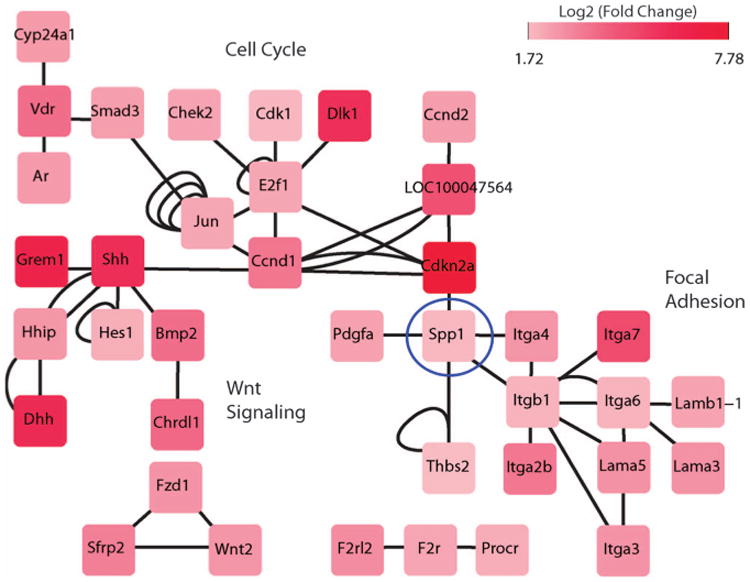
Gene/protein interaction network of differentially expressed genes in NSynR53 in comparison with neurons. An interaction network was constructed from publicly available databases comprising of protein–protein interactions (KEGG, STRING, BioGRID) and genetic interactions (TRANSFAC). The differentially expressed genes were projected into this network that revealed a module showing association of genes related to the focal adhesion, cell cycle and the Wnt signaling pathways. The edges represent the interactions observed in various databases.
As neurons and astrocytes were infected with the same lentiviral vector and the dedifferentiated cell types were phenotypically similar, we postulated that the genes involved in the functional network of the dedifferentiated neurons should have conserved expression in the dedifferentiated astrocytes. Indeed, the vast majority of these genes were upregulated in AGR53 in comparison with mature astrocytes (Supplementary Figure S5).
OPN is expressed in transformed neurons, astrocytes and glioma tumor samples
To gain further insight into the biological relevance of the network described in the previous section, we decided to focus on one specific gene, Spp1, which encodes for the protein OPN. This gene is at a node that connects all three pathways, so we postulated that perturbing its expression might compromise the capacity of the transformed neurons or astrocytes to dedifferentiate to a stem/progenitor state.
We first confirmed by qPCR analysis that Spp1/OPN is over-expressed in NSynR53 and AGR53 cells compared with their parental mature cell types (Figure 3a). Furthermore, previous microarray analysis of our mouse glioma tumor samples,4 as well as data from the TCGA on human glioma tumors revealed upregulation of Spp1 (13-fold increase in mouse GBMs over normal brain tissue, and upregulated in 6% of human GBM cases (http://www.cbioportal.org/public-portal/)).
Figure 3.
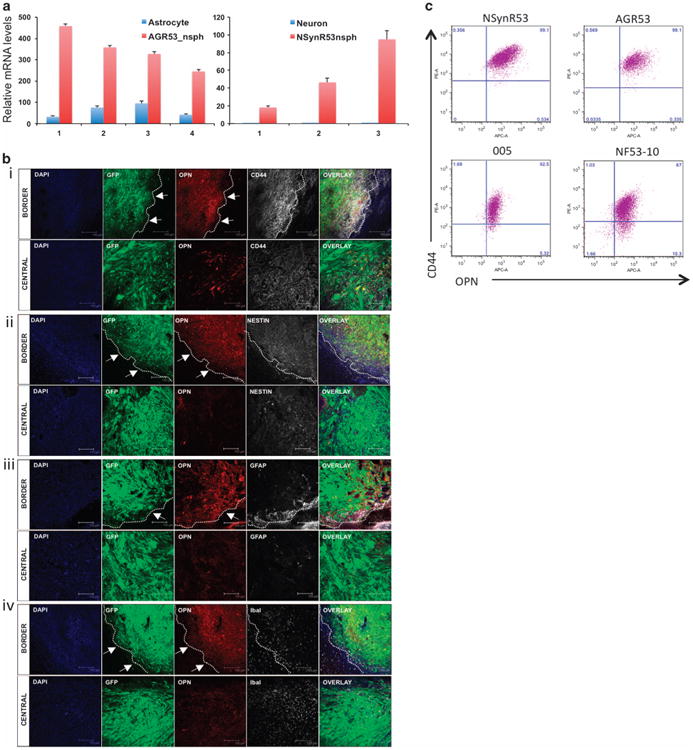
OPN is highly expressed in dedifferentiated neurons and astrocytes, GBM tumors and glioma stem cells (GSCs). (a) Expression of OPN was validated by qPCR on paired normal/transformed astrocytes (n = 4) and neurons (n = 3). AGR53-nsph = dedifferentiated AGR53 astrocytes, NSynR53-nsph = dedifferentiated NSynR53 neurons. (b) Confocal analysis of HRas-shp53-induced tumors in the cortex of SynapsinI-Cre mice. Sections of the brain were stained with (i) OPN/CD44, (ii) OPN/Nestin, (iii) OPN/GFAP and (iv) OPN/Iba-1. GFP = tumor cells, DAPI = cell nuclei. For each panel, a representative image of the border and central part of the tumor is depicted. Arrows point to the border (dashed line) of the tumor. Scale bar = 150 μm. (c) Flow cytometry analysis of OPN and CD44 expression in dedifferentiated astrocytes (AGR53) and neurons (NSynR53) as well as in two GSC lines derived from the murine GBM model: 005 and NF53-10.
