

Abstract
Objectives:
Parkinson disease (PD), multiple system atrophy (MSA), and pure autonomic failure (PAF) involve cytoplasmic deposition of α-synuclein and are considered to be synucleinopathies. Approximately 40% of patients with PD, most patients with MSA, and all patients with PAF have neurogenic orthostatic hypotension (OH). This study compared long-term survival in these synucleinopathies.
Methods:
In this prospective cohort study, survival data were obtained for 97.6% of 206 referred patients evaluated between 1994 and 2014 (47 PD + OH, 54 PD no OH, 15 cerebellar MSA [MSA-C], 57 parkinsonian MSA [MSA-P], 28 PAF). Individual diagnoses were confirmed by clinical criteria and results of pharmacologic, neurochemical, and neuroimaging tests of sympathetic noradrenergic innervation. The Cox proportional hazard model was used to calculate hazard ratios (HRs) from symptom onset and from time of evaluation to death.
Results:
Patients with MSA-C or MSA-P had shorter survival from symptom onset than did patients with PD + OH (age- and sex-adjusted HR = 6.1, 5.6; p < 0.0001 each), PAF (HR = 10.8, 9.9; p < 0.0001 each) or PD no OH (HR = 14.9, 13.6; p < 0.0001 each). Among parkinsonian patients who died, median times from motor onset to death were 7.5 years in MSA-P, 11.6 years in PD + OH, and 15.8 years in PD no OH. Probabilities of survival for 10 years from onset of relevant symptoms were 0.39 in MSA-C, 0.33 in MSA-P, 0.74 in PD + OH, 0.87 in PAF, and 0.93 in PD no OH.
Conclusions:
In synucleinopathies, survival depends on the particular disease, with the risk of death greater in MSA-P than in PD + OH and in PD + OH than in PD no OH.
The synucleinopathies are a group of diseases in which the protein α-synuclein is deposited in neurons or glial cells.1 Examples of the clinical conditions are the Lewy body diseases Parkinson disease (PD), pure autonomic failure (PAF), and dementia with Lewy bodies and the non–Lewy body disease multiple system atrophy (MSA). In PD and PAF, α-synuclein is deposited in Lewy bodies in neuronal perikarya and in Lewy neurites in axons; in MSA, α-synuclein is deposited in cytoplasmic inclusions in oligodendrocytes.2 All patients with PAF, most with MSA, and 30% to 40% of patients with PD3,4 have neurogenic orthostatic hypotension (OH). MSA has been classified into parkinsonian (MSA-P) and cerebellar (MSA-C) subtypes.
Survival rates among synucleinopathies have not been compared in the same study. Filling this gap in knowledge is challenging, for a few reasons. First, MSA is uncommon, PAF is rare, and although PD is the second most common neurodegenerative disease of the elderly, and OH occurs frequently in PD,4 PD + OH has not usually been studied as a separate entity. Second, for assessing survival rates in these diseases, accurate diagnostic assignments are essential, and differential diagnosis among these diseases by clinical criteria alone is imperfect.5 Third, although testing of central and peripheral catecholaminergic innervation can aid the differential diagnosis of autonomic synucleinopathies,6 this is not done routinely at most centers.
This study assessed long-term survival in cohorts of synucleinopathy patients evaluated at the NIH Clinical Center between 1994 and 2014. Diagnostic assignments were based both on clinical consensus definitions and assessments of catecholaminergic innervation by pharmacologic, neurochemical, or neuroimaging tests. From the clinical and laboratory findings at the time of initial evaluation at the NIH Clinical Center, we assigned patients to diagnostic groups (PD + OH, PD without OH [termed PD no OH here], MSA-C, MSA-P, or PAF).
Survival in these groups was assessed both from the time of onset of relevant symptoms (locomotor dysfunction in PD and MSA, orthostatic intolerance in PAF) and from the time of initial diagnostic testing at the NIH. Since the pattern of central or peripheral catecholaminergic denervation might be relevant to survival probability, we also examined whether results of putamen dopaminergic and cardiac noradrenergic neuroimaging in patients with neurogenic OH predict survival.
Therefore, the objective of this prospective cohort study was to compare long-term survival in synucleinopathies. Based on the available literature, we predicted that survival would be poorer in MSA-C and MSA-P than in PD + OH, PD no OH, or PAF.
