

Abstract
Mycobacterium bovis, the agent of bovine tuberculosis, causes an estimated $3 billion annual losses to global agriculture due, in part, to the limitations of current diagnostics. Development of next-generation diagnostics requires a greater understanding of the interaction between the pathogen and the bovine host. Therefore, to explore the early response of the alveolar macrophage to infection, we report the first application of RNA-sequencing to define, in exquisite detail, the transcriptomes of M. bovis-infected and non-infected alveolar macrophages from ten calves at 2, 6, 24 and 48 hours post-infection. Differentially expressed sense genes were detected at these time points that revealed enrichment of innate immune signalling functions, and transcriptional suppression of host defence mechanisms (e.g., lysosome maturation). We also detected differentially expressed natural antisense transcripts, which may play a role in subverting innate immune mechanisms following infection. Furthermore, we report differential expression of novel bovine genes, some of which have immune-related functions based on orthology with human proteins. This is the first in-depth transcriptomics investigation of the alveolar macrophage response to the early stages of M. bovis infection and reveals complex patterns of gene expression and regulation that underlie the immunomodulatory mechanisms used by M. bovis to evade host defence mechanisms.
Bovine tuberculosis is a chronic infectious disease of domestic livestock and wildlife caused by infection with Mycobacterium bovis, a pathogenic mycobacterial species belonging to the Mycobacterium tuberculosis complex (MTBC)1. As a zoonotic agent, M. bovis infection also has serious implications for human health2. Previous studies have shown that the aetiology and host immune response to M. bovis infection in cattle is nearly identical to M. tuberculosis infection in humans3,4,5.
M. bovis is primarily transmitted via inhalation of aerosolized bacteria, with the primary site of infection being the lung6. Following inhalation, the pathogen is phagocytosed by host alveolar macrophages, which serve as key effector innate immune cells and usually can kill intracellular bacilli or contain infection via the activity of inflammatory cytokines7. M. bovis, however, can persist and replicate within alveolar macrophages via diverse mechanisms that subvert or exploit host immune responses8. These mechanisms include prevention of macrophage phagosome-lysosome fusion, inhibition of apoptosis, suppression of antigen presentation and signalling mechanisms within the macrophage, and induction of necrosis, culminating in immunopathology and ultimately shedding of the pathogen from the host9,10,11,12.
We hypothesised that mechanisms used by pathogenic mycobacteria to overcome innate immunity and establish infection would be revealed through analysis of the gene expression changes in the macrophage response to infection. Previous transcriptomics studies of the bovine macrophage response to M. bovis infection, performed by us and others13,14,15, have yielded important insights into the molecular mechanisms and cellular pathways that govern mycobacteria-macrophage interplay. Here, we have extended this earlier work by performing the first in-depth analysis of the primary innate immune cell, the alveolar macrophage, response to infection with M. bovis using high-throughput RNA-sequencing (RNA-seq). We have analysed gene expression changes in purified bovine alveolar macrophages from ten age-matched unrelated Holstein-Friesian male calves infected with M. bovis across a 48 h time course.
The work described here provides important new information regarding mammalian host responses to mycobacterial pathogens for the following reasons. Firstly, we have used M. bovis to infect bovine alveolar macrophages–the primary host cell type that encounters M. bovis following inhalation16,17; earlier studies have generally used blood-derived macrophages13,14,15,18. Secondly, culture of M. bovis was performed in the absence of the detergent Tween 80 as previous research has shown that Tween 80 alters the cell wall composition and morphology of mycobacteria which may affect the interaction of mycobacteria with macrophages during in vitro infection studies19,20,21,22. Thirdly, to the best of our knowledge, all previous investigations of the alveolar macrophage transcriptome in infection with tubercle bacilli have involved the analysis of microarray data; none have used RNA-seq, which offers unprecedented opportunities for global gene expression analysis, including unbiased whole-transcriptome gene expression profiling, cataloguing of sense and antisense transcription, and discovery of novel RNA transcripts23,24.
Materials and Methods
The laboratory methods have previously been described by us25 and therefore a summary is provided below. Detailed materials and methods are provided in Supplementary Methods and the complete bioinformatics pipeline is accessible online26.
Ethics Statement
All animal procedures were performed according to the provisions of the Cruelty to Animals Act of 1876 and ethical approval was obtained from the University College Dublin Animal Ethics Committee (protocol number AREC-13-14-Gordon).
Animal work, lung lavages and alveolar macrophage purification
Fourteen unrelated, age-matched Holstein-Friesian male calves were selected from a tuberculosis-free herd and total lung cells were harvested by pulmonary lung lavage via tracheal infusion of calcium- and magnesium-free Hank’s Balanced Salt Solution (HBSS; Invitrogen™, Life Technologies Corp., Paisley, UK). All animals tested negative after microbial screening. The HBSS-cell suspension was washed by centrifugation, resuspended in foetal bovine serum (FBS) with 10% dimethyl sulfoxide (Sigma-Aldrich Ltd.), aliquoted and stored at −140 °C using Mr. Frosty® Cryo 1 °C Freezing Containers (Nalgene®, Thermo Fisher Scientific, Waltham, MA, USA).
Approximately 1.5 × 108 total lung cells from each animal were thawed in a 37 °C water bath for 1 min and transferred into R10+ media (RPMI 1640 medium [Invitrogen™] supplemented with 10% FBS, 2.5 μg/ml amphotericin B, 2 mM L-glutamine, 100 μg/ml ampicillin and 25 μg/ml gentamycin [all from Sigma-Aldrich Ltd.]). Cells were centrifuged, resuspended in R10+ media, placed in a vented culture flask (CELLSTAR®, Greiner Bio-One Ltd., Stonehouse, UK) and incubated for 24 h at 37 °C, 5% CO2. After incubation, media was removed and adherent cells were dissociated by adding 1× non-enzymatic cell dissociation solution (Sigma-Aldrich Ltd.). Cells were centrifuged, washed, seeded at 5 × 105 viable cells/well in culture plates (Sarstedt Ltd.) and incubated for 24 h at 37 °C, 5% CO2. The purity of the seeded macrophages was confirmed by flow cytometry with anti-CD14 antibody.
