

Abstract
Pain is a hallmark of almost all bodily ailments and can be modulated by agents, including analgesics and anesthetics that suppress pain signals in the central nervous system. Defects in the modulatory systems, including the endogenous pain‐inhibitory pathways, are a major factor in the initiation and chronicity of pain. Thus, pain modulation is particularly applicable to the practice of medicine. This review summarizes the existing literature on pain modulation. Here, we critically reviewed the literature from PubMed on pain modulation published primarily within the past 5 years in high impact journals. Specifically, we have discussed important anatomical landmarks of pain modulation and outlined the endogenous networks and underlying mechanisms of clinically relevant pain modulatory methods. The Gate Control Theory is briefly presented with discussion on the capacity of pain modulation to cause both hyper‐ and hypoalgesia. An emphasis has been given to highlight key areas in pain research that, because of unanswered questions or therapeutic potential, merit additional scientific scrutiny. The information presented in this paper would be helpful in developing novel therapies, metrics, and interventions for improved patient management.
Keywords: pain modulation, Gate Control Theory, opioids, inhibitory amino acids, cannabinoids, electroanalgesia, periaqueductal gray, rostral ventromedial medulla
Introduction
Pain modulation refers to the process by which the body alters a pain signal as it is transmitted along the pain pathway and explains, at least in part, why individual responses to the same painful stimulus sometimes differ. Modulation can also explain why the activation of pain neurons and the sensory experience of pain do not always coincide. Most importantly, pain modulation elucidates the mechanisms of action underlying clinical analgesia. In this paper, we have critically reviewed pain modulation literature by searching PubMed for primary research papers that elucidate therapeutically significant mechanisms in pain modulation. This review focuses on the following key questions: (i) does pain modulation have an analgesic effect, hyperalgesic effect, or both? (ii) What is the Gate Control Theory (GCT), and how does it impact our understanding of pain modulation? (ii) What are the clinically important pain modulation types? (iv) what are the outstanding questions in pain modulation research that could lead to new therapeutic approaches?
Does Pain Modulation Have an Analgesic Effect, Hyperalgesic Effect, or Both?
Opioids are widely recognized as the “gold standard” in pain control. Indeed, the use of opiates can cause hyperalgesia.1 Watanabi2 made a paradoxical observation: giving limited amounts of morphine to rats relieved the symptoms of pain; however, high doses of morphine led to pain‐related responses in the rats. Interestingly, opioids can cause recipients to become hypersensitive to certain painful stimuli. While opioid‐induced hyperalgesia is not the emphasis of this review, opiates offer a valuable example of pain modulation: they are capable of both increasing and decreasing the experience of pain. He et al.3 showed that, in rats, inflammatory markers, particularly HMGB 1, contribute to neuropathic pain. These changes in pain sensation were implemented via modulatory pathways that could both increase and decrease the sensation of pain via the HMGB1 and HMGB1‐RAGE pathways.3 In a review of pain modulatory mechanisms, Heinricher et al.4 concluded that descending modulation could be both “facilitatory” and “inhibitory.” Additionally, these investigators noted that a single modulatory structure in the brain can often mediate both “facilitatory” and “inhibitory” modulation of pain.4 Although, the term “modulation” is commonly assumed to have an exclusively analgesic connotation, pain modulation can lead to both analgesia and hyperalgesia.
Gate Control Theory
In a landmark paper, Wall and Melzack5 proposed the GCT. While some details of the GCT have been shown to be incorrect or incomplete, it has proven to be a powerful tool for guiding pain research.6, 7, 8 The GTC proposes that nociceptive and nonnociceptive signals are summated within the substantia gelatinosa (spinal cord).6, 7, 8 If nociceptive signals outweigh nonnociceptive signals, a pain signal is propagated.6,8 Wall and Melzack5 also proposed that descending afferent fibers could modulate pain signals within the substantia gelatinosa. A visual representation of the pain circuit proposed by Wall and Melzack is shown in Figure 1.
Figure 1.
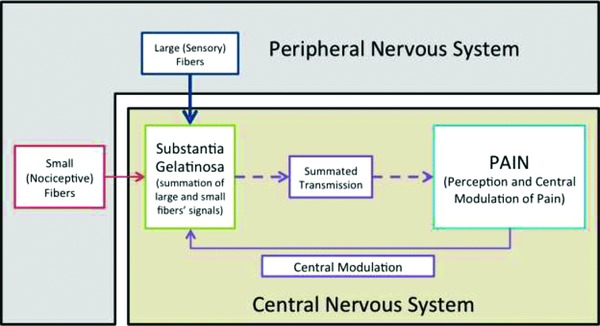
Gate Control Theory. Both small and large fibers from the periphery come into the substantia gelatinosa. Generally, larger fibers carry general somatosensation information while smaller fibers carry nociceptive information. The two fiber types summate in the substantia gelatinosa. If the signal carried by the nociceptive fibers is stronger than the general sensation signal, a pain stimulus can be passed from the substantia gelatinosa toward the brain. Descending modulatory fibers interact with pain signals in the substantia gelatinosa.
The GCT broadly suggests that large nerves conduct nonnociceptive information and that smaller fibers conduct nociceptive information.6,8 After the proposition of the theory, researchers tested it by electrically stimulating large fibers.6 In a variety of studies, this type of stimulation has been found to provide pain relief.6 Researchers continue to use the GTC rationale to propose new methods for achieving clinical pain relief. For example, Kessler and Hong invoked the GTC in explaining their investigation of whole body vibration as a potential therapy for diabetic neuropathy.9 Similarly, Fournier and Elman10 tested the use of pneumatic skin flattening as an analgesic technique. Their study emphasized its effect on pain transmission within the circuits described by the GTC.10 Often, those who injure themselves instinctively rub the affected area. Within the context of the GCT, this natural response is unsurprising: the GCT provides a scientific rationale for an instinctive response to painful stimuli.
Clinically Relevant Pain Modulation
In 1969, Reynolds11 placed electrodes into the brain of a rat and applied a current. In Reynolds’11 subjects, exploratory laparotomy could be performed without the use of anesthetics. It was not until after the removal of the electrodes that the rats responded to painful stimuli.11 Whether stimulated by electrodes, pills, or other interventions, pain modulatory systems underlie analgesic treatments. In this section, we reviewed the following pain modulatory mechanisms: (i) endogenous opioid, (ii) autonomic (serotonergic, dopaminergic, and noradrenergic), (iii) inhibitory amino acid (cholecystokinin [CCK], galinin, and gamma amino butyric acid [GABA]), (iv) placebo, (v) nontraditional, (vi) exogenous opioid, (vii) cannabinoid, and (viii) electrical.
Endogenous Opioid Modulation of Pain
The phenomenon is familiar: an individual undergoes a traumatic injury without demonstrating pain‐related behaviors. Endogenous opioid modulation gives important clues to explain this phenomenon. Feng et al.12 identified “at least ten” endogenous opioids in the brain. Table 1 lists the important endogenous opiates and their preferred receptors. Busch‐Dienstfertig and Stein1 identified three “major representative opioid peptides”: β‐endorphins, Met‐enkephalin, and dynorphin A. They further showed that most of the endogenous opioids are derived from three precursor proteins: pro‐opiomelanocortin (POMC), proenkephalin (PENK), and prodynorphin.1
Table 1.