METHODS
Patients.
Patients with PD, MSA, or PAF were referred to the Clinical Neurocardiology Section for evaluation of autonomic function at the NIH Clinical Center.
Standard protocol approvals, registrations, and patient consents.
All of the patients participated in institutional review board–approved protocols of intramural National Institute of Neurological Disorders and Stroke after giving written informed consent.
We sought survival data for all patients with MSA, PAF, PD + OH, or PD no OH studied between 1994 and 2014 (n = 206).
Diagnostic assignments.
For each patient, a diagnosis was assigned based on the medical and neurologic history and physical examination and by a set of tests conducted at the NIH Clinical Center. The diagnostic assignments made at the time of the initial inpatient evaluation were kept for the follow-up study.
MSA was diagnosed clinically using previously published and updated consensus statements.7,8 A diagnosis of “possible MSA” was assigned if a patient had autonomic failure, parkinsonism, or a cerebellar syndrome, and at least 1 of 13 listed additional features. A diagnostic assignment of “probable MSA” was given if a patient had autonomic failure and either parkinsonism poorly responsive to levodopa or a cerebellar syndrome. In addition, the findings of normal or only slightly decreased olfactory function by the University of Pennsylvania Smell Identification Test and normal myocardial 18F-dopamine–derived radioactivity were taken as supportive of MSA as opposed to PD.6 Patients with MSA and both parkinsonism and cerebellar ataxia were considered to have MSA-P.
A diagnosis of PD was assigned based on clinical criteria supplemented by laboratory test results. To be assigned to the PD group, the patient must have had at least 3 of the following 4 clinical criteria: bradykinesia, resting “pill roll” tremor, “cogwheel” rigidity, and excellent response of the movement disorder to levodopa treatment. The finding of cerebellar ataxia was taken as exclusionary for PD. In addition, the findings of anosmia by the University of Pennsylvania Smell Identification Test and low myocardial 18F-dopamine–derived radioactivity were taken as supportive of PD as opposed to MSA.6
A diagnosis of PAF was assigned based on chronic, persistent, consistent OH, identified as neurogenic by clinical laboratory testing,9 without an identified cause (e.g., diabetic autonomic neuropathy, autoimmune autonomic ganglionopathy) and by clinical laboratory evidence of loss of sympathetic noradrenergic innervation (low myocardial 18F-dopamine–derived radioactivity, low plasma levels of 3,4-dihydroxyphenylglycol, or both10).
OH was defined by a decrease in systolic blood pressure of at least 20 mm Hg or in diastolic pressure at least 10 mm Hg between supine rest for at least 15 minutes and upright posture for 5 minutes (unless symptomatic or rapid hypotension necessitated return to the supine position before 5 minutes upright). Neurogenic OH was defined by OH coupled with abnormal beat-to-beat blood pressure during and after performance of the Valsalva maneuver.10
Survival prediction by catecholaminergic neuroimaging.
We assessed survival in groups of patients with neurogenic OH stratified in terms of 2 neuroimaging findings: normal or low cardiac 18F-dopamine–derived and putamen 18F-DOPA–derived radioactivity, regardless of the clinical diagnosis (figure 1). Patients with low cardiac 18F-dopamine–derived radioactivity and normal putamen to occipital cortex ratios of 18F-DOPA–derived radioactivity were defined to have a “low heart, normal putamen” pattern, with low cardiac 18F-dopamine–derived radioactivity and low putamen to occipital cortex ratios of 18F-DOPA–derived radioactivity a “low heart, low putamen” pattern, with normal cardiac 18F-dopamine–derived radioactivity and a low putamen to occipital cortex ratio of 18F-DOPA–derived radioactivity a “normal heart, low putamen” pattern, and with normal cardiac 18F-dopamine–derived radioactivity and a normal putamen to occipital cortex ratio of 18F-DOPA–derived radioactivity a “normal heart, normal putamen” pattern.
Figure 1. Patterns of brain and cardiac catecholaminergic abnormalities.