Culture of M. bovis
M. bovis AF2122/97 strain was cultured to late logarithmic phase in Middlebrook 7H9 media (Difco™, Becton, Dickinson Ltd., Oxford, UK) enriched with 1× Middlebrook albumin-dextrose-catalase (ADC; Difco™) and 10 mM final concentration of sodium pyruvate (Sigma-Aldrich Ltd.) and then stored at −80 °C. Prior to infection, 1 ml M. bovis culture stock was thawed and cultured until mid-logarithmic phase. Further culturing was performed statically in vented Corning™ Erlenmeyer flasks (Thermo Fisher Scientific Inc.) until late-logarithmic phase. On the day of infection, the M. bovis culture was centrifuged, resuspended in R10− media (R10+ media without antibiotics) and sonicated for 1 min. The cell number was calculated from OD600nm and adjusted to 5 × 106 bacterial cells/ml using R10− media.
M. bovis-infection of alveolar macrophages and macrophage RNA extraction
For infections, the media from all wells containing alveolar macrophages was removed and replaced with 1 ml R10− media containing M. bovis, yielding a multiplicity of infection of 10 bacilli per macrophage. Parallel non-infected control alveolar macrophage received 1 ml R10− media only. Alveolar macrophages were then incubated at 37 °C, 5% CO2 for 2, 6, 24 and 48 h. After 2 h post-infection (hpi), the media from all infection experiments was replaced with fresh R10− media and culture plates were reincubated at 37 °C, 5% CO2 until cells were required for harvesting. Alveolar macrophages were lysed by adding 250 μl/well RLT buffer supplemented with 1% β-mercaptoethanol (Qiagen Ltd., Crawley, UK) and stored at −80 °C until required for RNA extraction.
All RNA extractions were performed using an RNeasy® Plus Mini kit (Qiagen Ltd.) according to the manufacturer’s instructions. RNA quantity and quality was assessed and all samples displayed a 260/280 ratio greater than 2.0 and RNA integrity numbers greater than 8.5. RNA samples were stored at −80 °C.
Strand-specific RNA-seq library preparation and sequencing
The samples used for RNA-seq library preparation comprised M. bovis-infected and non-infected samples from each post-infection time points across 10 animals (except for one animal that did not yield sufficient macrophages for the 48 hpi time point).
Approximately 200 ng of total RNA from each sample was used to prepare individually barcoded strand-specific RNA-seq libraries. Two rounds of poly(A)+ RNA purification were performed for all RNA samples using the Dynabeads® mRNA DIRECT™ Micro Kit (Invitrogen™) according to the manufacturer’s instructions. The poly(A) + RNA was then used to generate strand-specific RNA-seq libraries using the ScriptSeq™ v2 RNA-Seq Library Preparation Kit, the ScriptSeq™ Index PCR Primers (Sets 1 to 4) and the FailSafe™ PCR enzyme system (all from Epicentre®, Illumina® Inc., Madison, WI, USA) according to the manufacturer’s instructions. RNA-seq libraries were purified using the Agencourt® AMPure® XP system (Beckman Coulter Genomics, Danvers, MA, USA) according to the manufacturer’s instructions for double size selection (0.75× followed by 1.0 × ratio). Individually barcoded RNA-seq libraries were pooled in equimolar quantities and assessed for quantity and quality.
Cluster generation and paired-end 2 × 90 nucleotide read sequencing of the pooled RNA-seq libraries were performed by BGI (BGI–Hong Kong, Hong Kong, China) using an Illumina® HiSeq™ 2000 sequencer. All RNA-seq data are accessible from the NCBI GEO database27 (accession number GSE62506).
Bioinformatics and differential expression analyses of RNA-seq data
Computational analyses consisted of an initial quality check of the raw reads data files using the FastQC software28. Subsequently, a Perl script was used to deconvolute barcoded reads into individual libraries, filter out adapter sequence reads, and remove poor quality reads. Paired-end reads from individual libraries were then aligned to the B. taurus reference genome (UMD3.1.71) using the STAR aligner software29.
For each library, uniquely aligned paired-end reads were used to obtain raw counts for all bovine genes (B. taurus UMD3.1.71 Ensembl genome annotation30) based on sense strand data with the featureCounts software31. Subsequently, the gene counts were used for differential expression analysis with the edgeR package32 and the following steps: lowly expressed gene filtering, normalisation using the trimmed mean of M-values method, estimation of dispersion using the Cox-Reid method, evaluation of differential gene expression between M. bovis-infected versus non-infected samples at each time point (i.e., paired-sample statistical model) using a negative binomial generalised linear model, and correction for multiple testing using the Benjamini-Hochberg method33.
For antisense gene analyses, the uniquely aligned paired-end reads that were unassigned to any sense genes were used to generate raw counts for antisense genes using the featureCounts software. For this, the B. taurus UMD3.1.71 genome annotation was configured to include a promoter and terminator region34 and to exclude proximal genes. In addition, to remove antisense gene artefacts35, we modified the method of Perocchi and colleagues36. This involved generation of additional raw counts with featureCounts, a low gene expression filtering step and the computation of count ratios as shown below.
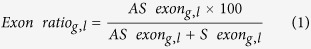 |
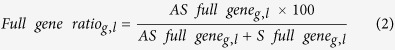 |
AS = antisense count; S = sense count; g = gene; l = library.
A greater-sided pairwise Wilcoxon signed-rank test was then used to assess significant differences between the full gene ratio and the exon ratio for each gene. Antisense genes found to be statistically significant for this ratio filtering step were then used for differential antisense gene expression analysis with the edgeR package as described for the sense genes.
For novel genes analyses, aligned reads were processed with the Cufflinks software37 using the reference annotation-based transcript assembly method38 and the B. taurus reference genome annotation (UMD3.1.71) to generate a de novo transcripts assembly. The reciprocal best hit (RBH) method39 was then used to annotate putative novel genes by orthology to Homo sapiens reference protein sequences (H. sapiens GRCh37.73) using standalone BLAST40 and Perl scripts. Subsequently, the uniquely aligned paired-end reads not assigned to any sense or antisense gene were used to compute raw counts for novel genes using the featureCounts software and the de novo B. taurus genome annotation. Then raw counts for each novel gene were used for differential expression analysis with the edgeR package as described for the sense and antisense genes.