Endogenous opiates with receptors.51
| Endogenous opiate | Preferred receptor |
|---|---|
| Dynorphins | κ receptor |
| Endomorphins | μ receptor |
| Endorphins | μ receptor |
| Enkephalins | δ receptor |
| Morphiceptin | μ receptor |
| Nociceptin/orphanin FQ | NOP/ORL receptors |
Martikainen et al.13 showed that individuals with chronic lower back pain have decreased endogenous opioid receptor availability relative to healthy controls. Decreased receptor availability may result from the downregulation of opioid receptors in response to persistent activation.13 Martikainen et al.13 proposed that the population of endogenous opioid receptors could be clinically relevant for diagnosing and treating lower back pain.
Significant studies have examined associations between the exogenous and endogenous opioid systems. Indeed, endogenous opioids do not contribute to the side effects associated with exogenous opiates14 or opioid‐induced hyperalgesia.15 One key difference between endogenous and exogenous opioids is associated with the side effects. Because both endogenous and exogenous opioids act on the same receptors, it would be reasonable to expect their similar central nervous system effects. However, unlike exogenous opioids, endogenous opioids are delivered to their specific sites of action by immune cells.16, 17 Targeted delivery generally prevents exogenous‐like side effects on the central nervous system.16 In fact, Rittner et al.18 observed a relationship between the number of leukocytes in tissue and the amount of endogenous pain relief.17
Autonomic Modulation of Pain
Pain research literature has established a link between autonomic function and pain. For example, Evans et al.19 submitted children to noxious stimuli while measuring autonomic responses. Children with chronic pain, unlike healthy counterparts, demonstrated only minimal autonomic response to acutely painful stimuli.19 In another study conducted by Chalaye et al.20 autonomic dysfunction was associated with fibromyalgia and irritable bowel syndrome (IBS). Chalaye et al.20 showed that the hyperalgesia of fibromyalgia and IBS corresponded to a state of sympathetic hyperactivity. This observation stood in stark contrast to healthy controls that, when exposed to the same stimulus, showed increases in parasympathetic function.20 In the following section, we discussed autonomic modulation via dopaminergic, noradrenergic, and serotonergic pain modulation.
Dopaminergic modulation of pain
While relatively little is known about the mechanisms of dopaminergic pain modulation, dopamine appears to affect the sensation of pain both directly and indirectly. For example, pain disorders are particularly common in individuals that have diseases that affect dopamine (these include Parkinson's disease and restless leg syndrome).7, 21 De la Mora et al.22 noted an important function for dopamine in fear and anxiety. The effect of dopamine on the amygdala can lead to different behavioral results, based on dopaminergic‐influenced, amygdaloid pain processing.22 This observation is consistent with the action of dopamine as an indirect modulator of pain. Triester et al.21 used the nonspecific dopamine agonist apomorphine to show a direct action of dopamine on conditioned pain modulation. While studying the actions of the descending dopaminergic pathway, Taniguchi et al.23 found that dopamine activated D2‐like receptors and K+ channels and decreased glutamate release. Figure 2 illustrates the result of glutamate release on nociception, demonstrating a potential pathway by which dopamine action could mediate pain modulation. The result is a decrease in nociceptive transmission from the substantia gelatinosa.23 Dopamine has been shown to be important in pain modulation, however, the specific mechanisms by which dopamine modulates pain are generally unclear. Thus, dopamine represents an important area in analgesia research.
Figure 2.
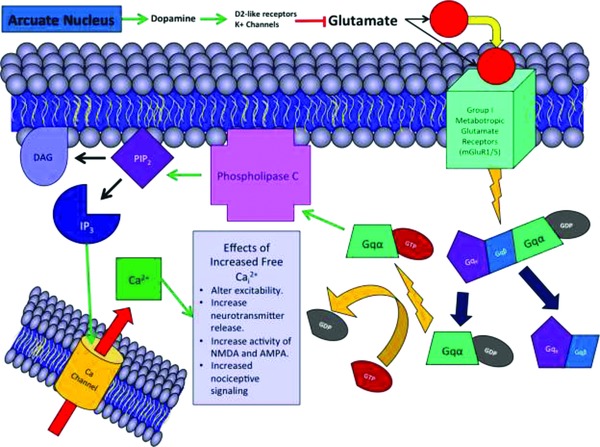
Nociceptive signaling in the amygdala. Dopamine acts within the amygdala. Via K+ channels and D2‐like receptors, dopamine leads to decreased glutamate secretion. Glutamate activates group 1 metabotropic glutamate receptors, leading to the activation of G‐proteins, phospholipase C, cleaving of PIP2 into DAG and IP3, and the opening of intracellular calcium channels. Once open, calcium channels release calcium into amygdala cells, leading to a variety of excitatory effects that cause increased nociception.
One interesting implication of the involvement of dopamine as a pain modulator involves the myriad of pharmaceutical agents whose actions affect dopaminergic receptors. Most antipsychotic drugs, for example, work via antagonism of dopamine receptors. As dopamine and its receptors are important in circuits ranging from pain to emotion to reward processing, treatments that affect dopamine could potentially lead to clinically undesirable results, including addictive behaviors. Opioids, like dopamine, act in the reward pathway and can influence “wanting”.24 Further research is needed to evaluate the interaction of pain medications, like opioids, with dopamine‐mediated pathways. Additional scholarship regarding the effect of analgesics on dopamine‐regulated reward pathways, sleep cycles, depression, psychosis, and underlying causes of pain would have direct clinical applicability.
Noradrenergic modulation of pain
Like other modulating pathways, norepinephrine has both analgesic and hyperalgesic effects. Wu et al.25 found that dezocaine mediated an analgesic effect in rats, which was, at least in part, mediated by the ability of the drug to block norepinephrine and serotonin reuptake. Albrecht et al.26 found changes in sympathetic innervation in the skin of those with fibromyalgia. These changes led to decreased norepinephrine signaling relative to normal, healthy controls.26 Individuals with fibromyalgia, have an imbalance of pain fibers and norepinephrine‐sensitive sympathetic innervation.26 In an interesting study of pain in those suffering from depression, Jaracz et al.27 found selective noradrenergic (and serotonergic) antidepressants to mediate the physical pain symptoms of depression, and found efficacy for dual‐action drugs in the treatment of depression and pain.
While the role of noradrenergic pain modulation in the action of opioids is debated,28 noradrenergic pain modulation has been shown to be involved in descending pain modulatory circuits, including the rostral ventromedial medulla (RVM) and periaqueductal gray (PAG).29 De Felice et al.30 showed that the RVM and PAG integrate descending pain modulation via two cell types: OFF (pain inhibitory) and ON (pain excitatory). Research in rats with induced allodynia showed that lidocaine injection into the RVM reversed the allodynia.31 However, when normal rats received the same injections, the lidocaine caused allodynia.31 De Felice et al.30 concluded that the development of neuropathy might depend on RVM modulation. Thus, descending modulation from the RVM may be a factor that explains why, after injury, some progress from acute to chronic pain while others do not.30 Given the direct action of norepinephrine on pain and a possible role for norepinephrine in establishing the chronicity of pain, agonists and antagonists of norepinephrine receptors could be useful in both research and clinical settings.