(A) Decreased putamen 18F-DOPA–derived radioactivity and decreased left ventricular myocardial 18F-dopamine–derived radioactivity. (B) Normal putamen 18F-DOPA–derived radioactivity and normal left ventricular myocardial 18F-dopamine–derived radioactivity. (C) Decreased putamen 18F-DOPA–derived radioactivity and normal left ventricular myocardial 18F-dopamine–derived radioactivity. (D) Normal putamen 18F-DOPA–derived radioactivity and decreased left ventricular myocardial 18F-dopamine–derived radioactivity.
Myocardial 18F-dopamine–derived radioactivity was defined as decreased when the interventricular septal myocardial concentration of 18F-dopamine–derived radioactivity was less than 7,000 nCi-kg/cc-mCi at the midpoint of the 5-minute interval beginning 5 minutes after initiation of 3-minute IV injection of tracer. (Taking into account about 0.5-minute delay between initiation of the injection and the scan start, the midpoint of the scanning interval was about 8 minutes after initiation of the injection.) Putamen 18F-DOPA–derived radioactivity was defined as decreased when the average putamen to occipital cortex (PUT:OCC) ratio of 18F-DOPA–derived radioactivity for the 2 putamina was less than 2.5 during the 15-minute interval ending 120 minutes after initiation of 3-minute IV injection of tracer.
Data analysis.
Survival time was defined in 2 ways. The first was the time in years from the onset of relevant symptoms necessary to meet the diagnostic criteria (a movement disorder in PD, MSA-P, or MSA-C, orthostatic intolerance in PAF) to the date of death or to August 18, 2014. The second was the time in years from the clinical and laboratory testing at the NIH, during which the diagnostic assignment was made. The Kaplan-Meier method was used to construct survival plots. The Cox proportional hazard model was used to estimate hazard ratios (HRs) and 95% confidence intervals and survival probabilities at 5 and 10 years, with age and sex as covariates. The assumption of proportional hazards was verified. The survival analysis was performed using SAS version 9.2.
Mean differences between PD + OH and MSA-P groups were assessed by 2-sample t tests; differences in frequencies were assessed by the χ2 test with the Yates correction.
RESULTS
Survival data from first evaluation to death were obtained for 201 patients (47 PD + OH, 54 PD no OH, 15 MSA-C, 57 MSA-P, 28 PAF) of a total of 206 patients sought (97.6% follow-up). Survival data from relevant symptoms to death were obtained for 181 patients. To meet the proportional hazard assumption, data for 5 patients were excluded from the survival analysis. Thus, data for 176 patients were used in the analysis (45 PD + OH, 50 PD no OH, 11 MSA-C, 44 MSA-P, 26 PAF). The HRs and p values estimated from the 2 datasets were very similar.
Clinical and laboratory comparison of diagnostic groups.
Based on the criteria outlined in the methods section, patients were assigned to 1 of 5 diagnostic groups: PD + OH, PD no OH, MSA-C, MSA-P, or PAF. The 5 groups consisted mainly of middle-aged to elderly people, with somewhat more men than women. At the time of initial evaluation, mean ages in the PD + OH, PD no OH, MSA-C, MSA-P, and PAF groups were 68.7, 60.0, 56.5, 61.0, and 62.8 years, and percentages of males were 68%, 80%, 63%, 61%, and 61%, respectively.
The mean duration of motor symptoms at the time of diagnostic assignment was 8.6 ± 1.2 years in the PD + OH group, 6.5 ± 0.8 years in the PD no OH group, and 4.1 ± 0.3 years in the MSA-P group. The duration of motor symptoms was greater in PD + OH than in MSA-P (p = 0.0006 by Fisher protected least significant difference test after factorial analysis of variance). Mean scores on the Unified Parkinson's Disease Rating Scale (UPDRS) were 45 ± 5, 45 ± 5, and 62 ± 11 units in the 3 groups. The mean UPDRS score was higher in MSA-P than in PD no OH (p = 0.04). Otherwise, the PD + OH, PD no OH, and MSA-P groups did not differ in mean duration of motor symptoms or UPDRS scores.
