

Abstract
Oxylipins play important roles in various biological processes and are considered as mediators of inflammation for a wide range of diseases such as rheumatoid arthritis (RA). The purpose of this research was to study differences in oxylipin levels between a widely used collagen induced arthritis (CIA) mice model and healthy control (Ctrl) mice. DBA/1J male mice (age: 6-7 weeks) were selected and randomly divided into two groups, namely, a CIA and a Ctrl group. The CIA mice were injected intraperitoneally (i.p.) with the joint cartilage component collagen type II (CII) and an adjuvant injection of lipopolysaccharide (LPS). Oxylipin metabolites were extracted from plasma for each individual sample using solid phase extraction (SPE) and were detected with high performance liquid chromatography/tandem mass spectrometry (HPLC-ESI-MS/MS), using dynamic multiple reaction monitoring (dMRM). Both univariate and multivariate statistical analyses were applied. The results in univariate Student's t-test revealed 10 significantly up- or downregulated oxylipins in CIA mice, which were supplemented by another 6 additional oxylipins, contributing to group clustering upon multivariate analysis. The dysregulation of these oxylipins revealed the presence of ROS-generated oxylipins and an increase of inflammation in CIA mice. The results also suggested that the collagen induced arthritis might associate with dysregulation of apoptosis, possibly inhibited by activated NF-κB because of insufficient PPAR-γ ligands.
1. Introduction
Rheumatoid arthritis (RA) is a chronic, destructive autoimmune disease which involves primarily the joints in the extremities. The disease is characterized by the destruction of the cartilage in the joints and inflammation of the synovium. This local immune response is characterized by both cell-mediated and humoral immune factors. CD4+ T cells and activated B cells are present in the synovium together with cytokines such as interleukins (e.g., IL-1 and IL-6), tumor necrosis factor (TNFα), and interferon gamma (IF-γ) [1–3]. Recent studies have shown an important role of fibroblasts-like synovial cells in the pathophysiology of RA [4–6]. Upon proinflammatory stimuli and in combination with genetic and epigenetic/environmental factors, these cells, normally responsible for proper composition of the synovial fluid and extracellular matrix, transform into an aggressive phenotype. This phenotype is characterized by a reduced ability to undergo apoptosis [7–12], the production of extracellular enzymes like collagenase and metalloproteases responsible for the destruction of the joints [13, 14], and the secretion of (pro-/anti-) inflammatory cytokines, chemokines, proangiogenic factors, and oxylipins [15–17]. Due to local hypoxia, the formation of reactive oxygen and nitrogen species is promoted [18–21].
Although the role of cytokine/chemokine triggered signal transduction pathways such as MAP kinase and nuclear factor-kappa B (NF-κB) in the pathophysiology of RA has been subject of extensive research, the role of oxylipins is less well understood. Oxylipins are bioactive lipid mediators synthesized from omega-6 polyunsaturated fatty acid such as arachidonic acid (AA), linoleic acid (LA), and dihomo-gamma-linolenic acid (DGLA) and omega-3 polyunsaturated fatty acid like eicosapentenoic acid (EPA), docosahexanoic acid (DHA), and alpha-linolenic acid (ALA) upon liberation from membrane bound phospholipids by activation of phospholipase A2 and subsequent oxidation by cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 epoxygenase (CYP450) systems [22]. This leads to the formation of, over at least hundred, bioactive oxylipins such as prostaglandins (PG), leukotrienes (LT), thromboxanes (TBX), hydroxyeicosatetraenoic acids (HETEs), and epoxyeicosatrienoic acids (EpETrEs). They can act on both local and distant targets by secretion into the circulation system of body. AA is the substrate of proinflammatory lipid mediators while EPA and DHA derived lipid mediators are anti-inflammatory such as resolvins and protectins playing a role in the resolution of inflammation [23]. Nonenzymatic oxidation of polyunsaturated fatty acids produces the closely related bioactive lipids mediators like, for example, isoprostanes, HETEs, and HDoHEs, indicators of oxidative stress [24–29]. Therefore, investigation of the changes of oxylipins in RA animal models will certainly contribute to the understanding of biochemical events in RA research.
Metabolomics is an important and rapidly emerging field of technology enabling the comprehensive analysis of a large number of metabolites associated with disease phenotypes. We have applied a metabolomics approach using a LC-MS based platform combined with elaborate statistical methods to analyze oxylipins in a validated model of RA that is collagen induced arthritis in mice. Our results point to a diminished anti-inflammatory response and increased oxidative stress in the RA induced situation.
2. Materials and Method
2.1. Chemicals
Methanol (MeOH), acetonitrile (ACN), isopropanol (IPA), ethyl-acetate (EtOAC), and purified water were purchased from Biosolve (Netherlands). All reagents used during the HPLC-MS/MS experiments were ultra-performance liquid chromatography grade (UPLC). Acetic acid was purchased from Sigma-Aldrich (St. Louis, Mo). Standards were purchased from Cayman (Netherlands).
2.2. Animal Studies
DBA/1J male mice (6-7 weeks; Charles River Laboratories) were used in this study. Twenty mice were randomly divided in two groups (10 in CIA group, 10 in Ctrl group as healthy control). In the CIA group, immunization with collagen type II will provoke chronic polyarthritis by the induced autoimmune response. Each mouse was intraperitoneally induced (i.p.) with joint cartilage component collagen type II (CII; 100 μg diluted with a 100 μL volume 0.005 M acetic acid) which was extracted from bovine nasal cartilage (Funakoshi Co., Tokyo, Japan) at day 0 (T = 0). Thereafter, the CII injection was repeated i.p. on days 14, 28, 42, and 56. In the Ctrl mice, 100 μL of 0.005 M acetic acid alone was administered i.p. on the same days (0, 14, 28, 42, and 56).