We next stained sections of mouse glioma tumors induced by injection of HRas-shp53 in the cortex of SynapsinI-Cre mice for expression of OPN, CD44 (one of the receptors of OPN) and other cell-type markers by immunofluorescence (Figure 3b). We found that OPN is secreted mostly in the border regions of the tumor and expressed less in the central part of the tumor. In the border region, higher correlation between OPN and CD44 expression were observed in comparison with the central part of the tumor (Figure 3b, panel i). In the border region, the OPN-expressing cells are mostly tumor cells as indicated by dual GFP and OPN expression in these cells. The OPN+GFP+ cells were also positive for Nestin and the vast majority was negative for GFAP (astrocytes) and Iba-1 (microglia) markers (Figure 3b, panels ii, iii and iv). OPN was previously reported to be associated with invasion and migration of tumor cells.17 We believe that based on the pattern of OPN staining in the mouse glioma tumors, the invasive and more aggressive tumor cells located at the edge of the tumors are in fact expressing high levels of OPN. Flow cytometry analysis confirmed that all tumor cell lines were double positive for expression of OPN and CD44, thus validating the expression of OPN and CD44 in the dedifferentiated NSynR53 and AGR53 cells as well as two other glioma stem cell lines derived directly from the lentivirus-induced tumors (Figure 3c).
Blocking OPN compromises dedifferentiation of transformed neurons
To assess the possible role of OPN in tumor cell dedifferentiation, we blocked the expression of OPN in two lines of transformed neurons (NSynR53-1 and NSynR53-2). Out of four individual short hairpin RNAs designed for targeting OPN (shOPN), shOPN-1 and shOPN-3 constructs were most efficient in suppressing the OPN mRNA and protein expression (Supplementary Figure S6 and Figure 4a and b). A scrambled shRNA (shIRR) was used as a control throughout the study. To test the ability of NSynR53-shOPN cells to dedifferentiate in vitro, we switched the cell culture media from neuron-specific media to stem cell media supplemented with FGF-2 (NSC media). Both OPN-targeting shRNA constructs decreased the ability of NSynR53 to form the characteristic neurospheres (Figure 4c). On the other hand, control NSynR53-shIRR formed characteristic neurospheres when switched to NSC media. Similar results were obtained when NSynR53 cells were cultured in the presence of a neutralizing OPN antibody (Figure 4d). The levels of secreted OPN in the supernatant of cells either transduced with shOPN or blocked by addition of neutralizing OPN antibody to the media was confirmed by ELISA (Supplementary Figure S7). Confocal analysis of the neurospheres obtained in the control NSynR53 cells stained both for Nestin and Sox2, two markers of neural progenitor/stem cells (Figure 4e, panel i). The NSynR53 cells incubated in the presence of neutralizing OPN-Ab were unable to form neurospheres and retained their cell morphology and remained attached to the plate. These cells not only stained positive for the neuronal marker MAP-2 but also expressed low levels of Nestin and Sox2 (Figure 4e, panels ii and iii). qPCR analysis of well-defined stem cell markers on NSynR53-shOPN cells also showed reduced levels of expression of NSC markers compared with the NSynR53-shIRR cells capable of forming neurospheres in culture (Supplementary Figure S8).
Figure 4.
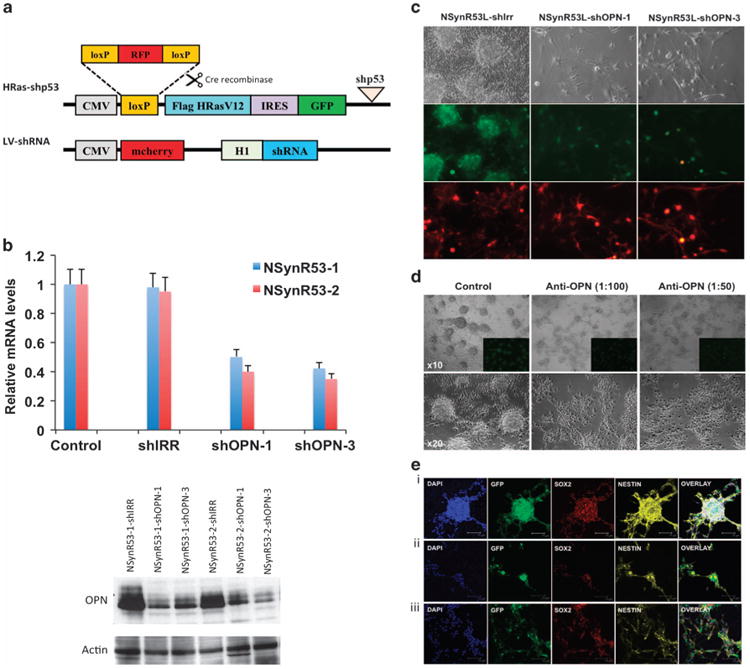
Blocking OPN compromises dedifferentiation of transformed neurons. (a) Schematic representation of the lentivectors. In the presence of Cre recombinase, the loxP-RFP-loxP cassette in the HRas-shp53 lentivector is cut out, and only GFP is expressed in the transformed cells (NSynR53). The LV-shRNA lentivector expresses the mcherry fluorescent reporter under the cytomegalovirus (CMV) promoter and the shRNA under the H1 promoter. (b) qPCR and western blot analysis of OPN silencing in NSynR53 cells. Two lines of NSynR53 cells were either transduced with a scrambled shRNA control (shIRR) or two different shRNAs targeting OPN (shOPN-1 and shOPN-3). (c) Bright and immunofluorescent images of NSynR53-shIRR and NSynR53-shOPN transduced cells. Green = GFP, fluorescent reporter in HRas-shp53 lentivector, Red = mcherry, fluorescent reporter in lenti-shIRR and lenti-shOPN vectors. Light microscopy magnification × 20. (d) Bright and immunofluorescent images of NSynR53 cultured in the absence (control) or presence of two different concentrations of neutralizing OPN antibody (stock 2 mg/ml; 1:100 and 1:50 dilution). Upper panels × 10 magnification and lower panels ×20 magnification. Insets in upper panels show GFP positive NSynR53 cells. (e) Confocal analysis of NSynR53 in the absence (panel i) or presence (panels ii and iii) of neutralizing OPN antibody cultured in NSC media. Blue = Dapi, Green = GFP, Red = Sox2 or Map2 and Yellow = Nestin. Scale bar=75 μm. The experiments were repeated three times and a representative result is shown in each section (c–e).