Systems analyses
Using the biomaRt package41, each B. taurus sense gene was annotated with its H. sapiens ortholog from Ensembl (GRCh37.71) to facilitate integration with the differentially expressed (DE) novel genes data set and to provide consistent data input across the systems biology analysis tools. The Ingenuity® Systems Pathway Analysis (IPA) software package (Qiagen Corp., Redwood City, CA, USA) was used to identify over-represented canonical pathways based on the Ingenuity® Knowledge Base. To further focus on the canonical pathways of most relevance to our study, the Pathway–Guide software package (Advaita Corp., Plymouth, MI, USA) was also used to perform signalling pathway impact analysis42; this method uses biological interaction data from the KEGG43 and Reactome44 databases. Finally, the Sigora package45 was used to perform signature over-representation analysis based on the KEGG and Reactome databases.
cDNA synthesis and qPCR analysis
For technical validation, cDNA was prepared from the same RNA used for the RNA-seq library preparations; for parallel biological validation, cDNA was prepared from RNA extracted from macrophage samples purified from four additional calves. The High Capacity cDNA Reverse Transcription Kit (Applied Biosystems®, Life Technologies Corp., Warrington, UK) was used to prepare cDNA from 60 ng total RNA according to the manufacturer’s instructions. Quantitative real-time PCR (qPCR) was performed using Fast SYBR® Green Master Mix (Applied Biosystems®) on a 7500 Fast Real-Time PCR System (Applied Biosystems®) according to the manufacturer’s instructions. Reactions contained 3 μl diluted cDNA samples (or appropriate controls), 10 μl of SYBR mix and 300 nM final concentration of each primer (Supplementary Data S1, worksheet 1). PCR cycling conditions comprised a 50 °C step for 2 min, a 95 °C step for 20 s, followed by 40 cycles at 95 °C for 3 s and 60 °C for 30 s.
The qbase+ software tool (Biogazelle NV, Zwijnaarde, Belgium)46 was used for qPCR normalisation (the GeNorm algorithm47 identified PPIA and H3F3A as suitable reference genes). Gene expression fold-changes were computed for M. bovis-infected versus non-infected samples at each time point using calibrated normalised relative quantities. Normal distribution of fold-change values was checked with the Shapiro-Wilk test in the SPSS statistical package (IBM Corp., Armonk, NY, USA). Two-tailed paired sample t-tests and pairwise Wilcoxon signed-rank tests were used to assess statistically significant gene expression fold-changes for normally and non-normally distributed data, respectively. The Pearson correlation (r) between RT-qPCR and RNA-seq gene expression fold-change was estimated.
Results and Discussion
RNA-seq summary statistics
In total, 78 individually barcoded strand-specific RNA-seq libraries (i.e., M. bovis-infected and non-infected macrophages from ten animals at 2, 6 and 24 hpi and nine animals at 48 hpi) were sequenced on an Illumina® HiSeq™ 2000 apparatus, generating a total of 2.2 billion paired-end (2 × 90 nucleotides) reads. Deconvolution and filtering of sequence reads yielded a mean of 22.7 million paired-end reads per individual library (Supplementary Data S2). These data satisfy previously defined criteria for RNA-seq experiments in terms of the number of independent biological replicates per treatment and the sequencing depth48,49. Alignment of the filtered paired-end reads to the B. taurus reference genome yielded mean values per library of 20.4 million reads (89.72%) mapping to unique locations. Gene count summarisation revealed mean values per library of 14.9 million reads (73.32%) assigned to Ensembl gene IDs based on sense strand sequence information, with 0.9 million reads (4.34%) assigned to gene IDs based on antisense strand sequence information and 0.5 million reads (2.27%) assigned to gene IDs based on novel gene annotation analysis. These three sets of sequence reads were used to separately derive gene expression values for sense gene data, antisense gene data and novel gene data, respectively.
Differential gene expression analyses
A stringent gene expression filtering criterion was applied to the sense strand expression data to remove lowly expressed genes and thereby reduce Type I error50. This yielded 11,928 sense genes (48.46% of total B. taurus reference genes) that were suitable for differential expression analysis. The data from all 11,928 filtered sense genes were used for multidimensional scaling analysis (Supplementary Figure S1), which revealed that biological samples could be differentiated according to treatment (M. bovis-infected or non-infected samples) on dimension 1 and 2 starting at 6 hpi, with further discrimination at later time points.
Statistical analysis identified 95 (48 up-, 47 downregulated), 1,290 (696 up-, 594 downregulated), 5,515 (2,674 up-, 2,841 downregulated), and 7,321 (3,592 up-, 3,729 downregulated) significantly DE genes (FDR-adjusted P ≤ 0.05) in the M. bovis-infected macrophages relative to the control non-infected macrophages at 2, 6, 24 and 48 hpi, respectively (Fig. 1a and Supplementary Data S3, worksheet 1). Among the top significantly upregulated sense genes were HCAR3, TNFAIP6, TNFAIP3, MAFF and NFKBIZ; while the top significantly downregulated sense genes were DUSP7, RNF169, EZH1, IL6R and CDC42ER3 (ranked by the average of FDR-adjusted P value across all time points). This increase in the number of DE genes over the alveolar macrophage infection time course has been reported in other studies using bovine MDM13 and human alveolar macrophages51; these studies also observed higher fold-changes in gene expression for upregulated compared to downregulated genes.
Figure 1. Number of significant DE genes at each time point post-infection.

The numbers of upregulated and downregulated genes, in the M. bovis-infected alveolar macrophages relative to the control non-infected cells at each time point, are shown for (a) sense, (b) antisense and (c) novel gene analyses (adjusted P-value ≤ 0.05).
The use of a strand-specific RNA-seq library preparation protocol also facilitated detection and quantification of transcripts that were located on the antisense DNA strand corresponding to an annotated sense gene. Natural antisense transcripts (NATs) have previously been reported in mammals with a wide range of functions, including transcriptional and post-transcriptional gene expression regulation, splicing event control and the regulation of allele-specific transcription52,53,54. Differential expression analyses of putative NATs involved both low expression and antisense ratio filtering criteria, which yielded 565 NATs suitable for downstream analyses. Statistical analysis identified 1 (1 upregulated), 58 (33 up-, 25 downregulated), 176 (85 up-, 91 downregulated), and 281 (139 up-, 142 downregulated) significantly DE NATs (FDR-adjusted P ≤ 0.05) in the M. bovis-infected relative to the control non-infected alveolar macrophages at 2, 6, 24 and 48 hpi, respectively (Fig. 1b and Supplementary Data S3, worksheet 2).