Next, to all experimental mice, 5 mg of lipopolysaccharide from E. coli 011:B4 (Chondrex, Redmond, USA) dissolved in 100 μL phosphate buffered saline (PBS) was given i.p. immediately after each injection of CII. In the Ctrl group, 100 μL PBS was similarly administered as a control. This protocol for arthritis induction is well established and extensively described [30]. All animals were maintained in a temperature and light controlled environment with free access to standard rodent chow and water. From day 71 to day 75, blood was taken from each animal of both groups (CIA mice (CIA1) died when sampling, leaving 9 animal blood samples in the CIA group) and collected in precooled tubes containing EDTA (ethylenediaminetetraacetic acid) as coagulant (BD Vacutainer, Plymouth, UK). After centrifugation at 3000 g for 10 minutes, the EDTA-plasma was collected and aliquots were stored at −80°C until further processing.
2.3. Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The experiments were performed with the approval of the Tohoku Institute of Technology Research Ethics Committee, Sendai, Japan (approval date 18 January 2009).
2.4. Oxylipin HPLC-MS/MS Analysis on Study Mouse Samples
The details of extraction and analysis of oxylipins species were adapted for the analysis of mouse plasma from a previously described oxylipin profiling method [31]. Antioxidant mixture (5 μL) (0.4 mg/mL BHT and 0.4 mg/mL EDTA mixed with volume ratio 1 : 1) and a mixture of internal-standard mixtures (ISTDs) (5 μL, 1000 nM) were added into each 50 μL aliquot of mouse plasma. Subsequently the samples were loaded on the activated SPE plates (Oasis-HLB 96-well plates, 60 mg, 30 μm) and eluted using ethyl acetate (1.5 mL). The dried eluate was redissolved in 50 μL acetonitrile/methanol (50 : 50 v/v) and 5 μL was analyzed by HPLC (Agilent 1290, San Jose, CA, USA) on an Ascentis Express column (2.1 × 150 mm, particle size of 2.7 μm) coupled to electrospray ionization on a triple quadrupole mass spectrometer (Agilent 6490, San Jose, CA, USA). Performance characteristics for the adapted method including recovery, linearity (R 2), linear dynamic range, and sensitivity (LOD/LOQ) were evaluated in a separate validation experiment and the results were comparable to those published before for human plasma by Strassburg et al. [31]. The data is included in the Supplementary Material (Table S1, Figure S1, available online at http://dx.doi.org/10.1155/2015/543541).
2.5. Data Processing and Statistical Analysis
Peak areas were exported from Mass Hunter software (Agilent Technologies, version B.05.01) and ratios to internal standards were computed (target compounds/ISTDs). Subsequently, an in-house developed QC tool [32, 33] was used to correct for instrument drift and batch effects. The reliability of the measurements was assessed by calculating the reproducibility of each metabolite in a QC pool which was measured after every 10 samples. Oxylipins which met the criteria RSD-QC lower than 35% were included in the final list for the further statistical analysis. Data were log transformed (Glog) and scaled by the standard deviation (autoscaling) in order to get a normal distribution [34, 35]. Univariate analysis (two-tailed unpaired Student's t-test) was employed to evaluate significant differences between groups for each metabolite (determined by p < 0.05). Principal component analysis (PCA) and partial least square discriminant analysis (PLSDA) were performed to further investigate the discrimination oxylipins between the two groups using tools provided in the metaboanalyst software package (http://www.metaboanalyst.ca/) [36]. Cross-validation was used in order to validate the performance of the PLS-DA model [37]. A permutation test with 100 iterations was performed to estimate the null distribution, by randomly permuting the class labels of the observations. p values of each pair of comparison in the permutation test were calculated to evaluate the null hypotheses. To select the potential important metabolites which contribute to group separation, Variable Importance in the Projection (VIP) scores based on PLS-DA analysis were used. The higher the VIP score of a metabolite is, the greater its contribution in the group clustering will be. VIP scores higher than 0.8 are considered as meaningful. Variables with VIP score higher than or equal to 1 were considered as significant important features [38, 39].
3. Results
In this study, the relative concentrations of a panel of oxylipins were determined in control and CIA mice. When evaluating the results from the LC-MS/MS analysis, lower responses of ISTDs peak areas were found in two samples, which lead to an extreme high peak area ratio compared with other study samples. Therefore, these two outliers from Ctrl group were excluded from statistical analysis. The list of detected endogenous oxylipins in mice plasma assigned by their precursors is given in Table 1 (details in supplementary table).
Table 1.
List of oxylipins detected in mice plasma, measured by multiple reaction monitoring (precursor ions → product ions) in LC-MS/MS analysis.
| Compounds | MS transitions (m/z) | p value | VIP | Regulation | Pathway |
|---|---|---|---|---|---|
| LA | |||||
| 9,10-DiHOME | 313.2 → 201.1 | 0.0002 | 1.86 | ↓ | CYP450 |
| 12,13-DiHOME | 313.2 → 183.2 | 0.006 | 1.51 | ↓ | CYP450 |