Blocking OPN affects cell proliferation and cell cycle
To test the effect of blocking OPN in tumor cell proliferation, we plated NSynR53-shOPN, NSynR53 in the presence of neutralizing OPN antibody and their respective controls for 72 h. Either silencing OPN or blocking its effect on NSynR53 cells significantly decreased their proliferative capacity (Figure 5a). The effect of blocking OPN on progression of NSynR53 through the cell cycle was determined by 7-aminoactinomycin D staining and flow cytometry analysis (Figure 5b). Silencing of OPN induced an accumulation of cells in the G2 phase of the cell cycle, increasing the proportion of cells in G2 from 32.9% in controls to 51% in cells depleted of OPN. There was also a corresponding decline in the G1 phase and S phase. Similar effects on cell cycle progression were previously reported for an effective OPN inhibitor, (-)-Agelastatin A, on breast cancer cell lines.18
Figure 5.
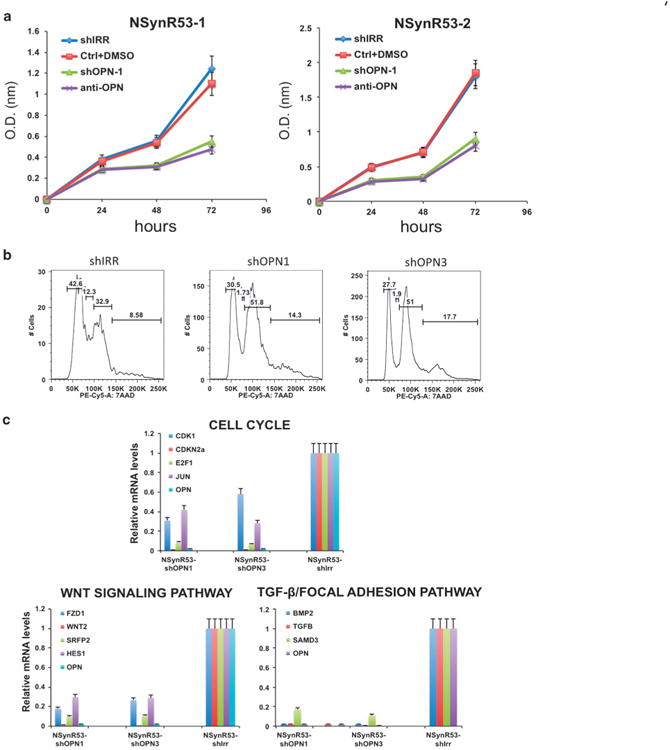
Blocking OPN affects NSynR53 proliferative capacity and decreases the expression of genes in the Wnt signaling, cell cycle and focal adhesion pathway. (a) Effect of OPN blocking on proliferation of NSynR53 cells. NSynR53-shIRR (shIrr), NSynR53 control (Ctrl+DMSO), NSynR53-shOPN1 (shOPN1) and NSynR53+OPN neutralizing antibody (anti-OPN) were seeded in culture plates (in triplicates) and WST-1 cell proliferation assay reagent was used to monitor cell proliferation at the indicated time points. The experiment was repeated three times and a representative result is shown. (b) Flow cytometry profile showing accumulation of NSynR53-shOPN in G2 phase (51 vs 32.9% in control cells). (c) qPCR analysis of representative genes of each of the three signaling pathways indicated in the graphs (n = 3).
Finally, to assess the overall effect of silencing OPN in NSynR53 cells and to validate the functional network defined before (Figure 2), we tested the expression of representative genes of each pathway by qPCR. As shown in Figure 5c, silencing OPN has major effects in representative genes from the cell cycle, Wnt and focal adhesion pathways validating the connectivity of the network through Spp1/OPN.
Specific inhibition of OPN in tumor cells prolongs mice survival
To selectively inhibit OPN expression in tumor cells in vivo, we transduced NSynR53 cells with an inducible version of the shOPN (miROPN; Supplementary Figure S9). NSynR53-miROPN cells were transplanted into NOD-SCID mice and 10 days after the transplantation, one group of mice (miROPN+Dox) received doxocyclin in their chow. The control group continued to feed on normal chow. As shown in Figure 6a, specific inhibition of OPN prolonged mice survival compared with control groups.
Figure 6.