Diagnostic group assignments were straightforward in the MSA-C and PAF groups (cerebellar ataxia and OH in MSA-C, no clinical evidence of central neurodegeneration in PAF); however, PD + OH and MSA-P are known to overlap clinically. A variety of clinical (table e-1 on the Neurology® Web site at Neurology.org) and laboratory (table e-2, figure 2) findings emerged that separated PD + OH from MSA-P. As expected from the use of the laboratory as well as clinical data to assign diagnoses, 18F-dopamine–derived radioactivity in the myocardial interventricular septum was universal in PD + OH, whereas most patients with MSA-P had normal radioactivity. In addition, two-thirds of the tested patients with PD + OH were anosmic, about 6 times the frequency of anosmia as in MSA-P.
Figure 2. Clinical laboratory distinctions among autonomic synucleinopathies.

In PAF, the putamen to occipital cortex ratio of 18F-DOPA–derived radioactivity (PUT:OCC) is normal, while interventricular septal myocardial 18F-dopamine–derived radioactivity is decreased. In PD + OH, the PUT:OCC ratio is normal, while interventricular septal myocardial 18F-dopamine–derived radioactivity is decreased. In MSA-P, the PUT:OCC ratio is decreased, while septal 18F-dopamine–derived radioactivity is normal. In MSA-C, both the PUT:OCC ratio and septal 18F-dopamine–derived radioactivity are normal. In the panel for MSA-P, the arrow indicates a patient with definite MSA-P who had putamen dopamine and myocardial norepinephrine depletion without Lewy bodies. MSA-C = cerebellar multiple system atrophy; MSA-P = parkinsonian multiple system atrophy; OH = orthostatic hypotension; PAF = pure autonomic failure; PD = Parkinson disease.
With few exceptions, patients with PD + OH had low 18F-dopamine–derived radioactivity and low values for the PUT:OCC ratio of 18F-DOPA–derived radioactivity; patients with PAF had low 18F-dopamine–derived radioactivity and normal PUT:OCC ratios; patients with MSA-P had normal 18F-dopamine–derived radioactivity and low PUT:OCC ratios; and patients with MSA-C had normal 18F-dopamine–derived radioactivity and normal PUT:OCC ratios (figure 2).
Survival in patient groups.
Kaplan-Meier curves (figure 3) and log-rank testing revealed clear differences in survival rates among the PD + OH, PD no OH, MSA-C, MSA-P, and PAF groups (log-rank test, p < 0.0001, df = 4).
Figure 3. Survival plots and numbers of at-risk patients in α-synucleinopathies.

Displayed are survival probabilities expressed as a function of years since symptom onset (A and B) or initial evaluation at the NIH Clinical Center (C and D). (A and C) Kaplan-Meier plots and numbers of at-risk living patients. (B and D) Survival plots after adjustment for age and sex. “Censored” at-risk patients were alive as of the indicated years. MSA-C = cerebellar multiple system atrophy; MSA-P = parkinsonian multiple system atrophy; OH = orthostatic hypotension; PAF = pure autonomic failure; PD = Parkinson disease.
Cox regression analysis, using age and sex as covariates, disclosed that the hazard for mortality from the time of symptom onset was 5 times worse in MSA-P than in PD + OH, 13.6 times worse than in PD no OH, and 9.9 times worse than in PAF (table 1). Survival in patients with PD + OH was shorter than in patients with PD no OH (HR = 2.4, p = 0.0055). PD + OH did not involve greater death risk than did PAF based on the time from relevant symptom onset (HR = 1.8, p = 0.2082) but did based on the time from initial evaluation at the NIH (HR = 2.7, p = 0.02; table e-3). Death risk in PAF did not differ from that in PD no OH.
Table 1.