Principles of noradrenergic pain modulation have found their way into both clinical practice and the analgesic pharmacopeia. For example, inhibitors of serotonin (selective serotonin reuptake inhibitors [SSRIs]) and norepinephrine reuptake (SNRIs), while traditionally used as anti‐depressants and anxiolytics, have clinical efficacy at alleviating pain. In a study using a rat model, Chu et al.32 found that duloxetine, an SNRI, decreased the firing of pain responsive neurons, and thus duloxetine effectively modulates the pain system in rats with spinal nerve ligations.32 Yarnitsky et al.33 evaluated the usefulness of duloxetine, an SNRI, in the treatment of neuropathic pain in patients with diabetic peripheral neuropathy. These investigators correctly hypothesized that patients with decreased endogenous descending pain inhibitory pathways would receive more efficacious pain management from SNRIs than patients with normal endogenous descending pain inhibitory pathways. This research highlights the analgesic value of SNRIs in certain predictable settings.33 Also, the clinical efficacy of SNRIs demonstrates the significance of norepinephrine as a mediator of pain.
Serotonergic modulation of pain
Serotonergic modulation of pain has been shown to contribute to both pro‐ and antinociceptive processes.34, 35 Ossipov et al.34 noted that, depending on the receptor subtype; the results of serotonin modulation could differ. Specifically, 5‐HT1A, 5‐HT1B, 5‐HT1D, and 5‐HT7 receptors tend to be antinociceptive.34 Conversely, 5‐HT2A and 5‐HT3 receptors tend to have a pronociceptive action.34 In an interesting genetic study, human subjects were tested for the functional variable tandem repeat polymorphisms for serotonin transporters, then subjected to painful stimuli.35, 36 The subjects with long alleles exhibited a higher magnitude of conditioned pain modulation than those with short alleles.32, 33 In response to these findings, Klintschar35 concluded that serotonin is particularly important in the process of endogenous analgesia.36 Aira et al.37 identified upregulation of serotonin receptors (5‐HT2A Receptor) and impairment of μ‐opioid receptors in neuropathic pain subjects.34 Moreover, Aira et al.37 observed that 5‐HT2A receptor agonists increased the potentials of pain‐transmitting C fibers in the dorsal horn. By evaluating specific receptors (TRPV1), Kim et al.38 showed that serotonergic modulation is a “central mechanism” in chronic pain, and that blockade of TRPV1 and 5‐HT3A receptors decreased central sensitization.38
As noted above in the section of noradrenergic pain modulation, serotonergic modulation of pain has proven to be of significant clinical efficacy. In a meta‐analysis on pain treatments, Dharmshaktu et al.39 evaluated the clinical efficacy of antidepressants as analgesics, and found that neuropathic pain is responsive to antidepressants. Further, tricyclic antidepressants (TCAs) were named as first‐line treatment for neuropathic pain.39 These investigators also evaluated other antidepressants like SNRIs and SSRIs. Specifically, it was noted that SSRIs are better tolerated than TCAs, but less effective at treating most types of persistent pain.39 Since TCAs, SSRIs, and SNRIs alter the reuptake of serotonin, the findings of the meta‐analysis highlight the importance of serotonin in pain modulation.
Inhibitory Amino Acids and Pain Modulation
Both inhibitory and excitatory neurotransmitters contribute to the sensation of pain. The opposing actions of these factors could be considered jointly as a pain modulatory mechanism, giving both inhibitory and excitatory agents true clinical value. The following section discusses important inhibitory amino acids, particularly CCK, GABA, and galanin.
Cholecystokinin
CCK is a gastrointestinal hormone released in response to food intake.40 Research, however, has elucidated other potential roles for CCK, including memory and pain.40 Cao et al.40 studied the role of CCK in both memory and visceral pain and concluded that CCK activates vagal afferent C fibers leading to “pain‐affective processing and memory.”40 This study has implications for human pain conditions, particularly IBS.40
Research findings have underscored the importance of centrally acting CCK in pain modulation. Marshall et al.31 suggest that CCK inhibits pain‐relieving modulation from the RVM. Marshall et al.31 injected CCK into the RVM of rats, eventually finding that the injections led to PGE2‐mediated pain hypersensitivity.30 The investigators also identified an “antiopioid” effect of CCK on descending modulation. Benedetti et al.41 studied the antiopioid effect of CCK specifically, showing that CCK agonists “completely disrupted” the placebo modulation of pain. Benedetti et al.41 hypothesized that CCK action may be a factor for placebo “nonresponders.” Lee at al.42 further examined the receptors associated with opioids, melanocortin, and CCK, and hypothesized that CCK and melanocortin antagonists could increase the effectiveness of opioids. Lee et al.42 synthesized “ligand 10,” which demonstrated biological activity at CCK, melanocortin, and opioid receptors. Mitchell et al.43 further elucidated the effects of CCK on descending pain modulation. Using the PAG of rats, these investigators showed that CCK1 receptors mediate the inhibitory effect of CCK on GABA transmission. Moreover, CCK was shown to affect cannabinoid pain modulation.43 Since it affects both cannabinoid and opioid pain modulation, CCK represents a potentially valuable avenue for clinical pain research.
GABA
GABA is the predominant inhibitory neurotransmitter in the central nervous system.44 Loss of GABA‐mediated inhibition of nociception may be a key process in the development of inflammatory and neuropathic pain.45,46 GABA is very important in descending pain modulation: most of the descending, modulatory projections of the central nervous system are either glycinergic or GABAergic.
Munro et al.45 studied GABA modulation in rats. Their study suggests that allosteric modulators for GABAA receptors could potentially serve to treat pain.45 Modulators with high selectivity for α2 and α3 GABAA receptors were found to mediate particularly powerful analgesia.45 In a study conducted by Reichl et al.,44 GABAA and GABAB receptors were activated in rats by administering agonists. Subsequently, the rats were subjected to surgical incisions.44 Intrathecal, but not peripheral, deposition of the agonists reduced hyperalgesia in the rats.44 Conversely, the administration of GABAA and GABAB antagonists had the opposite effect.44 This study highlights the potential for using GABA receptor agonists to provide postsurgical analgesia.44
Yowtak et al.47 examined the effect of radical oxygen species (ROS) on the pathogenesis of pain. Chronic pain was induced in mice followed by injections to increase or decrease the concentration of ROS in the subject.47 Increased ROS induced pain symptoms in the mice, while decreasing ROS produced an anti‐hyperalgesic effect.47 Yowtak et al.47 observed that ROS “selectively attenuate” GABAergic transmission.48 These results suggest that increased ROS may induce pain by reducing GABA inhibition of substantia gelatinosa neurons.47, 48
Drugs that target GABA receptors can be used to promote analgesia.46 Munro et al.46 noted that agonists of GABA receptors, including benzodiazepines, are not optimized for inducing analgesia. Munro et al.46 name GABA receptors as potential analgesic targets. Pain modulation by GABA represents an interesting avenue of investigation for pain‐relief treatment.