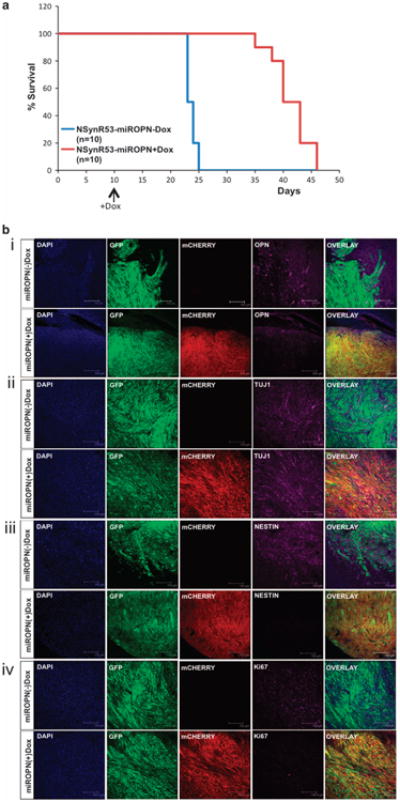
Silencing OPN extends survival in GBM-bearing mice. NSynR53 cells were transduced with an inducible shOPN construct (mirOPN) and were transplanted into NOD-SCID mice. (a) Kaplan–Meier plot of survival for mice fed with normal chow (miROPN(—)Dox, n = 10) and doxocycline containing chow (miROPN(+)Dox, n = 10). The (+)Dox group was fed with the special chow 10 days after transplantation of the NSynR53-mirOPN cells (arrow indicates beginning of +Dox chow diet). (b) Tumors from each group were collected at the end of the experiment and brain sections (40 nm) were immunostained with (i) OPN, (ii) Tuj1, (iii) Nestin and (iv) Ki67, and analyzed using confocal microscopy. Mcherry is only expressed in (+)Dox-fed mice (see lentivector scheme in Supplementary Figure S9A); GFP = tumor, DAPI = nuclei. Scale bar = 300)μm (panel i) and 150 μm (panels ii-iv).
Immunostaining and confocal microscopy analysis confirmed that inducible shOPN tumors (mirOPN(+)Dox) had less OPN expression than control mice (mirOPN(-)Dox) (Figure 6b, panel i). Although both tumors retained expression of the neuronal marker Tuj1 (higher expression is still observed in the shOPN tumors; Figure 6b, panel ii), the most striking difference was observed when staining for the NSC marker Nestin (Figure 6b, panel iii). shOPN tumors are not only negative for Nestin, but they also expressed lower levels of Ki67 (proliferation marker, Figure 6b, panel iv). Finally, we also noticed that shOPN tumors have smoother borders than control tumors, supporting the notion that OPN is involved in tumor invasion and migration (see Figure 6b panels i and iii). Altogether, the in vivo observations confirmed all our findings in vitro, and suggest a role of OPN in tumor cell dedifferentiation.
To determine the human relevance of our findings, we investigated the role of OPN/Spp1 in human GBM by assessing the expression of the gene in TCGA datasets. Spp1 gene is highly expressed in the majority of the GBM tumors (Supplementary Figure S10A). Furthermore, Spp1 was significantly upregulated in GBM tumors (>90% of the tumors) in comparison with pooled normal controls (Supplementary Figure S10B). This suggests an important role played by OPN/Spp1 in human GBM samples. To test the functional consequences of knocking down Spp1 (OPN) in vivo, we established orthotopic xenografts utilizing shIRR or shOPNh-expressing human glioma cell lines (U87-shIRR and U87-shOPNh, respectively; Supplementary Figure S11). U87-shIRR-transplanted mice readily developed intracranial tumors, whereas U87-shOPNh mice showed impaired tumor formation and increased survival (Figure 7a). When the first U87-shIRR mice succumbed to the disease, representative mice from the U87-shOPNh group were killed. U87-shIRR mice developed aggressive tumors that invaded both hemispheres (Figure 7b, panel i). In contrast, U87-shOPNh-transplanted mice had no clinical symptoms of the disease and no signs of tumor growth were identified at this time point (Figure 7b, panel ii). Eventually, all mice in the U87-shOPNh group developed symptoms of tumor growth and histology analysis of the collected brains revealed the presence of gliomas (Figure 7b, panel iii). Immunostaining and confocal microscopy analysis showed high expression of OPN in U87-shIRR but was undetectable in U87-shOPNh tumors (Figure 7c, panel i). In addition, U87-shOPNh tumors expressed lower levels of Ki67 (Figure 7c, panel ii).
Figure 7.
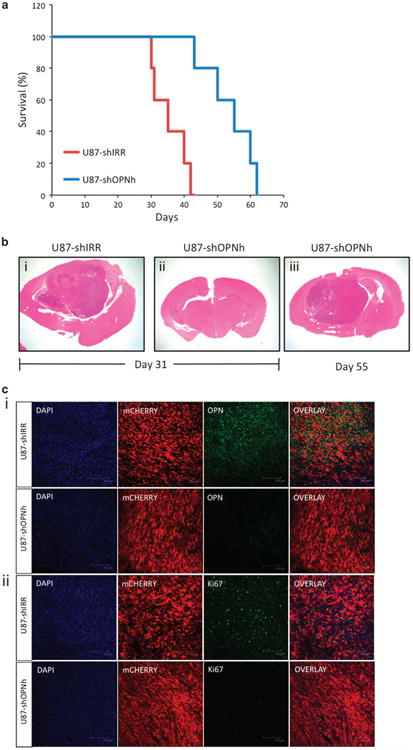
Targeting OPN in human GBM-bearing mice improves survival. U87 human cell line transduced with lentiviral shIRR or shOPNh were transplanted into the brain of immunocompromised NOD-SCID mice (3×105 cells per mouse). (a) Kaplan-Meier plot of survival for mice transplanted with U87-shIRR (n = 10) and U87-shOPNh (n= 10). (b) Representative images of brain sections (hematoxylin and eosin-stained) 31 days (when U87-shIRR mice started to show clinical symptoms; panels i and ii) and 55 days after transplantation (panel iii). (c) Tumors from each group were collected at the end of the experiment and brain sections (40 nm) were immunostained with (i) OPN and (ii) Ki67, and analyzed using confocal microscopy. mCHERRY is a fluorescent reporter expressed in the lentiviral shRNA construct, DAPI = nuclei. Scale bar= 150)μm.