Cox regression analysis for time from symptom onset to death (n = 176)
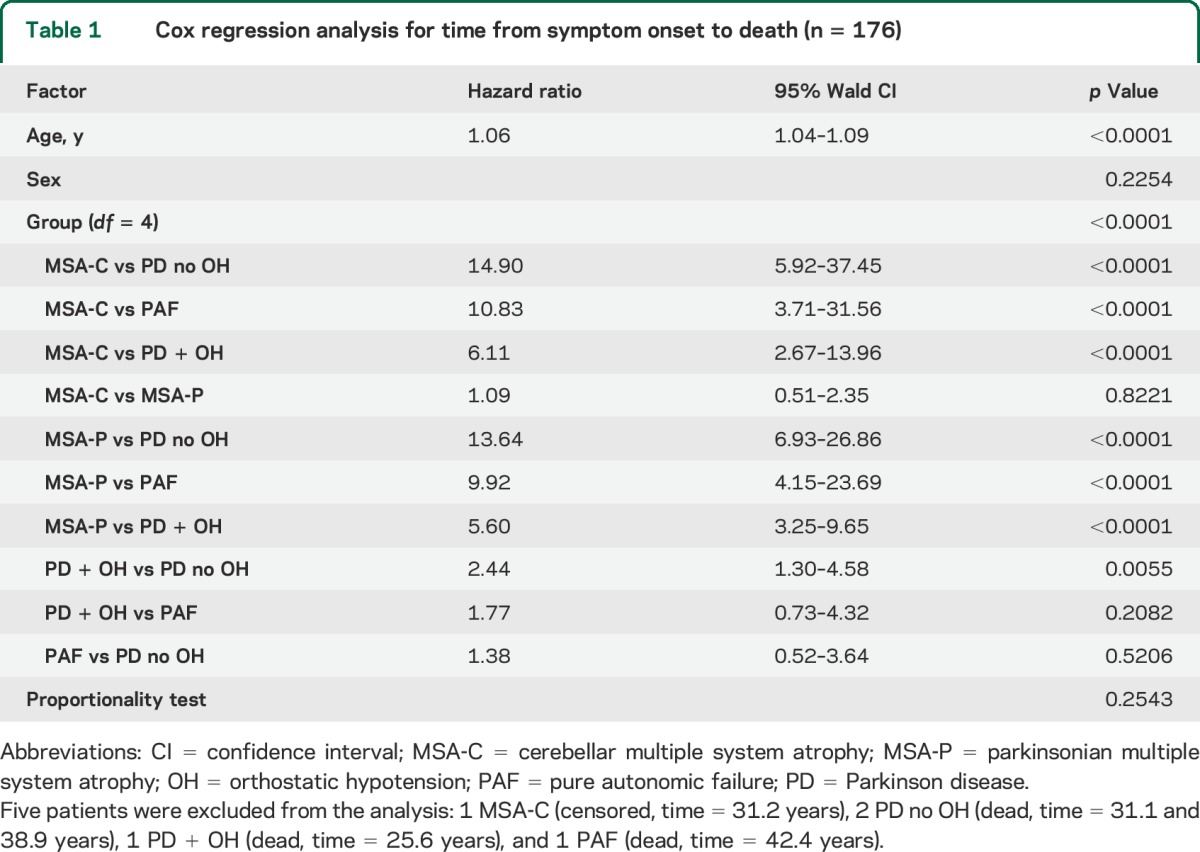
As indicated in figure 3B, after adjustment for age and sex, median survival in MSA-P from the time of symptom onset was about 8 years, much less than in PD + OH (about 15 years) and both PAF and PD no OH.
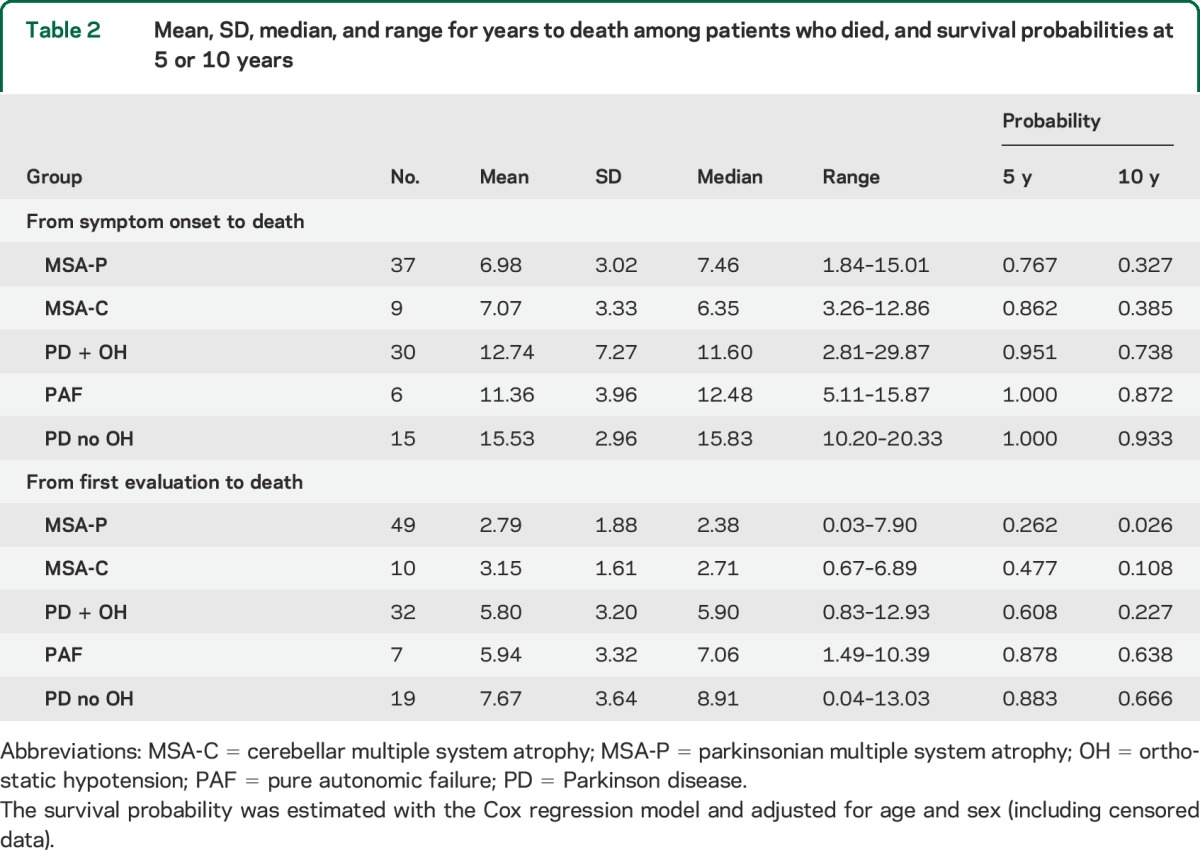
Among patients who died, the MSA-P and MSA-C groups stood out in terms of both short survival time in years from symptom onset and in years from initial evaluation to death (table 2).
Table 2.
Mean, SD, median, and range for years to death among patients who died, and survival probabilities at 5 or 10 years
When the pattern of cardiac and brain catecholaminergic neuroimaging findings alone was used to construct 4 groups (figure 1), the groups differed in survival (figure 4B, tables e-3–e-6). The hazard of mortality in the group with the C pattern (normal heart, abnormal putamen, generally corresponding to MSA-P) was 45 times that in the group with the D pattern (abnormal heart, normal putamen, generally corresponding to PAF) and 4.5 times that in the group with the A pattern (abnormal heart, abnormal putamen, generally corresponding to PD + OH) and did not differ from that in the group with the B pattern (normal heart, normal putamen, generally corresponding to MSA-C). Survival probability in the B pattern group did not differ from that in the C pattern group.
Figure 4. Survival plots and numbers of at-risk patients based on patterns of catecholaminergic abnormalities.

(A) Kaplan-Meier plots and numbers of at-risk living patients. (B) Survival plots after adjustment for age and sex.
DISCUSSION
This prospective cohort study compared prognoses among different forms of synucleinopathy. Survival in MSA (both parkinsonian and cerebellar types) was compared with that in PD + OH, PD no OH, and PAF. No previously published studies have made these comparisons. The relatively large numbers of patients in the MSA and PAF groups, extensive clinical laboratory testing in each patient that ensured a more accurate diagnosis than by clinical criteria alone, and 97.6% follow-up percentage were unique and strong features. The analysis also included differential survivals based on neuroimaging data alone.
The results show clearly that the long-term outlook in these diseases depends on the particular disease. Median survival is especially poor in MSA-P (7.5 years) and MSA-C (6.4 years). New findings in the present study are that age- and sex-adjusted survival in PD + OH is also decreased relative to PD no OH but is not as reduced as in either type of MSA. Whether survival was expressed as a function of the time from onset of motor symptoms or of the time from diagnostic confirmation at the NIH, the results were essentially the same: worse prognosis in MSA of both subtypes than in PD + OH and worse prognosis in PD + OH than in PD no OH.