Finally, RNA-seq was used to identify putative bovine novel genes and refine the incomplete B. taurus genome annotation. Differential expression analyses of novel genes—transcription occurring in intergenic location of the reference gene annotation as identified via de novo transcriptome reconstruction38—also involved a low expression filtering step and yielded 3,088 putative novel genes suitable for analysis. Statistical analysis identified 3 (1 up-, 2 downregulated), 179 (90 up-, 89 downregulated), 1,092 (590 up-, 502 downregulated), and 1,538 (788 up-, 750 downregulated) significantly DE putative novel genes (FDR-adjusted P ≤ 0.05) in the M. bovis-infected relative to the control non-infected alveolar macrophages at 2, 6, 24 and 48 hpi, respectively (Fig. 1c and Supplementary Data S3, worksheet 3). It is important to note that further analysis described below led to the removal of a large number of artefactual putative novel genes.
Correlation of sense and antisense gene expression
The DE NATs were compared to the DE sense genes according to direction of expression for each time point post-infection. In total, 1, 20, 114, and 195 genes showed the same direction of expression (i.e., up- or downregulated) on both the sense and the antisense strands at the 2, 6, 24 and 48 hpi time points, respectively. The ACPP, LOC782264, GAS1, MBP, MSC and RERE genes were among the top-ranking genes (based on NATs analysis FDR-adjusted P values), showing the same direction of expression for both sense genes and NATs at the 6, 24 and 48 hpi (Supplementary Figure S2). Furthermore, these genes have been shown to play a role in host defence mechanisms including immune development, resistance/susceptibility to infection and apoptosis55,56,57,58,59,60. In addition, we detected 3 and 10 genes at the 24 and 48 hpi time points, respectively, that exhibited opposing directions of expression on the antisense and sense strands (i.e., the NAT is up- and the sense gene is downregulated, or vice versa). These genes included ACAD10, RHOBTB3 and SLC30A4, which have been shown to mediate the response to infection including endocytosis, zinc homeostasis and bactericidal mechanisms61,62,63.
DE sense genes may be subject to regulation mediated by putative NATs, which carry out a large range of functions by acting in cis and/or trans through various transcriptional and post-transcriptional regulatory mechanisms52,53. Firstly, NATs can interact with DNA to induce chromatin remodelling via DNA methylation or histone-modifying enzyme recruitment, leading to (de)repression of sense transcription64; consequently, we hypothesise MSC gene expression is mediated via such a mechanism through its associated promoter-located NAT. Secondly, the interaction of a NAT with sense RNA can lead to the formation of an RNA duplex, which can result in sense RNA alternative splicing, change in sense RNA localisation, modification of sense RNA stability and formation of small interfering RNA65,66; therefore, the downregulation of the NAT for MBP may reduce the stability and abundance of its sense RNA. Thirdly, the process of NAT transcription can provoke transcriptional collision, where convergent transcription occurs on opposite strands resulting in collision of two RNA polymerases and aborted sense RNA transcription67; this suggests a regulatory mechanism whereby the NAT of SLC30A4 represses the sense gene. Sense gene expression regulation, modulated by NATs, may constitute host-mediated control of the immune response to fine-tune and tightly regulate the levels of proinflammatory cytokines and bactericidal molecules induced during infection65,66. Alternatively, such NAT-mediated regulation may comprise an immune evasion mechanism evoked by M. bovis to enable its survival within the macrophage. Finally, due to debate in the literature about the existence of functional NATs in vertebrates68,69,70, we implemented a stringent NAT detection filtering step to exclude artefactual NATs.
Annotation of novel genes
The de novo transcriptome analysis facilitated detection of novel bovine genes, which were subsequently examined for differential expression between M. bovis-infected and control non-infected alveolar macrophages. The RBH method was used to annotate putative novel genes based on H. sapiens protein reference sequences39. Of the 3,088 novel genes that passed the low expression filtering step, it was possible to annotate 103 novel genes by comparison to pre-existing H. sapiens orthologs (Supplementary Data S3, worksheet 3). This suggested that a large number of the putative novel genes we detected may be artefacts; therefore, we focused subsequently only on bovine novel genes with H. sapiens orthologs.
Irrespective of their differential expression, these annotated novel bovine genes are important for refining the B. taurus genome annotation. In the context of the present study, several annotated novel bovine genes may be of particular importance; for example, LILRA5 (upregulated at 24 and 48 hpi) encodes a leukocyte immunoglobulin-like receptor that induces production of IL-10 and other proinflammatory cytokines in macrophages71. In addition, KLF13 (downregulated at 6, 24 and 48 hpi) encodes a transcription factor that negatively regulates macrophage alternative activation (M2 polarisation); alternatively activated macrophages have diminished bactericidal activity but enhanced phagocytic activity72. Also, the KLF2 gene (upregulated at 48 hpi) encodes a negative regulator of proinflammatory cytokines and it is significant that macrophage migration and adhesion is reduced by increased expression of KLF273. In summary, LILRA5, KLF13 and KLF2 encode proteins with important immunoregulatory functions for macrophages upon infection (e.g., regulation of inflammation to avoid potential cell damage), but also enhance survival of intracellular bacteria (e.g., reduction of bactericidal activity and proinflammatory cytokine production).
RT-qPCR validation
We performed technical validation of the RNA-seq results by quantifying a panel of eight immune genes via RT-qPCR25 using the same RNA samples that were used for the RNA-seq analysis. Of the panel of genes analysed using RT-qPCR, six were significantly upregulated (CCL4, IL10, IL1B, IL6, TLR2 and TNF) and two were significantly downregulated (FOS and PIK3IP1) at one or more post-infection time points. The comparison of RT-qPCR and RNA-seq results yielded a concordance (in terms of both direction of fold-change and statistical significance) of 71.9% across all eight genes. Comparison of the mean log2 fold-change in gene expression between the RT-qPCR and RNA-seq results revealed an overall (across all genes) Pearson correlation (r) value of 0.98 (P-value ≤ 0.001); while six genes (CCL4, FOS, IL1B, IL6, PIK3IP1 and TNF) had r-values ≥ 0.97 (P-value ≤ 0.05) and two genes (IL10 and TLR2) had r-values between 0.92-0.94 (P-value ≤ 0.1). The RT-qPCR results for technical validation of the RNA-seq data are provided in Supplementary Data S1, worksheet 2.