To further evaluate whether the expression of OPN/Spp1 in GBM correlates with patient survival, we looked through the Rembrandt database and found that in human patients with glioma tumors, downregulation of OPN/Spp1 is also correlated with better survival (Supplementary Figure S12).
Discussion
We have used a systems biology approach together with an in vitro and in vivo cancer model system to investigate the mechanism by which transformed mature neurons or astrocytes dedifferentiate to a stem-like state to form recurrent aggressive glioma tumors. We used DP-seq to generate robust sequencing libraries from low amounts of mRNA obtained from cells derived from mouse models. DP-seq captured global transcripts expression changes and revealed that these dedifferentiated cell types acquired a stem-progenitor-like state, although they still retained memory of their parental cell types. Persistent memory of the original somatic cells has also been described in induced pluripotent stem cells19–22 where the genetic profiles of induced pluripotent stem cells were shown to be comparable but not identical with that of ESCs. The disparity in transcripts expression between the two populations may suggest retention of the gene expression of the donor cell in induced pluripotent stem cells. Parallels between reprogramming of somatic cells and dedifferentiation in cancer have been already found and described.23 Our findings suggest that retention of residual transcriptional memory of the parental somatic cell is an additional characteristic that the process of iPS generation shares with dedifferentiation in cancer.
At the transcriptome level, the dedifferentiated tumor cells mostly resemble NSC at least under the conditions used in vitro in this study, mainly NSC media, and they shared only a few genes with ESCs. The analysis of gene expression signatures associated with ESCs identity in human GBM24 supports a dedifferentiation process undergoing in these tumors. An attempt to elucidate the pathways involved in dedifferentiation of transformed neurons and astrocytes revealed a functional network, shared by these two populations undergoing dedifferentiation, involving three important signaling pathways: Wnt signaling, cell cycle and focal adhesion pathways. The Wnt signaling pathway and different genetic defects in this molecular pathway have been shown to cause and promote cancer.25 Considering the oncogene and tumor suppressor gene combination that we used to induce transformation in both neurons and astrocytes (HRas and loss of p53), it is not surprising to find cell cycle as one of the important pathways involved in dedifferentiation. The relevance of cell cycle deregulation in cancer has been extensively reported in the past decades (reviewed in Malumbres and Barbacid26). The pathway that warranted significant attention is the focal adhesion pathway, mainly because of the morphological changes the parental neurons and astrocytes undergo during the dedifferentiation process, from a cell flattened shape to the formation of clusters known as tumorspheres. We focused on Spp1, a gene located at a node that connects all three pathways represented in the functional network involved in the dedifferentiation process, which codes for a focal adhesion and secreted protein called OPN.
OPN is a glycophosphoprotein with an RGD-containing domain and multifunctional properties both as a cytokine and a chemokine. It is expressed intra- and extracellularly, and is produced by several types of cells including macrophages, epithelial cells, smooth muscle cells, osteoblasts and cancer cells.27,28 The RGD site mediates binding of OPN to multiple integrins, including αVβ3, αVβ5, αVβ1, α5β1, and to certain variant forms of CD44.29–31 Among the several postulated functions of OPN, its role in tumorigenesis includes several aspects such as remodeling the extracellular matrix, proliferation, cell invasion and migration, angiogenesis, and metastasis.32 In general, high OPN levels have been associated with aggressive tumors and poor prognosis in patients.33,34 Specifically in malignant gliomas, OPN has been shown to be involved in tumor growth and invasion,17 recruitment of macrophages and neutrophils,35 and tumor cell survival.36,37 A recent study reported OPN as a factor exclusively secreted in the perivascular niche of proneural GBM to promote stem cell properties of glioma cells in this niche.38 The study proposed that OPN secreted by stromal astrocytes confers radiation resistance and stemness properties to tumor cells. Our results suggest a new role of OPN in tumor cell dedifferentiation, whereby transformed astrocytes and neurons express high levels of OPN to promote dedifferentiation and acquisition of stem cell properties. As we have previously reported, tumors induced by transduction of cortical neurons and astrocytes are classified into the mesenchymal GBM subtype.4 We have confirmed high expression of OPN (Spp1 gene) and CD44 (one of its receptors) both in our mesenchymal GBM mouse tumors (microarray data4) as well as in human GBM samples (www.cbioportal.org). CD44 was identified as a GBM mesenchymal marker,39 and we confirmed its expression throughout our mesenchymal mouse GBM model. Interestingly, OPN is also expressed in these tumors but at higher levels in the border area compared with the central area of the tumor. This pattern of expression correlates well with the suggested role of OPN in invasion and migration in GBM.17 Furthermore, high levels of OPN in invasive GBM also correlated with high expression of vimentin, another mesenchymal marker, and low levels of GFAP. In this particular study, knocking down OPN reduced cell invasiveness, decreased vimentin and increased GFAP expression, the later used as a marker of differentiated astrocytes.17 Our conclusion is further supported by the observation that when blocking the effect of OPN in transformed neurons, these cells retained their parental cell morphology. Additionally, they were unable to form the typical neurospheres and expressed low levels of representative NSC makers while maintaining their differentiated state.