| 9,10-EpOME | 295.2 → 171.2 | 0.028 | 1.27 | ↓ | CYP450 |
| 12,13-EpOME | 295.2 → 195.2 | 0.096 | 1.00 | ↓ | CYP450 |
| 9-KODE | 293.2 → 185.2 | 0.003 | 1.61 | ↓ | 5-LOX |
| 9,12,13-TriHOME | 329.2 → 211.2 | 0.017 | 1.36 | ↓ | 5-LOX |
| 9,10,13-TriHOME | 329.2 → 171.1 | 0.026 | 1.29 | ↓ | 5-LOX |
| 9-HODE | 295.2 → 171.1 | 0.052 | 1.14 | ↓ | 5-LOX |
| 13-KODE | 293.2 → 113.1 | 0.082 | 1.04 | ↓ | 12/15-LOX |
| 13-HODE | 295.2 → 195.2 | 0.733 | 0.21 | — | 12/15-LOX |
| EPA | |||||
| 12-HEPE | 317.2 → 179.1 | 0.016 | 1.37 | ↑ | 12/15-LOX |
| DHA | |||||
| 14-HdoHE | 343.2 → 205.0 | 0.010 | 1.45 | ↑ | ROS |
| 13-HdoHE | 343.2 → 281.0 | 0.012 | 1.42 | ↑ | ROS |
| 10-HdoHE | 343.2 → 153.0 | 0.035 | 1.23 | ↑ | ROS |
| 17-HdoHE | 343.2 → 281.3 | 0.173 | 0.83 | — | 12/15 LOX |
| 19,20-DiHDPA | 361.2 → 273.3 | 0.509 | 0.41 | — | CYP450 |
| DGLA | |||||
| 6-keto-PGF1α | 369.2→ 163.1 | 0.390 | 0.53 | — | COX |
| 8-HETrE | 321.3 → 303.0 | 0.469 | 0.45 | — | 12/15 LOX |
| AA | |||||
| 8-HETE | 319.2 → 155.1 | 0.074 | 1.06 | ↑ | 12/15-LOX |
| 12-HETE | 319.2 → 179.2 | 0.116 | 0.95 | ↑ | 12/15 LOX |
| 15-HETE | 319.2 → 219.2 | 0.770 | 0.18 | — | 12/15-LOX |
| 5-HETE | 319.2 → 115.1 | 0.713 | 0.23 | — | 5-LOX |
| 13,14-dihydro-PGF2α | 355.2 → 275.3 | 0.112 | 0.96 | ↑ | COX |
| PGF2α | 353.2 → 193.1 | 0.176 | 0.82 | — | COX |
| 13,14-dihydro-15-keto-PGF2α | 353.2→ 183.1 | 0.618 | 0.31 | — | COX |
| 12S-HHTrE | 279.2 → 179.2 | 0.733 | 0.21 | — | COX |
| TXB2 | 369.2 → 169.1 | 0.900 | 0.08 | — | COX |
| 14,15-DiHETrE | 337.2 → 207.2 | 0.662 | 0.27 | — | CYP450 |
| 9-HETE | 319.2 → 167.1 | 0.408 | 0.51 | — | ROS |
| 11-HETE | 319.2 → 167.1 | 0.820 | 0.14247 | — | ROS |
The oxylipins are grouped based on the original polyunsaturated fatty acid precursor: linoleic acid (LA), eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), dihomo-γ-linolenic acid (DGLA), and arachidonic acid (AA). Their metabolic pathways include enzymatic pathways: cyclooxygenase (COX), lipoxygenase (LOX), cytochrome P450 (P450), and nonenzymatic reactive oxygen species (ROS) pathway. The significance of changes between two groups was illustrated by p value from univariate test (Student's t-test) and VIP score from multivariate test (PLS-DA). The important regulations in the CIA group were marked with “↓” or “↑” selected based on VIP scores.
↓: downregulated in CIA group.
↑: upregulated in CIA group.
3.1. Univariate and Multivariate Analysis Results
From the QC corrected data, a total of 30 unique oxylipins out of a target list of 110 oxylipins included in the metabolomics platform met the criteria RSD-QC <35%. In order to generally visualize the variance of the samples, a principal components analysis (PCA), as an unsupervised multivariate analysis approach, was performed using these oxylipins. Figure 1 displays the PCA results in the form of a score plot. The first two principal components accounted for 60.1% of the total variance (PC1 35.6% and PC2 24.5%, resp.), which means the model explains well the variance of the samples. The score plot showed a natural distribution of samples between the CIA group and Ctrl group (consisting of the symbols “△” or “+” plots). All 8 samples (100%) of Ctrl group clustered in PCA. Eight out of 9 mice (88.9%) of CIA group clustered as well, while one sample in CIA group was misclassified and clustered within the Ctrl group. This cluster indicates that there are some differences between the samples, which were mainly a reflection of the CIA/Ctrl groups.
Figure 1.
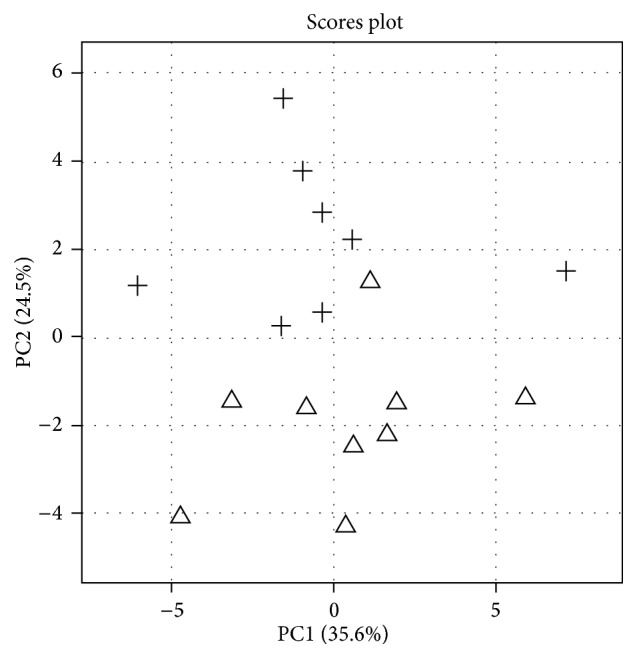
PCA plot of oxylipin data in study mice plasma. PCA score plot of plasma oxylipin data from all study samples revealed general clusters in CIA mice samples and Ctrl samples. The individual samples were marked with “△” or “+” to show the group (CIA versus Ctrl) clustering.
Determining the oxylipin species responsible for the differences between the CIA and Ctrl group is key to unraveling the biological role of this class of compounds in RA. Student's t-test is one of the most widely used methods to determine the statistical significance. In order to understand which of the detected oxylipins showed significant differences between the two groups, an unpaired Student's t-test analysis was evaluated in each individual metabolite. From the t-test, 10 out of the 30 detected oxylipins (percentage of 33.3%) showed significant differences (p < 0.05), namely, 9,10-DiHOME, 9-KODE, 12,13-DiHOME, 14-HDoHE, 13-HDoHE, 12S-HEPE, 9,12,13-TriHOME, 9,10,13-TriHOME, 9,10-EpOME, and 10-HDoHE. In order to show the effect size and variance among the samples, a comparison of individual metabolite levels measured for CIA and control mice is displayed in Figure 2, in the form of boxplots, with “∗” indicating statistical significance between groups. In the boxplot, lines extended from the boxes (whiskers) showed the variabilities outside from the upper and lower quartiles of the data.
Figure 2.

Changes in metabolite levels between Ctrl and CIA mice. Individual metabolite levels for the two groups are illustrated using box-plots with the whisker drawn, after logarithmic transformation for normalization. Boxplot colored: white box: metabolites in Ctrl group; grey box: metabolites in CIA group. The metabolites which differed significantly based on Student's t-test (p < 0.05) are marked with “∗”.