Galanin
Galanin is a neuropeptide capable of both facilitation and inhibition of nociception.49, 50 Galanin has been linked to anti‐nociception in mouse models of chronic pain.50 Galanin levels in the dorsal horn have been shown to increase in response to peripheral nerve damage.49, 50 In a study of peripheral modulation of pain by galanin, Hulse et al.49 observed that peripheral interaction between galanin and galanin receptor 2 (GalR2) could be a potential target for analgesic drugs. Hulse et al.49 conclude that GalR2, when activated by galanin (or other agonists), inhibits the activity of primary nociceptive afferents, reducing nociceptive transmission into the spinal cord.
In a different study, Hulse et al.50 probed the effect of galanin on mechanical‐ and cold‐pain. They showed that galanin 1 receptors (GalR1) mediate cold allodynia and GalR2 mediates mechanical allodynia.50 Since mechanical‐ and cold‐pain are common in neuropathies, this finding underscores the importance of galanin in treating neuropathy.50 Lemons and Wiley51 conducted an interesting study that explored the role of galanin in thermal pain modulation. In rats, Lemons and Wiley51 destroyed GalR1 to examine rats in vivo and their spinal cords post mortem. Observations from these studies suggest an important role of GalR1 in thermal modulation: loss of these neurons produced thermal hypoalgesia.51
Reed and Blackshaw52 report that GalR1 and GalR3 are pain inhibitory and GalR2 is pain excitatory, and identified galanin as an eventual player in gut‐pain. One study, conducted by Yu et al.,53 hypothesized that galanin is important in explaining differences in pain thresholds for those that are obese. The change in nociceptive processing among the obese could be tied to galanin and its activation of GalR1 and GalR2.53
Placebo Modulation of Pain
The effectiveness of placebo treatment in pain has been well documented.54 Levine et al.55 hypothesized that the placebo effect is tied to the release of endorphins. To test this hypothesis, Levine et al.55 administered naloxone (opioid receptor antagonist) or placebo to postoperative dental patients. Those given naloxone reported significantly more pain than those given placebo.55 The results of this research suggest that the release of endogenous opiates underlie the placebo effect.
Ellingsen et al.56 took a different approach to evaluating the clinical value of placebo treatments. Ellingsen et al.56 probed the role of placebo in mediating an increase in pleasant experiences rather than eliminating negative experiences, and concluded that the placebo effect is partly mediated by decreases in neural processing, suggesting that the neural structures that carry pain fibers experience a decrease in processing in response to placebo interventions. Ellingsen et al.56 further observed that placebo modulation can change the way that brain structures “appraise” a potentially painful stimulus, causing it to be less painful.
In a review of the role of placebo in back pain, Puhl et al.54 investigated potential differences in the effectiveness of “sham” treatments. This study aggregated the results of several studied that used placebo treatments for low back pain. The results of the analysis suggest that sham medications are a potentially valuable tool for clinicians, despite potential ethical objections.54 Due to these conclusions, Puhl et al.54 advocate for using placebo medications preferentially over interventions that are potentially hazardous or addictive.
Nontraditional Pain Modulation
Nontraditional methods have, at times, been shown to provide significant pain relief. In one study by Zeidan et al.57 individuals given 4 days of meditation training were tested with functional MRI while meditating in the presence of noxious stimuli. The meditators were found to experience significantly less “unpleasantness” (reduced by 57% based on the responses by the participants) and “pain intensity” (reduced by 40%) than those who were simply at rest.57 Decreases in cortical thickness, including the prefrontal cortex, can be predictive of chronic pain.58 Interestingly, studies have also shown that those who meditate have thicker frontal cortices.58
While exercise may not be correctly categorized as “nontraditional,” literature exists to support an important role for exercise in pain relief. For example, Meeus et al.59 explored the benefits of exercise in patients with rheumatoid arthritis, chronic fatigue syndrome, and fibromyalgia, comparing them to healthy controls. In this study, exercise was found to be important in pain suppression, particularly in rheumatoid arthritis patients.59 Meeus et al.59 concluded that a combination of centrally acting drugs and exercise therapy could both prevent pain onset and lessen pain intensity.
Another nontraditional pain‐relief intervention is acupuncture. In a paper that explored connections between acupuncture and autonomic function, Beissner et al.60 suggested that acupuncture might have therapeutic potential since these investigators found that acupuncture could activate or inactivate the sympathetic nervous system. Changes in sympathetic and vagal activity that occur with acupuncture are theoretically capable of impacting autonomic pain modulatory pathways.60 Additionally, as with all pain reducing interventions, nontraditional treatments could provide analgesia via the same endogenous opioid pathways that provide placebo pain relief.
Exogenous Opioid Modulation of Pain
Opioids are known to be particularly powerful, extraordinarily useful analgesics. Busch‐Dienstfertig and Stein1 characterized opioids the “most powerful” analgesic drugs. They also mentioned the downside of opioid therapy: opioid side effects (including addiction, breathing depression, constipation, nausea, and tolerance).1 Opioids work through inhibition of calcium and potassium channels, preventing the release of vesicles that contain pain neurotransmitters.1, 61 The pathways by which opioids inactivate calcium channels are illustrated in Figure 3.
Figure 3.
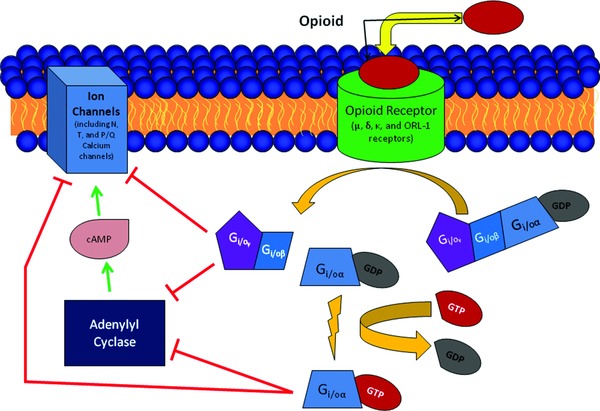
Opioid inactivation of calcium channels. Opioids act on opioid receptors (μ, κ, δ, and opioid receptor‐like receptor [ORL]) leading to the activation of G‐proteins and both direct and indirect closing of ion channels. Activated G‐proteins can directly close ion channels. Activated G‐proteins also inactive adenylate cyclase, which, when activated, opens ion channels.
There are various receptor types on which opiates have been found to work: μ‐, δ‐, and κ‐opioid receptors, nociception or orphan FQ receptors (NOP), and opioid receptor‐like orphan receptors (ORL).62 Clinically, most opioids target μ receptors.62 Exogenous and endogenous opioids act on the same receptor types.62 Although there is evidence that immune cells respond to opioids, it does not appear that immune cells have any of the known opioid receptor types.62 This observation is suggestive of novel opioid receptors that are currently unknown.
One major frontier in the study of opioid pain modulation investigates methods of mitigating opioid side effects. Research has shown that opioids are powerful analgesics, especially in cases of inflammation.63, 64 Peripherally, activation of opioid receptors on Aδ and C fibers, particularly in the dorsal root ganglia, leads to analgesia.64 The result of the peripheral action of opioids on ion channels is decreased excitability of nociceptors and decreased release of the vesicles that contain pain neurotransmitters.64 However, when exogenous opioid agonists act centrally, opioid side effects become a concern.63, 64 Sanchez‐Fernandez et al.65 conducted a study evaluating the effects of σ1‐receptor inhibition on μ opioid receptors. The study showed that σ1 receptor inhibition could enhance peripheral opioid analgesia without increasing opioid‐induced constipation.65 This study illustrates the impetus for finding ways to mute opioid side effects.