RT-qPCR biological validation of the RNA-seq results was also performed using the same panel of eight genes. For this, total RNA was extracted and purified from M. bovis-infected and non-infected control alveolar macrophages obtained from four additional animals. A concordance (in terms of the direction of fold-change and statistical significance) of 56.3% and an r-value of 0.96 (P-value ≤ 0.001) was obtained between the RT-qPCR results and the RNA-seq data for the tested genes. In addition, we observed r-values ≥ 0.99 (P-value ≤ 0.01) for CCL4, IL1B, IL6, and TLR2, and an r-value of 0.93 (P-value ≤ 0.1) for TNF; while the other three genes (FOS, IL10 and PIK3IP1) were not significant, presumably due to the smaller sample size used for biological validation. The RT-qPCR results for biological validation of the RNA-seq data are provided in Supplementary Data S1, worksheet 3.
Function and pathway enrichment analyses
To gain a better understanding of the macrophage cellular pathways modulated following M. bovis infection, we performed canonical pathway analyses using the IPA (Supplementary Data S4), Pathway-Guide (Supplementary Data S5) and Sigora (Supplementary Data S6) software tools with the DE genes identified using RNA-seq at each post-infection time point. We considered only canonical pathways flagged as significant by at least two of these tools. For the most part, these cellular pathways are involved in macrophage recognition of pathogens and subsequent signalling cascades that culminate in the activation of innate and adaptive immune processes.
Recognition of mycobacteria by host macrophages
Two of the enriched canonical pathways involved in the recognition of mycobacteria by host macrophages were Toll-like receptor signalling and RIG-I-like receptor signalling. Further inspection of these pathways revealed upregulation of Toll-like receptor (TLR) genes previously shown to have important roles in mycobacterial recognition and activation of immune responses74. These included TLR2 (encodes TLR2, which recognises bacterial peptidoglycan, lipoprotein and lipoarabinomannan), TLR3, TLR4 and CD14 (encodes a co-receptor in lipopolysaccharide recognition), all of which were upregulated at 6, 24 and/or 48 hpi. Notably, the MYD88 gene, which encodes a key adaptor protein that transduces intracellular TLR signalling, was downregulated at 24 and 48 hpi. However, all the upregulated TLR genes can engage the MYD88-independent signalling pathway via the alternative adapter molecules encoded by TICAM1 (upregulated at 6, 24 and 48 hpi) and TICAM2 (upregulated at 48 hpi). The transcriptional response we observe here represents M. bovis-mediated modulation of host immune responses to facilitate pathogen survival within the host macrophage. Indeed, earlier studies—using M. tuberculosis-infected macrophages—have shown that prolonged mycobacterial stimulation (24 to 48 h stimulation) of TLRs (particularly TLR2) gives rise to inhibition of several IFN-γ-induced immune responses, particularly the expression of MHC class II molecules, thereby repressing the antigen presentation process75,76. Such inhibition of IFN-γ-induced MHC class II expression has been shown to involve MYD88-dependent or -independent TLR signalling77. Conversely, the upregulation of both TICAM1 and TICAM2 may act to circumvent the suppression of MYD88-dependent TLR signalling by the host to activate the immune response to control infection.
We also observed that the RIG-I-like receptor (RLR) genes were upregulated at 6, 24 and/or 48 hpi; these were DDX58 (encodes the RIG-I protein that recognises 5′ triphosphate single-stranded RNA and short double-stranded RNA), IFIH1 (encodes a protein recognising long double stranded RNA) and DHX58 (encodes a protein that positively and/or negatively regulates DDX58 and IFIH1)78,79. Also, the MAVS gene, which encodes a key mitochondrial membrane-bound intermediary protein in the RLR signalling cascade, was upregulated at 48 hpi. In general, RLR signalling results in the activation of the IRF transcription factor complex (IRF3 and IRF7 were upregulated at 24 and/or 48 hpi), followed by type I interferons production that help control bacterial infection (e.g., enhancement of MHC class I antigen presentation)80,81 or can benefit bacterial survival (e.g., inhibition of the host immune response)82. Overall, the type I interferon-dependent immune response and outcome is largely pathogen- and tissue-specific; this is supported by the literature on models of M. tuberculosis infection83,84. In the current study, we did not observe expression of type I interferon genes and this could be due to a number of reasons. It is possible that alveolar macrophages respond to M. bovis infection by recruiting ATG5-ATG12 complex (genes upregulated at 24 and/or 48 hpi)—a key regulator of the autophagic process—that has been shown to interfere with RLR signalling85. Alternatively, M. bovis may actively inhibit induction of type I interferons, indeed M. tuberculosis can mediate such inhibition through TLR2 recognition84. Figure 2 shows the RIG-I-like receptor signalling pathway with gene expression values overlaid at all post-infection time points.
Figure 2. RIG-I-like receptor signalling pathway.

The RIG-I-like receptor signalling pathway is represented with gene expression (log2 fold-change) values overlaid at all post-infection time points; shown from left to right are the 2, 6, 24 and 48 hpi time points, respectively. The colour intensity corresponds to the level of upregulation (red) or downregulation (blue) in the M. bovis-infected versus the control non-infected alveolar macrophages. Genes coloured in grey were not significantly DE and genes coloured in white were filtered out due to low expression.