Given that compounds which showed nonsignificant changes from univariate approaches (such as t-tests) may also contribute to group clustering and provide useful information on biological interpretation, a PLS-DA model as a supervised clustering method was further applied to get a more focused view on the metabolites which contribute to group clustering. A PLS-DA scores plot using two components with total score of 43.5% (component 1 = 24.5%, component 2 = 19%) gives a reasonable group separation (figure in supplementary data). However, this model needs to be validated in order to prevent overfitting. Therefore, cross-validation and permutation test was performed. The predictive accuracy (0.88) accompanied with a goodness of fit R 2 (0.84) in cross-validation revealed a sound basis for the PLS-DA model. The permutation tests with an average of 4 misclassifications in 100 iterations (p = 0.04) showed robustness of the model. Thus classification of groups based on this approach can be considered as significant based on both cross-validation and 100 permutation tests.
For this model, the Variable Importance in the Projection (VIP) score was used to summarize the relative contributions of each individual metabolite to the group separation in the PLS-DA. The VIP score shows 14 variables which contributed to the group clustering (VIP > 1), including 5 upregulated oxylipins (14-HDoHE, 13-HDoHE, 12S-HEPE, 10-HDoHE, and 8-HETE) and 9 downregulated oxylipins (9,10-DiHOME, 9-KODE, 12,13-DiHOME, 9,12,13-TriHOME, 9,10,13-TriHOME, 9,10-EpOME, 9-HODE, 13-KODE, and 12,13-EpOME). The top ten of them are also detected in univariate t-test results, which confirmed the importance of these oxylipins.
Given that the oxylipins 13,14-dihydro-PGF2α and 12-HETE have been implicated in inflammatory regulation in disease and also given that they showed a meaningful VIP score close to 1 (0.96, 0.95, resp.) with increasing trend in the CIA group, changes in these metabolites can provide insight in the biological interpretation for CIA and are included in further biological interpretation. The detailed pieces of information of p value from Student's t-test, VIP scores from PLS-DA, and their direction of regulation are shown in Table 1.
3.2. Physiological Pathways of Altered Oxylipins
We grouped the detected oxylipins by their metabolic pathways in order to illustrate their biological roles in Figure 3. Color is used to indicate the up/downregulation (marked in yellow/blue boxes) in the CIA group.
Figure 3.

Overview of regulations of oxylipins in CIA mice compared with Ctrl, including metabolic pathways. Metabolites detected in mice plasma are grouped by metabolic pathways. Important metabolites which contribute most to group clustering based on PLS-DA are colored: yellow box: upregulated in the CIA group; blue box: downregulated in the CIA group.
Among these colored 16 metabolites, all the 9 downregulated oxylipins (9,10-DiHOME, 9-KODE, 12,13-DiHOME, 9,12,13-TriHOME, 9,10,13-TriHOME, 9,10-EpOME, 9-HODE, 13-KODE, and 12,13-EpOME) are derived via the LA group; 3 upregulated oxylipins (8-HETE, 13,14-dihydro-PGF2α, and 12-HETE) are derived from AA; 3 upregulated oxylipins (14-HDoHE, 13-HDoHE, and 10-HDoHE) are derived from DHA; and 1 upregulated oxylipin (12S-HEPE) is produced from EPA.
4. Discussion
Inflammation is a self-limiting innate mechanism under complex regulation with the purpose to recruit leukocytes and plasma proteins, trafficking these to the site of infection or tissue damage, supporting a robust adaptive immune response and subsequent resolution [40]. RA is the consequence of a systemic autoimmune activation/response within the synovial fluid in the joint triggering a dysregulated chronic inflammatory response, of which the exact underlying pathogenic mechanisms still remain largely unclear. RA is characterized with a strong inflamed cytokine phenotype with elevated levels of IL-1β, IL-6, and TNFα as well as increased levels of ROS [18, 41, 42], seen in Figure 4(a). Perturbations related to TNFα activation of the NF-κB pathway inhibiting apoptosis in activated antigen-presenting cells including neutrophils, macrophages, fibroblast-like cells, and B-cells form the general accepted pathological basis of RA [9, 10, 43, 44]. Hence we applied a comprehensive oxylipin metabolomics platform to the plasma of DBA/1J mice induced by a coadministration of type II collagen with lipopolysaccharide, to elucidate the role of these potent inflammatory mediators in RA.
Figure 4.

A systematic autoimmune activation in RA. Appearance of proinflammatory cytokines (IL-1β and IL-6, TNFα) as well as the appearance of ROS in RA. The cytokines normally induce the apoptosis via the caspase pathway but also inhibit apoptosis through degradation IκB activating nuclear factor-κB (NF-κB), which consequently translocate to the nucleus upregulating the antiapoptotic genes (BcL2 and BcL-xL). The activated NF-κB then can also further enhance the production of proinflammatory cytokines and chemokines as well as COX-II enzyme. (b) Upregulated oxylipin response. During RA increased levels of AA derived prostaglandins and HETEs are detected. 8- and 12-HETE are able to activate NF-κB exasperating RA. Due to increased levels of ROS, DHA derived peroxidation products are also found. (c) Dysregulated anti-inflammatory response. LA derived oxylipins including HODEs, KODEs, TriHOMEs, DiHOMEs, and EpOMEs are ligands of peroxisome proliferator-activated receptor- (PPAR-) γ. Due to decreased levels of these anti-inflammatory oxylipins, the ability of PPAR-γ to inhibit the activation of NF-κB and indirectly affect apoptosis is diminished.