Cannabinoid Modulation of Pain
Recent policy changes have brought cannabinoids into the public mind. Various states have legalized the medical use of marijuana.66 The states of Colorado and Washington have legalized its recreational use.66 This paper will not explore marijuana policies; however, a significant body of scientific research evaluates cannabinoids as pain modulators.
Maione et al.67 demonstrated that cannabinoids could work by modulating TRP channels. In anesthetized rats, cannabinoids were injected into the PAG.67 After these injections, the rats demonstrated antinociceptive responses accompanied by a decrease in both ON (pain excitatory) and OFF cell (pain inhibitory) activity in the RVM.63 Maione et al.67 suggest that cannabinoids function by inhibition of adenosine and by enhancement of serotonin receptors. Using a mouse model, Toth et al.68 studied the effect of cannabinoids on neuropathic pain. Toth and coinvestigators68 noted that the accumulation of microglial cells in the dorsal spinal cord is associated with induction of a neuropathic pain state. Administration of cannabinoids was found to lower nociceptive signaling in a mouse model of microglial accumulation.68 These findings suggest potential use of cannabinoids as a treatment for patients with neuropathies.68 Using a rodent model, Xiong et al.69 provided evidence that cannabinoids can decrease nociceptive transmission by activating the α3 glycine receptor. Cannabinoids could potentially be used as a novel class of agents for the treatment and management of chronic pain.69
In a study conducted by Benedetti et al.,70 human subjects were given naltrexone (opioid receptor antagonist), rimonabant (cannabinoid receptor antagonist), placebo, or a combination of naltrexone and rimonabant. The subjects were then submitted to experimental pain induction under different settings.70 This study showed that changing the subjects’ understanding of the meaning of a painful stimulus changed their ability to tolerate pain.70 Specifically, while expecting a positive result, the subjects tolerated pain longer.70 The study also showed that increased pain tolerance could be reduced by the blocking opioid and cannabinoid receptors.70 Benedetti et al.70 concluded that opiate and cannabinoid modulation of pain mediates the interaction between pain perception and reward mechanisms.
At present, research into the potential cannabinoid analgesic treatments is a particularly active area of pain research. Although controversial, cannabinoids represent a potentially fruitful avenue for generating novel interventions for the treatment of pain.
Electrical Modulation of Pain (Electroanalgesia)
Electroanalgesia finds its theoretical underpinnings in the GCT.7, 71 One study evaluated the efficacy of Transcutaneous Electrical Nerve Stimulation (TENS) in alleviating shoulder pain.72 Subjects were monitored via functional MRI while TENS was administered.72 Researchers found a statistically significant decrease in perceived pain intensity and pain‐specific activation of pain processing structures.72 Vo and Drummond,73 after noting studies that examined a link between forehead analgesia and painful stimuli on the forearm, conducted experiments in central sensitization using UV and electrical stimulation. Electrical stimulation was found to induce central sensitization that was stronger than UV‐induced sensitization.73 DaSilva et al.74 studied transcranial direct current stimulation in patients with migraines, demonstrating positive (albeit delayed) analgesic results.74
Studies examining electrical modulation of pain are not universally successful. In one study, for example, Claydon et al.75 compared the efficacy of shock treatments to placebo and found no significant difference between them, and noted that the parameters of the study differed from many other TENS studies. Claydon et al.75 also observed that no scientific consensus exists to define electroanalgiesia parameters. A study by Vassal et al.71 compared high frequency electrical stimulation analgesia to placebo. TENS was found to significantly attenuate pain compared to Vassal's TENS placebo.71 While standards to guide the application of electrical analgesia are poorly defined, there is significant evidence that electrical modulation of pain is possible.
Expert Commentary and 5‐Year Review
Various mechanisms by which the interventions discussed in this review mediate analgesia are graphically illustrated in Figure 4. In regard to potential analgesic therapies, existing research highlights a variety of areas in which additional research may yield novel analgesics. Specific serotonin receptors mediate analgesia (5‐HT1A, 5‐HT1B, 5‐HT1D, and 5‐HT7), while others are pronociceptive (5‐HT2A and 5‐HT3).34 Interventions that specifically agonized antinociceptive receptors or antagonized pro‐nociceptive receptors (or both) could produce analgesic results.34, 37 Antagonists for central CCK receptors could potentially increase the viability of placebo analgesia and other analgesic interventions.43 This suggests that the power of the placebo effect is likely to be magnified with the administration of central CCK antagonists.43 This effect should be tested for clinical applicability. Galinin, similarly, could be a target of analgesic therapy, particularly in those that are obese.49, 53 Perhaps most surprisingly, GABA agonists are not currently optimized for pain relief, but could theoretically have significant pain‐relief efficacy.46 Cannabinoids have been shown to possess pain‐relieving qualities and are likely to underlie future clinical pain interventions because they affect pain pathways in a novel way.47 Truly, existing research has underscored a variety of agents with unexploited analgesic potential. These agents and interventions merit additional scrutiny.
Figure 4.
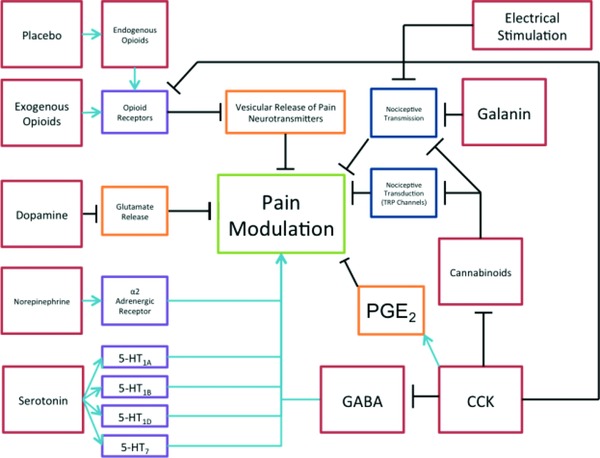
Schema for analgesia. This schematic diagram briefly illustrates the mechanism of analgesic action for a variety of modulatory processes. Starting from the top left and moving clockwise, the legend will briefly summarize each. Placebo modulation works via the endogenous opioid pathway. Endogenous opioids activate opioid receptors. The primary effect of activated opioid receptors is analgesia through inhibition of Ca++ and K+ channels, thus preventing the release of neurotransmitter vesicles. Electrical stimulation provides analgesia by increasing competitive, somatosensory signals, resulting in less nociceptive transmission. Galanin works by decreasing nociceptive transmission via GalR1, 2, and 3 receptors. Cannabinoids work by inhibiting TRP channels (pain transduction) and by decreasing nociceptive transmission via alpha‐3 receptors. Cholecystokinin (CCK) receptor activation decreases GABA and antagonizes opioid and cannabinoid receptors. Antagonizing CCK receptors can have an analgesic effect. GABA is an inhibitory amino acid. Agonistic activity of GABA receptors can diminish the sensation of pain. Serotonin mediates analgesia via a variety of 5‐HT receptors. Norepinephrine mediates analgesia via alpha‐2 receptors. Dopamine inhibits glutamate release, which decreases pain transmission. Exogenous opioids work via the same receptors and processes as endogenous opioids.