Our data further showed that blocking OPN compromises dedifferentiation of transformed neurons, affecting their proliferative capacity and causing cells to accumulate in the G2 phase of the cell cycle. Similar effects were observed when (-)-agelastatin A, a naturally occurring oroidin alkaloid, was used to treat human breast cancer cell lines.18 The authors claimed that (-)-agelastatin A increases the levels of Tcf-4 while reducing the expression of β-catenin and OPN. This supports the connectivity that we found between the focal adhesion pathway (through Spp1) and the Wnt signaling pathway. Our findings also show that lowering the levels of OPN (by shRNA silencing) decreases the levels of different components of the Wnt signaling pathway.
To mimic the therapeutic effect of a tumor-specific OPN inhibitor, we designed an inducible OPN shRNA (miROPN) to treat GBM-bearing mice. The results showed that OPN-treated mice survived longer compared with control mice. Confocal microscopy analysis confirmed low levels of OPN expression in these tumors and more interestingly, these tumors retained high expression levels of the neuronal marker Tuj1 and were negative for Nestin. This observation suggests that silencing OPN in tumor cells compromised the ability of these cells to dedifferentiate, hence retaining a more differentiated state. In this differentiated state, tumor cells proliferate slower (indicated by a significant decrease of cell proliferation marker Ki67 in miROPN(+)Dox tumors) and are less invasive and aggressive.
The human relevance of our findings was supported by high expression of OPN/Spp1 in human GBM patients (TCGA datasets), and in an in vivo experiment, which showed that targeting OPN in a GBM human cell line suppressed tumor growth and improved survival in GBM-bearing mice.
Eventually, all treated mice succumbed to the disease, and it is possible that as proposed previously, other non-tumor cells in the brain might be secreting OPN and supporting the dedifferentiation process of tumor cells in a paracrine manner. Other ways to neutralize the global levels of OPN or alternatively, blocking the receptors of OPN in brain tumors should be tested. CD44 is not the only receptor of OPN, and as shown recently, plateled-derived growth factor beta-induced murine gliomas in CD44 –/– background mice, lead to longer survival, but these mice also eventually developed tumors.38
Collectively, our systems biology approach identified the relevant pathways involved in the dedifferentiation of glioma cells and revealed the functional hubs of protein/gene interactions relevant for the disease phenotype. Early intervention of the disease may warrant therapies targeting these hubs. Using a GBM mouse model, we demonstrated that one of these targets, OPN, plays an important role in dedifferentiation of glioma cells and can be a potential therapeutic target in human tumor.
Materials and Methods
Cell culture
Primary cortical astrocytes and neurons were obtained from 11 days postnatal pups from GFAP-Cre and SynapsinI-Cre transgenic mice, respectively, and prepared according to published methods.40,41
Astrocytes were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and neurons were cultured in Neurobasal-A Medium (Gibco, Carlsbad, CA, USA) containing Glutamax (Gibco) and B-27 supplement (Gibco). Following transduction of either primary astrocytes or neurons with the lentivirus, at the early passages, cells were either cultured in the medium described above or in parallel cultured in NSCs medium containing FGF-2.4 U87 human glioma cell line was maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum.
Lentiviral vectors
shRNA hairpins were cloned into an NheI site of the p156RRLsin vector.42 The shRNAs used in this study were: shOPN-1 5′- AGGATGACTTTAAG CAAGAAA-3′ and shOPN-3 5′- TGACGAATCTCACCATTCGGATGAGTCTG-3′ to target mouse OPN, and shOPNh-1 5′- CCGAGGTGATAGTGTGGTTTA-3′, shOPNh-2 5′- CCACAAGCAGTCCAGATTATA-3′, shOPNh-3 5′- CACAAGCAG TCCAGATTATTT-3′ to target human OPN (shOPNh-1 was selected for in vivo experiments). To achieve inducible silencing of mouse OPN (miROPN), we designed a one-lentivector system based on the Tet-regulated miR30-shRNA technology,43 combined with expression of the transactivator (m2RtTA) under the EF1α promoter. Lentivirus was prepared as described previously.44
Mice and stereotaxic injections
hGFAP-Cre, SynapsinI-Cre and NOD-SCID mice were all purchased from the Jackson Laboratories (Bar Harbor, ME, USA). Mice were maintained in pathogen-free conditions in the Salk Institute and monitored daily for signs of illness and killed when moribund. All the procedures performed in this study were approved by the IACUC. Lentivirus and tumor cells were injected stereotaxically as described previously.4
RNA extraction and sequencing
Library PreparationTotal RNA was extracted from harvested cells using Trizol (Invitrogen, Carlsbad, CA, USA). About 1–3 μg of total RNA was later subjected to Oligo(dT) selection using Dynabeads mRNA Purification Kit (Invitrogen) according to the manufacturer's protocol. The first stand cDNA was synthesized using oligo dT primers (20 bp) and QuantiTect reverse transcription kit (Qiagen, Carlsbad, CA, USA) according to the manufacturer's protocol. The cDNA derived from the biological samples were later subjected to sequencing library preparation using DP-seq7 while minimizing technical variations observed in the amplification-based methods for sequencing library preparation.45 The number of amplification cycles was kept at 13 for all samples.