We detected an increased proinflammatory oxylipin response, which can be attributed to the activation of NF-κB and increased ROS (Figure 4(b)). NF-κB is the transcription factor for COX-II, and its activation during RA [45, 46] can explain the increased levels of the COX derived prostaglandin F2α measured via its downstream product 13,14-dihydro-PGF2α in CIA mice [47, 48]. Several hydroxyl-fatty acids were also implicated as role players in the chronic inflammatory phenotype of RA. Due to two possible de novo synthesis routes for hydroxyl-fatty acids, it implicates increased LOX activity concurrently with elevated oxidative stress within CIA mice [24–27]. Increased 12-LOX signaling mediators included 8-HETE and 12-HETE supporting a proinflammatory milieu [49, 50]. In an oral tolerance test in CIA rats, Ding et al. [51] measured elevated levels of EPA-derived 18-HEPE, while we detected increased level of a similar metabolite 12-HEPE. Overexpression of 12-LOX in RA has been published by Liagre and Kronke [52, 53], which can further mediate the activation of NF-κB [54–56], indicating the chronic nature of RA. However, 8-HETE, 12-HETE, and 12-HEPE together with the docosahexaenoic acid derived HDOHEs also provide a readout for ROS induced biologically active lipid peroxidation products [24–27]. Oxidative stress leading to increased free radicals as well as ROS levels has been reported in RA by Ozkan et al. [18], supporting this finding.
Alongside the increased pro-inflammatory oxylipins, we also identified significantly decreased LA derived oxylipins in CIA mice plasma. The decreased LA cytochrome P450 products (EpOMEs, DiHOMEs) and LA LOX products (TriHOMEs) implicate a fatty acid precursor perturbation and/or a possible oxylipin enzymatic impairment in RA. AA is the ELOVL mediated elongation product of LA, and the detected increasing trend in AA derived oxylipins indicate sufficient CYP and LOX activity to rule out enzyme activity as the cause of the LA oxylipin reductions. In addition, these LA derived oxylipins as well as the decreased HODEs and KODEs are ligands for nuclear hormone receptor peroxisome proliferator-activated receptor-gamma (PPAR-γ) activation [57–63], shown in Figure 4(c). PPAR-γ are anti-inflammatory regulators of immune cells and can inhibit the activation of NF-κB [44, 46, 61, 62, 64–70]. Therefore, the decreased LA-derived oxylipins and PPAR-γ ligands indicate a perturbation in mechanisms related to the resolution of inflammation, unable to inhibit NF-κB activation and its downstream inhibition of apoptosis.
As discussed above, our detected oxylipins indicate insufficient PPAR-γ ligands, as well as mechanisms leading to the activation of NF-κB, supporting and enhancing our understanding of the inhibition of apoptosis in CIA mice. Apoptosis plays an important role leading to the phagocytic clearances of damage cells stifling the development of chronic inflammation and autoimmunity [71]. The inhibition of apoptosis prevents the silencing of activated leukocytes, dysregulating clearance mechanisms contributing to chronic autoimmune inflammation in RA [72].
5. Conclusion
Using our comprehensive oxylipin method we were able to show that the CIA mice had an arachidonic acid dependent increased proinflammatory profile, with increased levels of oxidative stress. Several studies have been published advocating anti-inflammatory diets (the restriction of AA in the diet), leading to therapeutic benefits and ameliorating RA [73]. We also detected a significant decrease in potent anti-inflammatory oxylipins derived from linoleic acid capable of signaling via PPAR-γ to inhibit the activation of NF-κB, namely, the molecular basis for RA. Interestingly, PPAR-γ has been identified and reported as a therapeutic agent for arthritis [74]. The reduced levels of linoleic acid derived oxylipins implicated fatty acid precursor pools, shedding light on the unexplored routes of fatty acid elongation pathways in the pathogenicity of RA, and need further work. As additional metabolites have been reported to play a role in RA, a systems biology approach would complement the study of systematic autoimmune induced rheumatoid arthritis.
Supplementary Material
The supplementary material provides the methodology of oxylipin extraction and detection and reports performance characteristics of this method. Detailed results from supervised. PLS-DA analysis and VIP scores are also provided in order to demonstrate the important contributions of significant oxylipins to the group clusters.
Acknowledgments
M. He is awarded a scholarship under the support by Chinese Scholarship Council (CSC) during her study in Leiden University in Netherlands as a Ph.D. student (Scholarship File no. 20108220166). Therefore the author would like to give thanks for the support program from CSC. The authors would like to thank Lieke Lamont-de Vries for supporting the QC part of this study. Also thanks are given to Slavik Koval for supporting the statistical analysis part.
Abbreviations
- AA:
Arachidonic acid
- ALA:
α-arachidonic acid
- CIA:
Collagen induced arthritis
- CII:
Collagen type II
- COX:
Cyclooxygenase
- CYP 450:
Cytochrome P450 epoxygenases
- DGLA:
Dihomo-γ-linolenic acid
- DHA:
Docosahexaenoic acid
- DiHETrE:
Dihydroxyeicosatrienoic acid
- DiHOME:
Dihydroxyoctadeca (mono) enoic acid
- EPA:
Eicosapentaenoic acid
- EpETrE:
Epoxyeicosatrienoic acids
- EpOME:
Epoxyoctadecenoic acid
- HDoHE:
Hydroxydocosahexaenoic acid
- HEPE:
Hydroxyeicosapentaenoic acid
- HETE:
Hydroxyeicosatetraenoic acid
- HETrE:
Hydroxyeicosatrienoic acid
- HHTrE:
Hydroxyheptadecatrienoic acid
- HODE:
Hydroxyoctadecadienoic acid
- HOTrE:
Hydroxyoctadecatrienoic acid
- ISTDs:
Internal standards
- KETE:
Ketoeicosatetraenoic acid
- KODE:
Ketooctadecadienoic acid
- LA:
Linoleic acid
- LOX:
Lipoxygenase
- LPS:
Lipopolysaccharide
- LT:
Leukotrienes
- LX:
Lipoxins
- NF-κB:
Nuclear factor-kappa B
- PG:
Prostaglandin
- PPAR:
Peroxisome proliferator-activated receptor
- RA:
Rheumatoid arthritis