In regard to potential diagnostic methods to measure or evaluate pain, currently, no objective measures are available. While a flawless objective measure of pain is unlikely, several biomarkers of physiological and chemical changes could act as objective “clues.” For example, measurements of cortical thickness could potentially be used to predict and diagnose chronic pain.58 Other studies suggest that the availability of opioid receptors could be linked to pain severity.13 Similarly, a relationship has been observed between the population of white cells in an area and the degree of endogenous pain relief.18 Genetically, the presence of certain alleles could explain differences in pain thresholds.35, 36 These effects should be studied for generalizability and clinical significance. Collectively, these observations point to the potential for developing objective, albeit imperfect, measures of clinical pain. Additional research is needed to define the sensitivity, specificity, and cost effectiveness of using these techniques.
Conclusion and Future Direction
Indeed, objective evaluation of pain remains a tremendous clinical challenge. Despite a vast body of research, many significant questions remain unanswered. These include: (i) the investigators debate the usefulness of electroanalgesia, but use different parameters in their research.71 What parameters maximize the efficacy of electroanalgesia? (ii) Endogenous opioids lack central side effects because they are delivered to their site of action by immune cells.16, 18 Could exogenous opioids be delivered in this way? (iii) Dopamine underlies a variety of different pharmaceutical interventions, including pain. What are the specific interactions of pain treatments with pathologies caused by a derangement of dopamine and its receptors? How do agents that act on dopamine receptors interact with each other? When given in combination, how do these agents affect clinical outcomes? (iv) The RVM (and mediators that affect its action) has been named as an anatomical site tied to the chronicity of pain.43 Could further research into RVM function elucidate the process of pain chronicity? (v) Allele polymorphisms may be predictive of pain susceptibility.35, 36 Do those that possess these alleles account for an outsized portion of the chronic pain population? (vi) Could tests be devised to estimate risk in an individual to develop chronic pain? Combined with questions regarding the generation of novel analgesic treatments and the potential for objectives tools to measure pain in a clinical setting, these questions represent key areas of inquiry for the improvement in pain modulation and developing better therapeutic approaches.
Conflict of Interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Acknowledgments
This work was supported by research grants R01HL116042, R01HL112597, R01HL120659 from the National Institutes of Health, USA to DK Agrawal. The content of this review is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
References
- 1. Busch‐Dienstfertig M, Stein C. Opioid receptors and opioid peptide‐producing leukocytes in inflammatory pain—basic and therapeutic aspects. Brain Behav Immun. 2010; 24(5): 683–694. [DOI] [PubMed] [Google Scholar]
- 2. Watanabi C. Mechanism of spinal pain transmission and its regulation. Yakugaku Zasshi. 2014; 134(12): 1301–1307. [DOI] [PubMed] [Google Scholar]
- 3. He Z, Guo Q, Xiao M, He C, Zou W. Intrathecal lentivirus‐mediated transfer of interleukin‐10 attenuates chronic constriction injury‐induced neuropathic pain through modulation of spinal high‐mobility group box 1 in rats. Pain Physician. 2013; 16(5): E615–625. [PubMed] [Google Scholar]
- 4. Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain Res. 2009; 60(1): 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Melzack R, Wall PD. Pain mechanisms: a new theory. Science 1965; 150(3699): 971–978. [DOI] [PubMed] [Google Scholar]
- 6. Mendell L. Constructing and deconstructing the gate theory of pain. Pain 2014; 155(2): 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moayedi M, Davis KD. Theories of pain: from specificity to gate control. J Neurophysiol. 2012; 109(1): 5–12. [DOI] [PubMed] [Google Scholar]
- 8. Braz J, Solorzano C, Wang X, Basbaum A. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 2014; 82(3): 522–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kessler NJ, Hong J. Whole body vibration therapy for painful diabetic peripheral neuropathy: a pilot study. J Bodyw Mov Ther. 2013; 17(4): 518–522. doi: 10.1016/j.jbmt.2013.03.001. Epub 2013 Apr 30. [DOI] [PubMed] [Google Scholar]
- 10. Fournier N, Elman M. Reduction of pain and side effects in the treatment of solar lentigines with pneumatic skin flattening (PSF). J Cosmet Laser Ther. 2007; 9(3): 167–172. [DOI] [PubMed] [Google Scholar]
- 11. Reynolds D. Surgery in the rat during electrical analgesia induced by focal brain stimulation. Science 1969; 164(3878): 444–445. [DOI] [PubMed] [Google Scholar]
- 12. Feng Y, He X, Yang Y, Chao D, Lazarus LH, Xia Y. Current research on opioid receptor function. Curr Drug Targets. 2012; 13(2): 230–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martikainen IK, Pecina M, Love TM, Nuechterlein EB, Cummiford CM, Green CR, Harris RE, Stohler CS, Zubieta JK. Alterations in endogenous opioid functional measures in chronic back pain. J Neurosci. 2013; 33(37): 14729–14737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta RK, Bruehl S, Burns JW, Buvanendran A, Chont M, Schuster E, France CR. Relationship between endogenous opioid function and opioid analgesic adverse effects. Region Anesth Pain M. 2014; 39(3): 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chu LF, Dairmont J, Zamora AK, Young CA, Angst MS. The endogenous opioid system is not involved in modulation of opioid‐induced hyperalgesia. J Pain. 2011; 12(1): 108–115. [DOI] [PubMed] [Google Scholar]
- 16. Kapitzke D, Vetter I, Cabot P. Endogenous opioid analgesia in peripheral tissues and the clinical implications for pain control. Ther Clin Risk Manag. 2005; 1(4): 279–297. [PMC free article] [PubMed] [Google Scholar]
- 17. Mousa SA, Shaqura M, Brendl U, Al‐Khrasani M, Fürst S, Schäfer M. Involvement of the peripheral sensory and sympathetic nervous system in the vascular endothelial expression of ICAM‐1 and the recruitment of opioid‐containing immune cells to inhibit inflammatory pain. Brain Behav Immun. 2010; 24(8): 1310–1323. [DOI] [PubMed] [Google Scholar]
- 18. Rittner HL, Brack A, Machelska H, Mousa SA, Bauer M, Schäfer M, Stein C. Opioid peptide expressing leukocytes. Identification, recruitment and simultaneously increasing inhibition of inflammatory pain. Anesthesiology. 2001; 9(5): 500–508. [DOI] [PubMed] [Google Scholar]