Activation and recruitment of bactericidal immune mechanisms
The recognition and uptake of M. bovis by alveolar macrophages leads to induction/repression of several immune-related transcription factors (e.g., the NF-κB, STAT and IRF complexes) and culminates in the activation and recruitment of bactericidal immune processes. In the present study, the macrophage bactericidal immune mechanisms enriched in response to M. bovis infection were the Apoptosis and Lysosome canonical pathways. Inspection of these pathways revealed upregulation at 6, 24 and 48 hpi of CASP8, CASP7, BID and CYCS, which encode several pro-apoptotic proteins. However, a larger number of genes encoding inhibitors of apoptosis were upregulated at 2, 6, 24 and/or 48 hpi, such as BCL2A1, CFLAR, BCL2, BCL2L1, BIRC2, BIRC3, XIAP, MCL1 and PRKX. Furthermore, nearly all genes encoding apoptotic endonucleases or effector molecules—which are key features for effective apoptosis—were downregulated at 24 and/or 48 h, including ENDOG, DFFA and AIFM186. Several studies have reported reduced mycobacterial survival resulting from apoptotic cell death, which actively destroys the host cell and its contents, including intracellular bacteria such as M. bovis86,87. However, it is possible that virulent M. bovis actively represses host cell apoptotic processes in vitro, thereby facilitating persistence within host cells88,89; the upregulation of anti-apoptotic genes observed here supports this hypothesis.
We also observed downregulation at 6, 24 and/or 48 hpi of many genes encoding proteins involved in the transport and activation of lysosomal enzymes, such as SUMF1, GNPTAB, IGF2R, AP3B1, AP4M1, GGA1 and the AP-1 assembly complex genes. Furthermore, the majority of genes encoding lysosomal enzymes were downregulated at 6, 24 and/or 48 hpi, notably the glycosidases, lipases, nucleases, sphingomyelinases, ceramidase, and prosaposin. Conversely, the genes encoding proteins mediating the acidification of lysosome compartments were mostly upregulated (i.e., TCIRG1 and V-ATPase family members), with the exception of ATP6V0D2 and ATP6V0A1 (downregulated at 24 and 48 hpi). Interestingly, a gene encoding a key transcription factor of the Lysosome pathway, TFEB, was also downregulated at 6, 24 and 48 hpi90. Previous studies have characterised the repression of phagosome-lysosome fusion as a hallmark of infection by mycobacteria91,92. Our results also show suppression of lysosomal function by M. bovis via downregulation of a majority of key effector lysosomal enzymes. Figure 3 shows the Lysosome pathway with gene expression values overlaid.
Figure 3. Lysosome pathway.

The Lysosome pathway is represented with gene expression (log2 fold-change) values overlaid at all post-infection time points; shown from left to right are the 2, 6, 24 and 48 hpi time points, respectively. The colour intensity corresponds to the level of upregulation (red) or downregulation (blue) in the M. bovis-infected versus the control non-infected alveolar macrophages. Genes coloured in grey were not significantly DE and genes coloured in white were filtered out due to low expression.
Comparison of the alveolar macrophage and monocyte-derived macrophage responses to tubercle bacilli
We next compared the transcriptomic responses for two different bovine host cell types in response to M. bovis infection: alveolar macrophages and monocyte-derived macrophages (MDM). RNA-seq expression data (accession number GSE45439) from M. bovis-infected and non-infected MDM were used from a study previously published by our group14. This experiment consisted of a single time point contrast (24 hpi), which was therefore compared to the contrast between M. bovis-infected and non-infected alveolar macrophages at 24 hpi.
The correlation between gene expression fold-changes (M. bovis-infected versus non-infected control groups) for the alveolar macrophage and MDM at 24 hpi was investigated for all genes that passed the low expression filtering in both experiments (number of genes = 10,314). A Spearman rank correlation coefficient of 0.352 (P-value ≤ 1 × 10−15) was observed, indicating a moderate correlation of gene expression fold-changes between the two experiments. A much larger number of DE genes (FDR adjusted P-value ≤ 0.05) were detected for the alveolar macrophage contrast at 24 hpi (2,674 up-, 2,841 downregulated) compared to the MDM experiment (1,392 up-, 1,192 downregulated). Of these DE genes, 1,220 were identical and exhibited the same direction of expression in both experiments (638 up-, 582 downregulated). Conversely, 240 genes were DE in both experiments but displayed an opposite direction of expression (Supplementary Figure S3). The concordant genes (1,220) between both experiments represented 22% of the total number of DE genes for the alveolar macrophage and 47% for the MDM. We also detected 307 and 195 enriched canonical pathways at 24 hpi for the alveolar macrophage and MDM, respectively; of these enriched canonical pathways, 52 were common to both cell types. Taken together, these findings demonstrate that primary alveolar macrophages, as the target host cell, provide a better model for studies of infection with tubercle bacilli. Notwithstanding this, the canonical pathways that overlapped between the two cell types related to pattern recognition receptors, NF-κB activation, apoptosis and cytokine-mediated cell signalling, suggesting that MDM can also provide meaningful, but arguably more limited, immunobiological information if used for in vitro studies of mycobacterial infection. Other groups have reported similar gene expression results for comparisons between alveolar macrophages and MDM infected with M. tuberculosis or stimulated with lipopolysaccharides93,94.
It is important to highlight a number of experimental factors that may have also contributed to the differences observed between the two studies, including: (1) the number of biological replicates (n = 10 for the present study versus n = 6 for the MDM); (2) the age and sex of the animals (young male calves versus mature four year-old females); (3) the bacterial culture media (Tween− versus Tween+); (4) the MOI (10:1 versus 2:1); (5) differences in the RNA-seq library protocols used; (6) the high-throughput sequencing apparatus (Illumina® HiSeq™ 2000 versus Genome Analyzer IIx); and (7) differences in the computational pipelines used for analysis of the RNA-seq data.
In conclusion, we have, for the first time, applied RNA-seq technology to define the response of the mammalian host alveolar macrophage to infection with tubercle bacilli. Our results with M. bovis and the bovine alveolar macrophage have clear parallels to M. tuberculosis and tuberculosis disease in humans. Our analysis highlights the complex gene expression patterns and regulation underlying the early mammalian host response during infection with tubercle bacilli. Notably, we report the involvement of NATs in the innate immune response to intracellular bacilli, which may play a previously underappreciated role in the pathogen-mediated suppression of host defence mechanisms. Furthermore, we have dissected the innate immune response to M. bovis-infection in unprecedented detail through identification and inclusion of previously unannotated novel bovine genes. Our results show involvement of Toll-like and RIG-I-like receptors, apoptosis and lysosome signalling, as well as repression of these mechanisms that indicates subversion of protective host processes by M. bovis to promote its survival and persistence within the macrophage and ultimately establish infection.