- ROS:
Reactive oxygen species
- sEH:
Soluble epoxide hydrolase
- TNF:
Tumor necrosis factor
- TriHOME:
Trihydroxyoctadecenoic acid
- TX:
Thromboxane.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Choy E. H. S., Panayi G. S. Cytokine pathways and joint inflammation in rheumatoid arthritis. The New England Journal of Medicine. 2001;344(12):907–916. doi: 10.1056/nejm200103223441207. [DOI] [PubMed] [Google Scholar]
- 2.Firestein G. S. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 3.Ferraccioli G., Bracci-Laudiero L., Alivernini S., Gremese E., Tolusso B., de Benedetti F. Interleukin-1β and interleukin-6 in arthritis animal models: roles in the early phase of transition from acute to chronic inflammation and relevance for human rheumatoid arthritis. Molecular Medicine. 2010;16(11-12):552–557. doi: 10.2119/molmed.2010.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Connor A. M., Mahomed N., Gandhi R., Keystone E. C., Berger S. A. TNFα modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts. Arthritis Research and Therapy. 2012;14(2, article R62) doi: 10.1186/ar3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ottonello L., Cutolo M., Frumento G., et al. Synovial fluid from patients with rheumatoid arthritis inhibits neutrophil apoptosis: role of adenosine and proinflammatory cytokines. Rheumatology. 2002;41(11):1249–1260. doi: 10.1093/rheumatology/41.11.1249. [DOI] [PubMed] [Google Scholar]
- 6.Bartok B., Firestein G. S. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunological Reviews. 2010;233(1):233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salmon M., Scheel-Toellner D., Huissoon A. P., et al. Inhibition of T cell apoptosis in the rheumatoid synovium. Journal of Clinical Investigation. 1997;99(3):439–446. doi: 10.1172/jci119178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raza K., Scheel-Toellner D., Lee C.-Y., et al. Synovial fluid leukocyte apoptosis is inhibited in patients with very early rheumatoid arthritis. Arthritis Research and Therapy. 2006;8(4, article R120) doi: 10.1186/ar2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinmann P., Moura R. A., Caetano-Lopes J. R., et al. Delayed neutrophil apoptosis in very early rheumatoid arthritis patients is abrogated by methotrexate therapy. Clinical and Experimental Rheumatology. 2007;25(6):885–887. [PubMed] [Google Scholar]
- 10.Ottonello L., Frumento G., Arduino N., et al. Delayed neutrophil apoptosis induced by synovial fluid in rheumatoid arthritis: role of cytokines, estrogens, and adenosine. Annals of the New York Academy of Sciences. 2002;966:226–231. doi: 10.1111/j.1749-6632.2002.tb04219.x. [DOI] [PubMed] [Google Scholar]
- 11.Pope R. M. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nature Reviews Immunology. 2002;2(7):527–535. doi: 10.1038/nri846. [DOI] [PubMed] [Google Scholar]
- 12.Liu H., Pope R. M. The role of apoptosis in rheumatoid arthritis. Current Opinion in Pharmacology. 2003;3(3):317–322. doi: 10.1016/s1471-4892(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 13.Siwik D. A., Colucci W. S. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Failure Reviews. 2004;9(1):43–51. doi: 10.1023/B:HREV.0000011393.40674.13. [DOI] [PubMed] [Google Scholar]
- 14.Ishiguro N., Ito T., Miyazaki K., Iwata H. Matrix metalloproteinases, tissue inhibitors of metalloproteinases, and glycosaminoglycans in synovial fluid from patients with rheumatoid arthritis. The Journal of Rheumatology. 1999;26(1):34–40. [PubMed] [Google Scholar]
- 15.Koch A. E., Volin M. V., Woods J. M., et al. Regulation of angiogenesis by the C-X-C chemokines interleukin-8 and epithelial neutrophil activating peptide 78 in the rheumatoid joint. Arthritis and Rheumatism. 2001;44(1):31–40. doi: 10.1002/1529-0131(200101)44:1&lt;31::AID-ANR5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 16.Kapoor M., Kojima F., Crofford L. J. Arachidonic acid-derived eicosanoids in rheumatoid arthritis: implications and future targets. Future Rheumatology. 2006;1(3):323–330. doi: 10.2217/17460816.1.3.323. [DOI] [Google Scholar]
- 17.Das U. N. Interaction(s) between essential fatty acids, eicosanoids, cytokines, growth factors and free radicals: relevance to new therapeutic strategies in rheumatoid arthritis and other collagen vascular diseases. Prostaglandins, Leukotrienes and Essential Fatty Acids. 1991;44(4):201–210. doi: 10.1016/0952-3278(91)90018-z. [DOI] [PubMed] [Google Scholar]
- 18.Ozkan Y., Yardým-Akaydýn S., Sepici A., Keskin E., Sepici V., Simsek B. Oxidative status in rheumatoid arthritis. Clinical Rheumatology. 2007;26(1):64–68. doi: 10.1007/s10067-006-0244-z. [DOI] [PubMed] [Google Scholar]
- 19.Ostrakhovitch E. A., Afanas'ev I. B. Oxidative stress in rheumatoid arthritis leukocytes: suppression by rutin and other antioxidants and chelators. Biochemical Pharmacology. 2001;62(6):743–746. doi: 10.1016/s0006-2952(01)00707-9. [DOI] [PubMed] [Google Scholar]
- 20.Jaswal S., Mehta H. C., Sood A. K., Kaur J. Antioxidant status in rheumatoid arthritis and role of antioxidant therapy. Clinica Chimica Acta. 2003;338(1-2):123–129. doi: 10.1016/j.cccn.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 21.van Wijk E., Kobayashi M., van Wijk R., van der Greef J. Imaging of ultra-weak photon emission in a rheumatoid arthritis mouse model. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0084579.e84579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shearer G. C., Harris W. S., Pedersen T. L., Newman J. W. Detection of omega-3 oxylipins in human plasma and response to treatment with omega-3 acid ethyl esters. Journal of Lipid Research. 2010;51(8):2074–2081. doi: 10.1194/M900193-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greene E. R., Huang S., Serhan C. N., Panigrahy D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins & Other Lipid Mediators. 2011;96(1–4):27–36. doi: 10.1016/j.prostaglandins.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kühn H., O'Donnell V. B. Inflammation and immune regulation by 12/15-lipoxygenases. Progress in Lipid Research. 2006;45(4):334–356. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Shishehbor M. H., Zhang R., Medina H., et al. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radical Biology and Medicine. 2006;41(11):1678–1683. doi: 10.1016/j.freeradbiomed.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pidgeon G. P., Lysaght J., Krishnamoorthy S., et al. Lipoxygenase metabolism: roles in tumor progression and survival. Cancer and Metastasis Reviews. 2007;26(3-4):503–524. doi: 10.1007/s10555-007-9098-3. [DOI] [PubMed] [Google Scholar]