- 19. Evans S, Seidman L, Naliboff B. Heart rate variability as a biomarker for autonomic nervous system response differences between children with chronic pain and healthy control children. J Pain Res. 2013; 6: 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chalaye P, Goffaux P, Bourgault P, Lafrenaye S, Devroede G, Watier A, Marchand S. Comparing pain modulation and autonomic responses in fibromyalgia and irritable bowel syndrome patients. Clin J Pain. 2012; 28(6): 519–526. [DOI] [PubMed] [Google Scholar]
- 21. Triester R, Pud D, Eisenberg E. The dopamine agonist apomorphine enhances conditioned pain modulation in healthy humans. Neurosci Lett. 2013; 548: 115–119. [DOI] [PubMed] [Google Scholar]
- 22. De La Mora MP, Gallegos‐Cari A, Arizmendi‐García Y, Marcellino D, Fuxe K. Role of dopamine receptor mechanisms in the amygdaloid modulation of fear and anxiety: Structural and functional analysis. Prog Neurobiol. 2010; 90(2): 198–216. [DOI] [PubMed] [Google Scholar]
- 23. Taniguchi W, Nakatsuka T, Miyazaki M, Yamada H, Takeda D, Fujita T, Kumamoto E, Yoshida M. In vivo patch‐clamp analysis of dopaminergic antinociceptive actions on substantia gelatinosa neurons in the spinal cord. Pain 2011; 152(1): 95–105. [DOI] [PubMed] [Google Scholar]
- 24. Pecina S, Berridge K. Dopamine or opioid stimulation of nucleus accumbens similarly amplify cue‐triggered ’wanting’ for reward: entire core and medial shell mapped as substrates for PIT enhancement. Eur J Neurosci. 2013; 37(9); 1529–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu FX, Pan RR, Yu WF, Liu R. The anti‐nociception effect of dezocine in a rat neuropathic pain model. Transl Perioper Pain Med. 2014; 1(1): 5–8. [PMC free article] [PubMed] [Google Scholar]
- 26. Albrecht PJ, Hou Q, Argoff CE, Storey JR, Wymer JP, Rice FL. Excessive peptidergic sensory innervation of cutaneous arteriole‐venule shunts (AVS) in the palmar glabrous skin of fibromyalgia patients: implications for widespread deep tissue pain and fatigue. Pain Med. 2013; 14(6): 895–915. [DOI] [PubMed] [Google Scholar]
- 27. Jaracz J, Gattner K, Moczko J, Hauser J. Comparison of the effects of escitalopram and nortriptyline on painful symptoms in patients with major depression. Gen Hosp Psychiat. 2014; 37(1): 36–39. [DOI] [PubMed] [Google Scholar]
- 28. Kimura M, Obata H, Saito S. Peripheral nerve injury reduces analgesic effects of systemic morphine via spinal 5‐hydroxytryptamine 3 receptors. Anesthesiology. 2014; 121(2): 362–371. [DOI] [PubMed] [Google Scholar]
- 29. Cui M, Feng Y, McAdoo DJ, Willis WD. Periaqueductal gray stimulation‐induced inhibition of nociceptive dorsal horn neurons in rats is associated with the release of norepinephrine, serotonin, and amino acids. J Pharmacol Exp Ther. 1999; 289(2): 868–876. [PubMed] [Google Scholar]
- 30. De Felice M, Price TJ, Wang R, Sanoja R, Porreca F, Fields FL, Dussor GO, Lai J, Vanderah TW, Ossipov MH, et al. Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain 2011; 152(12): 2701–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marshall TM, Herman DS, Largent‐Milnes TM, Badghisi H, Zuber K, Holt SC, Lai J, Porreca F, Vanderah TW. Activation of descending pain‐facilitatory pathways from the rostral ventromedial medulla by cholecystokinin elicits release of prostaglandin‐E2 in the spinal cord. Pain 2012; 153(1): 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chu KL, Xu J, Frost J, Li L, Gomez E, Dart MJ, Jarvis MF, Meyer MD, McGaraughty S. A selective α2 B adrenoreceptor agonist (A‐1262543) and duloxetine modulate nociceptive neurons in the medial prefrontal cortex, but not in the spinal cord of neuropathic rats. Eur J Pain 2014; 19: 649–660. [DOI] [PubMed] [Google Scholar]
- 33. Yarnitsky D, Granot M, Nahman‐Averbuch H, Khamaisi M, Granovsky Y. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain 2012; 153(6): 1193–1198. [DOI] [PubMed] [Google Scholar]
- 34. Ossipov M, Morimura K, Porreca F. Descending pain modulation and chronification of pain. Curr Opin Support Palliat Care. 2014; 8(2): 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klintschar M. The association between polymorphisms in serotonin‐related genes and pain modulation might be of importance for the pathogenesis of sudden infant death syndrome. J Pain 2012; 13: 516. [DOI] [PubMed] [Google Scholar]
- 36. Treister R, Pud D, Ebstein RP, Laiba E, Raz Y, Gershon E, Haddad M, Eisenberg E. Association between polymorphisms in serotonin and dopamine‐related genes and endogenous pain modulation. J Pain 2011; 12(8): 875–883. [DOI] [PubMed] [Google Scholar]
- 37. Aira ZM, Zimmermann J, Bilbao DR, Mingo J, Mendiable N, Garcia Del Cano G, Gallego M, Buesa I, Azkue JJ. Time‐dependent cross talk between spinal serotonin 5‐HT2A receptor and mGluR1 subserves spinal hyperexcitability and neuropathic pain after nerve injury. J Neurosci. 2012; 32(39): 13568–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim YS, Wei F, Li M, Han L, Chu Y, Dong X, Dubner R, Ren K, Caterina MJ, Park K, et al. Central terminal sensitization of TRPV1 by descending serotonergic facilitation modulates chronic pain. Neuron 2014; 81(4): 873–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dharmshaktu P, Tayal V, Kalra BS. Efficacy of antidepressants as analgesics: a review. J Clin Pharmacol. 2013; 52(1): 6–17. [DOI] [PubMed] [Google Scholar]
- 40. Cao B, Zhang X, Yan N, Chen S, Li Y. Cholecystokinin enhances visceral pain‐related affective memory via vagal afferent pathway in rats. Molec Pain. 2012; 5: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benedetti F, Amanzio M, Thoen W. Disruption of opioid‐induced placebo responses by activation of cholecystokinin type‐2 receptors. Psychopharmacol. 2011; 213(4): 791–797. [DOI] [PubMed] [Google Scholar]
- 42. Lee YS, Porreca F, Hruby VJ, Lai J, Gillies RJ, Brown K, Ma SW, Davis P, Mayorov A, Kulkarani K, et al. Design and synthesis of trivalent ligands targeting opioid, cholecystokinin, and melanocortin receptors for the treatment of pain. Bioorg Med Chem Lett. 2010; 20(14): 4080–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mitchell VA, Jeong HJ, Drew GM, Vaughan CW. Cholecystokinin exerts an effect via the endocannabinoid system to inhibit GABAergic transmission in midbrain periaqueductal gray. Neuropsychopharmacol. 2011; 36(9): 1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reichl S, Augustin M, Zahn PK, Pogatzki‐Zahn EM. Peripheral and spinal GABAergic regulation of incisional pain in rats. Pain 2012; 153(1): 129–141. [DOI] [PubMed] [Google Scholar]
- 45. Munro G, Erichsen HK, Rae MG, Mirza NR. A question of balance—positive versus negative allosteric modulation of GABAA receptor subtypes as a driver of analgesic efficacy in rat models of inflammatory and neuropathic pain. Neuropharmacol. 2011; 61(1‐2): 121–132. [DOI] [PubMed] [Google Scholar]