Additional Information
How to cite this article: Nalpas, N. C. et al. RNA sequencing provides exquisite insight into the manipulation of the alveolar macrophage by tubercle bacilli. Sci. Rep. 5, 13629; doi: 10.1038/srep13629 (2015).
Supplementary Material
Acknowledgments
This work was supported by Investigator Grants from Science Foundation Ireland (Nos: SFI/01/F.1/B028 and SFI/08/IN.1/B2038), a Research Stimulus Grant from the Department of Agriculture, Food and the Marine (No: RSF 06 405), a European Union Framework 7 Project Grant (No: KBBE-211602–MACROSYS), the Irish Research Council (IRC) funded UCD Bioinformatics and Systems Biology PhD Programme (http://bioinfo-casl.ucd.ie/PhD) and the UCD Wellcome Trust funded Computational Infection Biology PhD Programme (Grant no: 097429/Z/11/Z). We thank Dr. Eddie Jordan (UCD Lyons Research Farm), Dr. Joseph Cassidy (UCD) and Mr. Brian Cloak (UCD) for assistance with animal work and lung lavages. We also thank Dr. George Cloughley, Ms. Bernadette Legget, Ms. Yvonne Abbott and Ms. Maureen McCullough (all UCD) for assistance with microbial testing of samples.
Footnotes
Author Contributions Conceived and designed the experiments: N.C.N., D.A.M., K.M.C., J.A.B., E.G., S.V.G. and D.E.M. Performed the experiments: N.C.N., D.A.M., K.M.C., J.A.B., C.H. and K.E.M. Analysed the data: N.C.N., D.A.M., K.R., K.E.K. and P.A.M. Prepared and edited the manuscript: N.C.N., D.A.M., S.V.G. and D.E.M. All authors read and approved the manuscript.
References
- Brosch R. et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. USA 99, 3684–3689, 10.1073/pnas.052548299 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier T. et al. The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. USA 100, 7877–7882, 10.1073/pnas.1130426100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy J. P. The pathogenesis and pathology of bovine tuberculosis with insights from studies of tuberculosis in humans and laboratory animal models. Vet. Microbiol. 112, 151–161, 10.1016/j.vetmic.2005.11.031 (2006). [DOI] [PubMed] [Google Scholar]
- Van Rhijn I., Godfroid J., Michel A. & Rutten V. Bovine tuberculosis as a model for human tuberculosis: advantages over small animal models. Microbes Infect. 10, 711–715, 10.1016/j.micinf.2008.04.005 (2008). [DOI] [PubMed] [Google Scholar]
- Waters W. R., Maggioli M. F., McGill J. L., Lyashchenko K. P. & Palmer M. V. Relevance of bovine tuberculosis research to the understanding of human disease: historical perspectives, approaches, and immunologic mechanisms. Vet. Immunol. Immunopathol. 159, 113–132, 10.1016/j.vetimm.2014.02.009 (2014). [DOI] [PubMed] [Google Scholar]
- Menzies F. D. & Neill S. D. Cattle-to-cattle transmission of bovine tuberculosis. Vet. J. 160, 92–106, 10.1053/tvjl.2000.0482 (2000). [DOI] [PubMed] [Google Scholar]
- Bowden D. H. The alveolar macrophage. Environ. Health Perspect. 55, 327–341 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock J. M. & Neill S. D. Mycobacterium bovis infection and tuberculosis in cattle. Vet. J. 163, 115–127, S1090023301906550 (2002). [DOI] [PubMed] [Google Scholar]
- Kumar D. & Rao K. V. Regulation between survival, persistence, and elimination of intracellular mycobacteria: a nested equilibrium of delicate balances. Microbes Infect. 13, 121–133, 10.1016/j.micinf.2010.10.009 (2011). [DOI] [PubMed] [Google Scholar]
- de Chastellier C. The many niches and strategies used by pathogenic mycobacteria for survival within host macrophages. Immunobiology 214, 526–542, 10.1016/j.imbio.2008.12.005 (2009). [DOI] [PubMed] [Google Scholar]
- Ehrt S. & Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 11, 1170–1178, 10.1111/j.1462-5822.2009.01335.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abebe M. et al. Modulation of cell death by M. tuberculosis as a strategy for pathogen survival. Clin. Dev. Immunol. 2011, 678570, 10.1155/2011/678570 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee D. A. et al. Global gene expression and systems biology analysis of bovine monocyte-derived macrophages in response to in vitro challenge with Mycobacterium bovis. PLoS ONE 7, e32034, 10.1371/journal.pone.0032034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalpas N. C. et al. Whole-transcriptome, high-throughput RNA sequence analysis of the bovine macrophage response to Mycobacterium bovis infection in vitro. BMC Genomics 14, 230, 10.1186/1471-2164-14-230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caimi K., Blanco F., Soria M. & Bigi F. Transcriptional response of bovine monocyte-derived macrophages after the infection with different Argentinean Mycobacterium bovis isolates. Biomed. Res. Int. 2013, 458278, 10.1155/2013/458278 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez L. E. & Goodman J. Mycobacterium tuberculosis invades and replicates within type II alveolar cells. Infect. Immun. 64, 1400–1406 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Crevel R., Ottenhoff T. H. & van der Meer J. W. Innate immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 15, 294–309 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. Effects of Mycobacterium bovis on monocyte-derived macrophages from bovine tuberculosis infection and healthy cattle. FEMS Microbiol. Lett. 321, 30–36, 10.1111/j.1574-6968.2011.02304.x (2011). [DOI] [PubMed] [Google Scholar]
- Masaki S., Sugimori G., Okamoto A., Imose J. & Hayashi Y. Effect of Tween 80 on the growth of Mycobacterium avium complex. Microbiol. Immunol. 34, 653–663 (1990). [DOI] [PubMed] [Google Scholar]
- Ortalo-Magne A. et al. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J. Bacteriol. 178, 456–461 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler T. H. & Youmans G. P. The Effect of “Tween 80,” Bovine Albumin, Glycerol, and Glucose on the Growth of Mycobacterium tuberculosis var. hominis (H37Rv). J. Bacteriol. 56, 235–243 (1948). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boxtel R. M., Lambrecht R. S. & Collins M. T. Effect of polyoxyethylene sorbate compounds (Tweens) on colonial morphology, growth, and ultrastructure of Mycobacterium paratuberculosis. APMIS 98, 901–908 (1990). [DOI] [PubMed] [Google Scholar]
- Ozsolak F. & Milos P. M. RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98, 10.1038/nrg2934 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettigan P. A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 17, 4–11, 10.1016/j.cbpa.2012.12.008 (2013). [DOI] [PubMed] [Google Scholar]
- Magee D. A. et al. Innate cytokine profiling of bovine alveolar macrophages reveals commonalities and divergence in the response to Mycobacterium bovis and Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 94, 441–450, 10.1016/j.tube.2014.04.004 (2014). [DOI] [PubMed] [Google Scholar]
- Nalpas N. C. A complete RNA-sequencing analysis pipeline. ZENODO v. 1.0.0, http://dx.doi.org/10.5281/zenodo.10668, doi: 10.5281/zenodo.10668 (2014).