- 27.Derogis P. B. M. C., Freitas F. P., Marques A. S. F., et al. The development of a specific and sensitive LC-MS-based method for the detection and quantification of hydroperoxy- and hydroxydocosahexaenoic acids as a tool for lipidomic analysis. PLoS ONE. 2013;8(10) doi: 10.1371/journal.pone.0077561.e77561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee C.-Y. J., Huang S. H., Jenner A. M., Halliwell B. Measurement of F2-isoprostanes, hydroxyeicosatetraenoic products, and oxysterols from a single plasma sample. Free Radical Biology and Medicine. 2008;44(7):1314–1322. doi: 10.1016/j.freeradbiomed.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 29.Halliwell B., Lee C. Y. J. Using isoprostanes as biomarkers of oxidative stress: some rarely considered issues. Antioxidants and Redox Signaling. 2010;13(2):145–156. doi: 10.1089/ars.2009.2934. [DOI] [PubMed] [Google Scholar]
- 30.Yoshino S., Sasatomi E., Ohsawa M. Bacterial lipopolysaccharide acts as an adjuvant to induce autoimmune arthritis in mice. Immunology. 2000;99(4):607–614. doi: 10.1046/j.1365-2567.2000.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strassburg K., Huijbrechts A. M. L., Kortekaas K. A., et al. Quantitative profiling of oxylipins through comprehensive LC-MS/MS analysis: application in cardiac surgery. Analytical and Bioanalytical Chemistry. 2012;404(5):1413–1426. doi: 10.1007/s00216-012-6226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Kloet F. M., Tempels F. W. A., Ismail N., et al. Discovery of early-stage biomarkers for diabetic kidney disease using ms-based metabolomics (FinnDiane study) Metabolomics. 2012;8(1):109–119. doi: 10.1007/s11306-011-0291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Der Kloet F. M., Bobeldijk I., Verheij E. R., Jellema R. H. Analytical error reduction using single point calibration for accurate and precise metabolomic phenotyping. Journal of Proteome Research. 2009;8(11):5132–5141. doi: 10.1021/pr900499r. [DOI] [PubMed] [Google Scholar]
- 34.van den Berg R. A., Hoefsloot H. C. J., Westerhuis J. A., Smilde A. K., van der Werf M. J. Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics. 2006;7, article 142 doi: 10.1186/1471-2164-7-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Greef J., Stroobant P., van der Heijden R. The role of analytical sciences in medical systems biology. Current Opinion in Chemical Biology. 2004;8(5):559–565. doi: 10.1016/j.cbpa.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Xia J., Sinelnikov I. V., Han B., Wishart D. S. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Research. 2015;43(1):W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westerhuis J. A., Hoefsloot H. C. J., Smit S., et al. Assessment of PLSDA cross validation. Metabolomics. 2008;4(1):81–89. doi: 10.1007/s11306-007-0099-6. [DOI] [Google Scholar]
- 38.Chong I.-G., Jun C.-H. Performance of some variable selection methods when multicollinearity is present. Chemometrics and Intelligent Laboratory Systems. 2005;78(1):103–112. doi: 10.1016/j.chemolab.2004.12.011. [DOI] [Google Scholar]
- 39.Cassotti M., Grisoni F. Molecular Descriptors. 2012. Variable selection methods: an introduction; p. p. 10. [Google Scholar]
- 40.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 41.Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology. 2012;51(supplement 5):v3–v11. doi: 10.1093/rheumatology/kes113. [DOI] [PubMed] [Google Scholar]
- 42.Hitchon C. A., El-Gabalawy H. S. Oxidation in rheumatoid arthritis. Arthritis Research & Therapy. 2004;6(6):265–278. doi: 10.1186/ar1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simmonds R. E., Foxwell B. M. Signalling, inflammation and arthritis: NF-κB and its relevance to arthritis and inflammation. Rheumatology. 2008;47(5):584–590. doi: 10.1093/rheumatology/kem298. [DOI] [PubMed] [Google Scholar]
- 44.Eguchi J., Koshino T., Takagi T., Hayashi T., Saito T. NF-κB and I-κB overexpression in articular chondrocytes with progression of type II collagen-induced arthritis in DBA/1 mouse knees. Clinical & Experimental Rheumatology. 2002;20(5):647–652. [PubMed] [Google Scholar]
- 45.Makarov S. S. NF-kappa B in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Research. 2001;3(4):200–206. doi: 10.1186/ar300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tak P. P., Firestein G. S. NF-κB: a key role in inflammatory diseases. The Journal of Clinical Investigation. 2001;107(1):7–11. doi: 10.1172/jci11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inazu N., Kogo H., Aizawa Y. Effect of 13,14-dihydroprostaglandin F2alpha on steroid biosynthesis in rat ovary. Japanese Journal of Pharmacology. 1981;31(2):301–303. doi: 10.1254/jjp.31.301. [DOI] [PubMed] [Google Scholar]
- 48.Aizawa Y., Inazu N., Kogo H. Catabolism of prostaglandin F2α in rat ovary: differences between ovarian and uterine tissues. Prostaglandins. 1980;20(1):95–103. doi: 10.1016/0090-6980(80)90009-x. [DOI] [PubMed] [Google Scholar]
- 49.Ahnfelt-Rønne I., Bramm E., Arrigoni-Martelli E. Chronic inflammation in adjuvant arthritic rats correlates with enhancement of 12-l-HETE-synthesis. Agents and Actions. 1981;11(6-7):587–589. doi: 10.1007/bf01978753. [DOI] [PubMed] [Google Scholar]
- 50.Puri P., Wiest M. M., Cheung O., et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009;50(6):1827–1838. doi: 10.1002/hep.23229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding X., Hu J., Li J., et al. Metabolomics analysis of collagen-induced arthritis in rats and interventional effects of oral tolerance. Analytical Biochemistry. 2014;458:49–57. doi: 10.1016/j.ab.2014.04.035. [DOI] [PubMed] [Google Scholar]