Quantification of the sequencing library
Quantitative real-time PCR was used to determine the concentration of the sequencing libraries prepared by our protocol. The standard curve for various dilutions of phiX control library was generated using the adapter-specific primers recommended by Illumina (San Diego, CA, USA). We later used the standard curve to determine the molarity of our sequencing libraries. The concentration of sequencing library loaded into the flowcell was calibrated by the sequencing facility. We typically obtained good cluster density with 5 pM of library concentration on HiSeq v3 kit.
Mapping reads
All libraries were sequenced by Illumina's HiSeq2000 systems (TruSeq SR Cluster Kit v3-cBot-HS and TruSeq SBS Kit v3-HS). The libraries were sequenced as 50-bp single-end reads, except for astrocytes samples, which were sequenced as 100-bp single-end reads. The first seven sequencing cycles of the biological samples, excluding cortical astrocytes and dedifferentiated astrocytes, were truncated as they came from our defined set of 44 heptamer primers that were used for targeted amplification using DP-seq. In case of astrocyte samples, the first 14 sequencing cycles were truncated. The next 32-bp reads were aligned to the mouse NCBI RefSeq mRNA database (Version 41 mRNA RefSeq database, 9 May 2010) using in-house mapping software while allowing up to 2 mismatches. The 44 DP-seq primers were designed for the same version of the NCBI RefSeq mRNA database where about 26 566 transcripts with NM and NR ids were selected. The transcripts with XM and XR ids were removed from the database. The reads that did not map to the mRNA database were further aligned to mouse genomic locations including intronic and intergenic locations (Build 37) using Bowtie46 while allowing ≤2 mismatches.
Differential gene expression analysis
Unique reads obtained from different samples were quantile-normalized using limma library in R statistical package. Differentially expressed genes were identified using local pooled error test.47 A P-value cutoff of 0.05 was used to assign significance to the differentially expressed genes. Additional cutoffs of minimum fold change of 1.5 and an average expression of ≥8 unique reads were used to identify differentially expressed genes.
Single sample gene set enrichment analysis
The sequencing data were arranged into a matrix, where rows and column represented the transcripts and different cell types, respectively. The sequencing measurement of biological replicates were averaged and assigned to each column. There were four primary cell types (mESC, NSC, neuron and astrocyte), two transformed cell types (dedifferentiated neuron and dedifferentiated astrocytes) and one in vivo cancer sample. The count data were quantile-normalized and parameters including mean, standard deviation and mean absolute deviation were calculated for each transcript based on four primary cell types:
where, x̄, xmad and σx are mean, mean absolute deviation and standard deviation, respectively. The transcripts were ordered based on this statistics. An arbitrary median absolute deviation cutoff of ≥20 was applied to eliminate transcripts with low expression. The rescaled gene rankings for each cell type (column) were used for single sample gene set analysis.
qRT-PCR analysis
qPCR reactions were carried out with the 7900HT Fast Real-Time PCR System using the PerfeCTa SYBR Green SuperMix (Quanta Biosciences, Gaithersburg, MD, USA). Data are presented after normalization with cyclophilin. The primers are listed in Supplementary Table 6.
Western blotting
Western blot assays were performed as previously described48 using goat anti-OPN (R&D Systems, Minneapolis, MN, USA) and actin (Sigma, St Louis, MO, USA).
Flow cytometry and fluorescent staining
For flow cytometry analysis, cells were stained with the antibodies listed in Supplementary Table 7 and analyzed on a BD LSR II flow cytometer (Beckton Dickinson, Franklin Lakes, NJ, USA) and FlowJo software. For fluorescent staining, coronal sections (40 μm) were cut on a microtome and images were obtained using a confocal laser-scanning microscope (Leica TCS SP2 ABS, Mannheim, Germany). All the antibodies are listed in Supplementary Table 7.