Among the 4 diseases typically involving neurogenic OH (PD + OH, MSA-C, MSA-P, and PAF), results of catecholaminergic neuroimaging in the brain by 18F-DOPA PET scanning and the heart by 18F-dopamine PET scanning also predicted survival. This was mainly because the patients with neuroimaging evidence of intact cardiac sympathetic innervation had MSA. Between the MSA subgroup with intact putamen dopaminergic innervation (generally corresponding to MSA-C) and the subgroup with decreased putamen dopaminergic innervation (generally corresponding to MSA-P), there was no difference in survival.
Since PAF involves OH without signs of central neurodegeneration, and PD no OH involves parkinsonism without OH, shortened survival in MSA and PD + OH seems to reflect combined effects of central neurodegeneration and neurogenic OH. However, worse survival in MSA than in PD + OH, which agrees with a study of neurogenic OH groups,11 suggests that additional factors besides central neurodegeneration and OH determine the poor survival seen in MSA. Clinical findings that patients with MSA had higher frequencies of slurred speech and urinary retention than did patients with PD + OH would be consistent with reported increased risks of respiratory and infectious causes of death in MSA.11
The present finding of poor survival in patients diagnosed with MSA is confirmatory and not new,12 and the finding of approximately equally poor survivals in MSA-P and MSA-C confirms that in a recent report.13 Other studies have noted poorer survival in MSA than in PD14 but without stratification of PD in terms of OH or other nonmotor manifestations.
Recent studies have noted different survival rates in PD according to clinical subtypes, with the worst survival associated with subtypes involving nonmotor manifestations such as cardiovascular autonomic dysfunction,15,16 which would include OH. Our results agree with the notion that distinct PD subgroups have different clinical manifestations and survivals. Nonmotor manifestations of PD, such as OH, supine hypertension, sympathetic noradrenergic denervation, anosmia, baroreflex failure, cognitive dysfunction, and REM behavior disorder tend to cluster together. The present findings fit with the view that this constellation is associated with poor survival.17 Survival in PD no OH seems to have improved substantially since the classic report by Hoehn and Yahr18 in 1967.
Study limitations.
Too few patients were autopsied to confirm the diagnosis of the specific form of synucleinopathy; however, in all of the 8 cases who were autopsied (2 PD + OH, 6 MSA) and in 1 case of PD + OH due to triplication of the gene encoding α-synuclein, the diagnostic assignment based on clinical and laboratory investigations during life was found to be correct.19
We did not evaluate other signs of autonomic failure besides OH as potential risk factors. Because of clustering of OH with other autonomic and nonmotor aspects of PD, the present results do not enable assessment of relative contributions of OH vs other nonmotor aspects to survival. This is a matter for future research.
The numbers of patients in the PD + OH, PD no OH, MSA-P, MSA-C, and PAF groups should not be taken as representative of the prevalence of these diseases in the general population, because of the pattern of referrals and acceptances for clinical evaluation at the NIH.
Implications.
There is no effective treatment to retard the progressive neurodegeneration that occurs in the synucleinopathies. Nevertheless, the present results highlight the importance of distinguishing among synucleinopathies because of the very different prognoses depending on the particular disease. In PD no OH and in PAF, the outlook is relatively benign, whereas in MSA-P and MSA-C the outlook is bleak.
Experimental treatments vary in potential risks. This concern applies especially to immunomodulatory, stem cell, and gene therapy approaches. Because of the poor prognoses in MSA-P and MSA-C compared with other forms of synucleinopathy, these patient groups seem most suitable for relatively high-risk experimental therapeutic trials.20–23
PD is a prevalent neurodegenerative disease. Since OH occurs fairly often in PD,4 and based on the present findings is associated with substantially decreased survival, PD + OH may pose a disproportionate public health burden.
Supplementary Material
GLOSSARY
- HR
hazard ratio
- MSA
multiple system atrophy
- MSA-C
cerebellar multiple system atrophy
- MSA-P
parkinsonian multiple system atrophy
- OH
orthostatic hypotension
- PAF
pure autonomic failure
- PD
Parkinson disease
- PUT:OCC
putamen to occipital cortex ratio
- UPDRS
Unified Parkinson's Disease Rating Scale
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
David S. Goldstein: study conception, data analysis, manuscript writing. Courtney Holmes: data collection, data analysis, manuscript editing. Yehonatan Sharabi: data analysis, manuscript editing. Tianxia Wu: data analysis, statistics, manuscript editing.