- 52.Liagre B., Vergne P., Rigaud M., Beneytout J. L. Expression of arachidonate platelet-type 12-lipoxygenase in human rheumatoid arthritis B synoviocytes. FEBS Letters. 1997;414(1):159–164. doi: 10.1016/s0014-5793(97)00904-6. [DOI] [PubMed] [Google Scholar]
- 53.Kronke G., Katzenbeisser J., Uderhardt S., et al. 12/15-Lipoxygenase counteracts inflammation and tissue damage in arthritis. The Journal of Immunology. 2009;183(5):3383–3389. doi: 10.4049/jimmunol.0900327. [DOI] [PubMed] [Google Scholar]
- 54.Kandouz M., Nie D., Pidgeon G. P., Krishnamoorthy S., Maddipati K. R., Honn K. V. Platelet-type 12-lipoxygenase activates NF-κB in prostate cancer cells. Prostaglandins & Other Lipid Mediators. 2003;71(3-4):189–204. doi: 10.1016/s1098-8823(03)00042-x. [DOI] [PubMed] [Google Scholar]
- 55.Wong B. C. Y., Wang W. P., Cho C. H., et al. 12-Lipoxygenase inhibition induced apoptosis in human gastric cancer cells. Carcinogenesis. 2001;22(9):1349–1354. doi: 10.1093/carcin/22.9.1349. [DOI] [PubMed] [Google Scholar]
- 56.Vonach C., Viola K., Giessrigl B., et al. NF-κB mediates the 12(S)-HETE-induced endothelial to mesenchymal transition of lymphendothelial cells during the intravasation of breast carcinoma cells. British Journal of Cancer. 2011;105(2):263–271. doi: 10.1038/bjc.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edwards L. M., Lawler N. G., Nikolic S. B., et al. Metabolomics reveals increased isoleukotoxin diol (12,13-DHOME) in human plasma after acute intralipid infusion. Journal of Lipid Research. 2012;53(9):1979–1986. doi: 10.1194/jlr.p027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sisemore M. F., Zheng J., Yang J. C., et al. Cellular characterization of leukotoxin diol-induced mitochondrial dysfunction. Archives of Biochemistry and Biophysics. 2001;392(1):32–37. doi: 10.1006/abbi.2001.2434. [DOI] [PubMed] [Google Scholar]
- 59.Thompson D. A., Hammock B. D. Dihydroxyoctadecamonoenoate esters inhibit the neutrophil respiratory burst. Journal of Biosciences. 2007;32(2):279–291. doi: 10.1007/s12038-007-0028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayakawa M., Sugiyama S., Takamura T., et al. Neutrophils biosynthesize leukotoxin, 9, 10-epoxy-12-octadecenoate. Biochemical and Biophysical Research Communications. 1986;137(1):424–430. doi: 10.1016/0006-291X(86)91227-1. [DOI] [PubMed] [Google Scholar]
- 61.Daynes R. A., Jones D. C. Emerging roles of PPARs in inflammation and immunity. Nature Reviews Immunology. 2002;2(10):748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 62.Duez H., Fruchart J.-C., Staels B. PPARs in inflammation, atherosclerosis and thrombosis. European Journal of Preventive Cardiology. 2001;8(4):187–194. doi: 10.1177/174182670100800402. [DOI] [PubMed] [Google Scholar]
- 63.Itoh T., Fairall L., Amin K., et al. Structural basis for the activation of PPARγ by oxidized fatty acids. Nature Structural and Molecular Biology. 2008;15(9):924–931. doi: 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Han Z., Boyle D. L., Manning A. M., Firestein G. S. AP-1 and NF-kB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity. 1998;28(4):197–208. doi: 10.3109/08916939808995367. [DOI] [PubMed] [Google Scholar]
- 65.Teng F. Modulation of inflammation, apoptosis, and oncogenesis by the nuclear transcription factor, NF-κB. Chinese Dental Journal. 2006;25(1):12–24. [Google Scholar]
- 66.Okamoto H., Iwamoto T., Kotake S., Momohara S., Yamanaka H., Kamatani N. Inhibition of NK-κB signaling by fenofibrate, a peroxisome proliferator-activated receptor-α ligand, presents a therapeutic strategy for rheumatoid arthritis. Clinical and Experimental Rheumatology. 2005;23(3):323–330. [PubMed] [Google Scholar]
- 67.Ji J. D., Cheon H., Jun J. B., et al. Effects of peroxisome proliferator-activated receptor-gamma (PPAR-gamma) on the expression of inflammatory cytokines and apoptosis induction in rheumatoid synovial fibroblasts and monocytes. Journal of Autoimmunity. 2001;17(3):215–221. doi: 10.1006/jaut.2001.0542. [DOI] [PubMed] [Google Scholar]
- 68.Kawahito Y., Kondo M., Tsubouchi Y., et al. 15-Deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. The Journal of Clinical Investigation. 2000;106(2):189–197. doi: 10.1172/JCI9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang C., Ting A. T., Seed B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391(6662):82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 70.Straus D. S., Glass C. K. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends in Immunology. 2007;28(12):551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 71.Hochreiter-Hufford A., Ravichandran K. S. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harbor Perspectives in Biology. 2013;5(1) doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Erwig L.-P., Henson P. M. Clearance of apoptotic cells by phagocytes. Cell Death and Differentiation. 2008;15(2):243–250. doi: 10.1038/sj.cdd.4402184. [DOI] [PubMed] [Google Scholar]
- 73.Adam O., Beringer C., Kless T., et al. Anti-inflammatory effects of a low arachidonic acid diet and fish oil in patients with rheumatoid arthritis. Rheumatology International. 2003;23(1):27–36. doi: 10.1007/s00296-002-0234-7. [DOI] [PubMed] [Google Scholar]
- 74.Giaginis C., Giagini A., Theocharis S. Peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands as potential therapeutic agents to treat arthritis. Pharmacological Research. 2009;60(3):160–169. doi: 10.1016/j.phrs.2009.02.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The supplementary material provides the methodology of oxylipin extraction and detection and reports performance characteristics of this method. Detailed results from supervised. PLS-DA analysis and VIP scores are also provided in order to demonstrate the important contributions of significant oxylipins to the group clusters.