- 46. Munro G, Hansen R, Mirza N. GABAa receptor modulation: potential to deliver novel pain medicines? Eur J Pharmacol. 2013; 716(1‐3): 17–23. [DOI] [PubMed] [Google Scholar]
- 47. Yowtak J, Lee KY, Kim HY, Wang J, Kim HK, Chung K, Chung JM. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain 2011; 152(4): 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Z, Ji G, Neugebauer V. Mitochondrial reactive oxygen species are activated by mGluR5 through IP3 and activate ERK and PKA to increase excitability of amygdala neurons and pain behavior. J Neurosci. 2011; 31: 1114–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hulse RP, Donaldson LF, Wynick D. Peripheral galanin receptor 2 as a target for the modulation of pain. Pain Res Treat. 2012; 2012: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hulse RP, Donaldson LF, Wynick D. Differential roles of galanin on mechanical and cooling responses at the primary afferent nociceptor. Mol Pain. 2012; 8: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lemons LL, Wiley RG. Galanin receptor‐expressing dorsal horn neurons: role in nociception. Neuropeptides 2011; 45(6): 377–383. [DOI] [PubMed] [Google Scholar]
- 52. Reed DE, Blackshaw LA. Inhibition of visceral nociceptors. Front Pharmacol. 2014; 5: 72. doi: 10.3389/fphar.2014.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu M, Fang P, Shi M, Zhu Y, Sun Y, Li Q, Bo P, Zhang Z. Galanin receptors possibly modulate the obesity‐induced change in pain threshold. Peptides 2013; 44: 55–59. [DOI] [PubMed] [Google Scholar]
- 54. Puhl A, Reinhart C, Rok E, Injeyan S. An examination of the observed placebo effect associated with the treatment of low back pain—a systematic review. Pain Res Manag. 2011; 16(1): 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Levine J D, Gordon NC, Fields HL. The mechanism of placebo analgesia. Lancet. 1978; 32(8091): 654–657. [DOI] [PubMed] [Google Scholar]
- 56. Ellingsen DM, Wessberg J, Eikemo M, Liljencrantz J, Endestad T, Olausson H, Leknes S. Placebo improves pleasure and pain through opposite modulation of sensory processing. Proc Natl Acad Sci U S A. 2013; 110(44): 17993–17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zeidan FK, Martucci T, Kraft RA, Gordon NS, Mchaffie JG, Coghill RC. Brain mechanisms supporting the modulation of pain by mindfulness meditation. J Neurosci. 2011; 31(14): 5540–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang FC, Chou KH, Fuh JL, Huang CC, Lirng JF, Lin YY, Lin CP, Wang SJ. Altered gray matter volume in the frontal pain modulation network in patients with cluster headache. Pain 2013; 154(6): 801–807. [DOI] [PubMed] [Google Scholar]
- 59. Meeus M, Hermans L, Nijs J, Ickmans K, Struyf F, Van Cauwenbergh D, Bronckaerts L, De Clerk L, Moorken G, Hans G, et al. Endogenous pain modulation in response to exercise in patients with rheumatoid arthritis, patients with chronic fatigue syndrome and comorbid fibromyalgia, and healthy controls: a double‐blind randomized controlled trial. Pain Pract. 2015; 15: 98–106. [DOI] [PubMed] [Google Scholar]
- 60. Beissner F, Deichmann R, Henke C, Bär KJ. Acupuncture —deep pain with an autonomic dimension? NeuroImage. 2012; 60(1): 653–660. [DOI] [PubMed] [Google Scholar]
- 61. Akins PT, Mccleskey EW. Characterization of potassium currents in adult rat sensory neurons and modulation by opioids and cyclic AMP. Neuroscience 1993; 56(3): 759–769. [DOI] [PubMed] [Google Scholar]
- 62. Al‐Hashimi MS, Scott WM, Thompson JP, Lambert DG. Opioids and immune modulation: more questions than answers. Brit J Anaesth. 2013; 111(1): 80–88. [DOI] [PubMed] [Google Scholar]
- 63. Sehgal N, Smith H, Manchikanti L. Peripherally acting opioids and clinical implications for pain control. Pain Physician 2011; 14: 249–258. [PubMed] [Google Scholar]
- 64. Stein C. Targeting pain and inflammation by peripherally acting opioids. Front Pharmacol. 2013; 4: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sanchez‐Fernandez C, Entrena JM, Montilla‐Garcia A, Cobos EJ, Baeyens JM, Merlos M, Fernandez‐Pastor B, Montes R, Artacho‐Cordon A, Romero L, et al. Modulation of peripheral μ‐opioid analgesia by Σ‐1 receptors. J Pharmacol Exp Ther. 2013; 348(1): 32–45. [DOI] [PubMed] [Google Scholar]
- 66. Couch K (Ed). Legalization of Marijuana: point/counterpoint. J Policy Anal Manage. 2013; 33: 211. [Google Scholar]
- 67. Maione S, Piscitelli F, Gatta L, Vita D, De Petrocellis L, Palazzo E, De Novellis V, Di Marzo V. Non‐psychoactive cannabinoids modulate the descending pathway of antinociception in anaesthetized rats through several mechanisms of action. Brit J Pharmacol. 2011; 162(3): 584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Toth CC, Jedrzejewski NM, Ellis CL, Frey WH. Cannabinoid‐mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol Pain 2010; 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xiong W, Xu Y, Zhang L, Ren K, Pan HL, Guan Y, Willenbring D, Chen SR, Yang F, Cheng K, Cui T. Cannabinoids suppress inflammatory and neuropathic pain by targeting 3 glycine receptors. J Exp Med. 2012; 209(6): 1121–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Benedetti F, Thoen W, Blanchard C, Vighetti S, Arduino C. Pain as a reward: changing the meaning of pain from negative to positive co‐activates opioid and cannabinoid systems. Pain 2013; 154(3): 361–367. [DOI] [PubMed] [Google Scholar]
- 71. Vassal F, Créach C, Convers PH, Laurent B, Garcia‐Larrea L, Peyron R. Modulation of laser‐evoked potentials and pain perception by transcutaneous electrical nerve stimulation (TENS): a placebo‐controlled study in healthy volunteers. Clin Neurophysiol. 2013; 124(9): 1861–1867. [DOI] [PubMed] [Google Scholar]
- 72. Kocyigit F, Akalin E, Gezer NS, Orbay O, Kocyigit A, Ada E. Functional magnetic resonance imaging of the effects of low‐frequency transcutaneous electrical nerve stimulation on central pain modulation. Clin J Pain 2012; 28(7): 581–588. [DOI] [PubMed] [Google Scholar]
- 73. Vo L, Drummond PD. High frequency electrical stimulation concurrently induces central sensitization and ipsilateral inhibitory pain modulation. Eur J Pain 2013; 17(3): 357–368. [DOI] [PubMed] [Google Scholar]
- 74. Dasilva AF, Bikson M, Datta A, Bajwa Z, Spierings EL, Dossantos MF, Lopes M, Zaghi S, Mendonca ME, Fregni F. tDCS‐induced analgesia and electrical fields in pain‐related neural networks in chronic migraine. Headache: Headache J Head Face Pain 2012; 52(8): 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Claydon LS, Chesterton LS, Barlas P, Sim J. Alternating‐frequency TENS effects on experimental pain in healthy human participants. Clin J Pain 2013; 29(6): 533–539. [DOI] [PubMed] [Google Scholar]