STUDY FUNDING
The funding organization for this study was the Division of Intramural Research (DIR), National Institute of Neurological Disorders and Stroke (NINDS), NIH. The DIR, NINDS, NIH, supported the research reported here.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jellinger KA. Neuropathological spectrum of synucleinopathies. Mov Disord 2003;18(suppl 6):S2–S12. [DOI] [PubMed] [Google Scholar]
- 2.Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 1998;249:180–182. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein DS. Dysautonomia in Parkinson's disease: neurocardiological abnormalities. Lancet Neurol 2003;2:669–676. [DOI] [PubMed] [Google Scholar]
- 4.Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RM. Prevalence of orthostatic hypotension in Parkinson's disease: a systematic review and meta-analysis. Parkinsonism Relat Disord 2011;17:724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajput AH, Rozdilsky B, Rajput A. Accuracy of clinical diagnosis in parkinsonism: a prospective study. Can J Neurol Sci 1991;18:275–278. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein DS, Holmes C, Bentho O, et al. Biomarkers to detect central dopamine deficiency and distinguish Parkinson disease from multiple system atrophy. Parkinsonism Relat Disord 2008;14:600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaufmann H. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure and multiple system atrophy. Clin Auton Res 1996;6:125–126. [DOI] [PubMed] [Google Scholar]
- 8.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein DS, Sharabi Y. Neurogenic orthostatic hypotension: a pathophysiological approach. Circulation 2009;119:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology 2003;60:1327–1332. [DOI] [PubMed] [Google Scholar]
- 11.Maule S, Milazzo V, Maule MM, Di Stefano C, Milan A, Veglio F. Mortality and prognosis in patients with neurogenic orthostatic hypotension. Funct Neurol 2012;27:101–106. [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Shlomo Y, Wenning GK, Tison F, Quinn NP. Survival of patients with pathologically proven multiple system atrophy: a meta-analysis. Neurology 1997;48:384–393. [DOI] [PubMed] [Google Scholar]
- 13.Roncevic D, Palma JA, Martinez J, Goulding N, Norcliffe-Kaufmann L, Kaufmann H. Cerebellar and parkinsonian phenotypes in multiple system atrophy: similarities, differences and survival. J Neural Transm 2014;121:507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wenning GK, Ben Shlomo Y, Magalhaes M, Daniel SE, Quinn NP. Clinical features and natural history of multiple system atrophy: an analysis of 100 cases. Brain 1994;117:835–845. [DOI] [PubMed] [Google Scholar]
- 15.de Lau LM, Verbaan D, van Rooden SM, Marinus J, van Hilten JJ. Relation of clinical subtypes in Parkinson's disease with survival. Mov Disord 2014;29:150–151. [DOI] [PubMed] [Google Scholar]
- 16.Fanciulli A, Strano S, Colosimo C, Caltagirone C, Spalletta G, Pontieri FE. The potential prognostic role of cardiovascular autonomic failure in alpha-synucleinopathies. Eur J Neurol 2013;20:231–235. [DOI] [PubMed] [Google Scholar]
- 17.Stubendorff K, Aarsland D, Minthon L, Londos E. The impact of autonomic dysfunction on survival in patients with dementia with Lewy bodies and Parkinson's disease with dementia. PLoS One 2012;7:e45451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442. [DOI] [PubMed] [Google Scholar]
- 19.Cook GA, Sullivan P, Holmes C, Goldstein DS. Cardiac sympathetic denervation without Lewy bodies in a case of multiple system atrophy. Parkinsonism Relat Disord 2014;20:926–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunwoo MK, Yun HJ, Song SK, et al. Mesenchymal stem cells can modulate longitudinal changes in cortical thickness and its related cognitive decline in patients with multiple system atrophy. Front Aging Neurosci 2014;6:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12:264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novak P, Williams A, Ravin P, Zurkiya O, Abduljalil A, Novak V. Treatment of multiple system atrophy using intravenous immunoglobulin. BMC Neurol 2012;12:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.May S, Gilman S, Sowell BB, et al. Potential outcome measures and trial design issues for multiple system atrophy. Mov Disord 2007;22:2371–2377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.