Acknowledgments
We thank Gabriela Estepa for her technical help and Ruben Alvarez Rodriguez for designing the inducible miRNA lentivector. IMV is an American Cancer Society Professor of Molecular Biology, and holds the Irwin and Joan Jacobs Chair in Exemplary Life Science. This work was supported in part by grants from the NIH (HL053670) (to IM Verma and D Friedmann-Morvinski), Cancer Center Core Grant (P30 CA014195-38), the H.N. and Frances C. Berger Foundation, and the Leona M. and Harry B. Helmsley Charitable Trust grant #2012-PG-MED002 (to IM Verma). S Subramaniam is supported by National Institutes of Health (NIH) National Heart, Lung and Blood Institute Grant HL087375, HL106579, HL108735, the National Science Foundation (NSF) grants DBI-0835541 and STC-0939370. V Bhargava was the recipient of a California Institute for Regenerative Medicine (CIRM) Graduate Fellowship (T1-00003 and TG2-01154, Interdisciplinary Stem Cell Training Program at UCSD). Accession Code. Gene Expression Omnibus: GSE64411 (sequencing read data).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 2.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 3.Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154:61–74. doi: 10.1016/j.cell.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. Direct reprogramming by oncogenic Ras and Myc. Proc Natl Acad Sci USA. 2013;110:3937–3942. doi: 10.1073/pnas.1219592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 7.Bhargava V, Ko P, Willems E, Mercola M, Subramaniam S. Quantitative transcriptomics using designed primer-based amplification. Sci Rep. 2013;3:1740. doi: 10.1038/srep01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao W, Ji X, Zhang F, Li L, Ma L. Embryonic stem cell markers. Molecules. 2012;17:6196–6236. doi: 10.3390/molecules17066196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4:a008052. doi: 10.1101/cshperspect.a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013;25:254–264. doi: 10.1016/j.ceb.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol. 2008;86:342–367. doi: 10.1016/j.pneurobio.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res. 2013;41:D816–D823. doi: 10.1093/nar/gks1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jan HJ, Lee CC, Shih YL, Hueng DY, Ma HI, Lai JH, et al. Osteopontin regulates human glioma cell invasiveness and tumor growth in mice. Neuro Oncol. 2010;12:58–70. doi: 10.1093/neuonc/nop013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mason CK, McFarlane S, Johnston PG, Crowe P, Erwin PJ, Domostoj MM, et al. Agelastatin A: a novel inhibitor of osteopontin-mediated adhesion, invasion, and colony formation. Mol Cancer Ther. 2008;7:548–558. doi: 10.1158/1535-7163.MCT-07-2251. [DOI] [PubMed] [Google Scholar]
- 19.Chin MH, Mason MJ, Xie W, Volinia S, Singer M, Peterson C, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh Z, Wilson KD, Wu Y, Hu S, Quertermous T, Wu JC. Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PloS One. 2010;5:e8975. doi: 10.1371/journal.pone.0008975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchetto MC, Yeo GW, Kainohana O, Marsala M, Gage FH, Muotri AR. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PloS One. 2009;4:e7076. doi: 10.1371/journal.pone.0007076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol. 2011;13:541–549. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 2014;15:244–253. doi: 10.1002/embr.201338254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 26.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 27.Denhardt DT, Noda M, O'Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–1061. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wai PY, Kuo PC. The role of Osteopontin in tumor metastasis. J Surg Res. 2004;121:228–241. doi: 10.1016/j.jss.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 29.Katagiri YU, Sleeman J, Fujii H, Herrlich P, Hotta H, Tanaka K, et al. CD44 variants but not CD44s cooperate with beta1-containing integrins to permit cells to bind to osteopontin independently of arginine-glycine-aspartic acid, thereby stimulating cell motility and chemotaxis. Cancer Res. 1999;59:219–226. [PubMed] [Google Scholar]
- 30.Weber GF, Ashkar S, Glimcher MJ, Cantor H. Receptor-ligand interaction between CD44 and osteopontin (Eta-1) Science. 1996;271:509–512. doi: 10.1126/science.271.5248.509. [DOI] [PubMed] [Google Scholar]
- 31.Weber GF. The metastasis gene osteopontin: a candidate target for cancer therapy. Biochim Biophys Acta. 2001;1552:61–85. doi: 10.1016/s0304-419x(01)00037-3. [DOI] [PubMed] [Google Scholar]
- 32.Rangaswami H, Bulbule A, Kundu GC. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol. 2006;16:79–87. doi: 10.1016/j.tcb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Bache M, Kappler M, Wichmann H, Rot S, Hahnel A, Greither T, et al. Elevated tumor and serum levels of the hypoxia-associated protein osteopontin are associated with prognosis for soft tissue sarcoma patients. BMC Cancer. 2010;10:132. doi: 10.1186/1471-2407-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sreekanthreddy P, Srinivasan H, Kumar DM, Nijaguna MB, Sridevi S, Vrinda M, et al. Identification of potential serum biomarkers of glioblastoma: serum osteopontin levels correlate with poor prognosis. Cancer Epidemiol Biomarkers Prev. 2010;19:1409–1422. doi: 10.1158/1055-9965.EPI-09-1077. [DOI] [PubMed] [Google Scholar]
- 35.Atai NA, Bansal M, Lo C, Bosman J, Tigchelaar W, Bosch KS, et al. Osteopontin is upregulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology. 2011;132:39–48. doi: 10.1111/j.1365-2567.2010.03335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamour V, Le Mercier M, Lefranc F, Hagedorn M, Javerzat S, Bikfalvi A, et al. Selective osteopontin knockdown exerts anti-tumoral activity in a human glioblastoma model. Int J Cancer. 2010;126:1797–1805. doi: 10.1002/ijc.24751. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi Y, Shao Z, Sharif S, Du XY, Myles T, Merchant M, et al. Thrombin-cleaved fragments of osteopontin are overexpressed in malignant glial tumors and provide a molecular niche with survival advantage. J Biol Chem. 2013;288:3097–3111. doi: 10.1074/jbc.M112.362954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14:357–369. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 40.McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 42.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011;29:79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikawa M, Tanaka N, Kao WW, Verma IM. Generation of transgenic mice using lentiviral vectors: a novel preclinical assessment of lentiviral vectors for gene therapy. Mol Ther. 2003;8:666–673. doi: 10.1016/s1525-0016(03)00240-5. [DOI] [PubMed] [Google Scholar]
- 45.Bhargava V, Head SR, Ordoukhanian P, Mercola M, Subramaniam S. Technical variations in low-input RNA-seq methodologies. Sci Rep. 2014;4:3678. doi: 10.1038/srep03678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B. Aligning short sequencing reads with Bowtie. Current Protoc Bioinformatics. 2010;Chapter 11 doi: 10.1002/0471250953.bi1107s32. Unit 11 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain N, Thatte J, Braciale T, Ley K, O'Connell M, Lee JK. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics. 2003;19:1945–1951. doi: 10.1093/bioinformatics/btg264. [DOI] [PubMed] [Google Scholar]
- 48.Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]