- Edgar R., Domrachev M. & Lash A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- FastQC A Quality Control tool for High Throughput Sequence Data (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, 2010).
- Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P. et al. Ensembl 2014. Nucleic Acids Res. 42, D749–755, 10.1093/nar/gkt1196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G. K. & Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, 10.1093/bioinformatics/btt656 (2014). [DOI] [PubMed] [Google Scholar]
- Robinson M. D., McCarthy D. J. & Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. & Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Method. 57, 289–300 (1995). [Google Scholar]
- Finocchiaro G. et al. Localizing hotspots of antisense transcription. Nucleic Acids Res. 35, 1488–1500, 10.1093/nar/gkm027 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Liberman L. M., Mukherjee N., Benfey P. N. & Ohler U. Integrated detection of natural antisense transcripts using strand-specific RNA sequencing data. Genome Res. 23, 1730–1739, 10.1101/gr.149310.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F., Xu Z., Clauder-Munster S. & Steinmetz L. M. Antisense artifacts in transcriptome microarray experiments are resolved by actinomycin D. Nucleic Acids Res. 35, e128, 10.1093/nar/gkm683 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515, 10.1038/nbt.1621 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A., Pimentel H., Trapnell C. & Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 27, 2325–2329, 10.1093/bioinformatics/btr355 (2011). [DOI] [PubMed] [Google Scholar]
- Tatusov R. L., Koonin E. V. & Lipman D. J. A genomic perspective on protein families. Science 278, 631–637 (1997). [DOI] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W. & Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410, 10.1016/S0022-2836(05)80360-2 (1990). [DOI] [PubMed] [Google Scholar]
- Durinck S., Spellman P. T., Birney E. & Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191, 10.1038/nprot.2009.97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca A. L. et al. A novel signaling pathway impact analysis. Bioinformatics 25, 75–82, 10.1093/bioinformatics/btn577 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. & Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G. et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 33, D428–432, 10.1093/nar/gki072 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- sigora: SIGNATURE OVERREPRESENTATION ANALYSIS v. R package version 0.9.2 (http://CRAN.R-project.org/package=sigora, 2012).
- Hellemans J., Mortier G., De Paepe A., Speleman F. & Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19, 10.1186/gb-2007-8-2-r19 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer P. L. & Doerge R. W. Statistical design and analysis of RNA sequencing data. Genetics 185, 405–416, 10.1534/genetics.110.114983 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhou J. & White K. P. RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30, 301–304, 10.1093/bioinformatics/btt688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard J. H., Purdom E., Hansen K. D. & Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics 11, 94, 10.1186/1471-2105-11-94 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R. F. et al. Human alveolar macrophage gene responses to Mycobacterium tuberculosis strains H37Ra and H37Rv. Am. J. Respir. Cell Mol. Biol. 40, 491–504, 10.1165/rcmb.2008-0219OC (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi M. A. & Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat. Rev. Mol. Cell Biol. 10, 637–643, 10.1038/nrm2738 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot M. & Pilpel Y. Genome-wide natural antisense transcription: coupling its regulation to its different regulatory mechanisms. EMBO Rep. 7, 1216–1222, 10.1038/sj.embor.7400857 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A. Biological functions of natural antisense transcripts. BMC Biol. 11, 31, 10.1186/1741-7007-11-31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari M. E. et al. Characterization of ABF-1, a novel basic helix-loop-helix transcription factor expressed in activated B lymphocytes. Mol. Cell. Biol. 18, 3130–3139 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. S. et al. Seminal plasma accelerates semen-derived enhancer of viral infection (SEVI) fibril formation by the prostatic acid phosphatase (PAP248-286) peptide. J. Biol. Chem. 287, 11842–11849, 10.1074/jbc.M111.314336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero I. B. et al. Prostatic acid phosphatase is not a prostate specific target. Cancer Res. 67, 6549–6554, 10.1158/0008-5472.CAN-07-1651 (2007). [DOI] [PubMed] [Google Scholar]
- Zarco N., Gonzalez-Ramirez R., Gonzalez R. O. & Segovia J. GAS1 induces cell death through an intrinsic apoptotic pathway. Apoptosis 17, 627–635, 10.1007/s10495-011-0696-8 (2012). [DOI] [PubMed] [Google Scholar]
- Waerner T., Gardellin P., Pfizenmaier K., Weith A. & Kraut N. Human RERE is localized to nuclear promyelocytic leukemia oncogenic domains and enhances apoptosis. Cell Growth Differ. 12, 201–210 (2001). [PubMed] [Google Scholar]
- Koul A. et al. Serine/threonine protein kinases PknF and PknG of Mycobacterium tuberculosis: characterization and localization. Microbiology 147, 2307–2314 (2001). [DOI] [PubMed] [Google Scholar]
- He M. et al. Identification and characterization of new long chain acyl-CoA dehydrogenases. Mol. Genet. Metab. 102, 418–429, 10.1016/j.ymgme.2010.12.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter R. D. & Huang L. Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch. 447, 744–751, 10.1007/s00424-003-1070-7 (2004). [DOI] [PubMed] [Google Scholar]
- Espinosa E. J., Calero M., Sridevi K. & Pfeffer S. R. RhoBTB3: a Rho GTPase-family ATPase required for endosome to Golgi transport. Cell 137, 938–948, 10.1016/j.cell.2009.03.043 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistri M., Faghihi M. A., St Laurent G. 3rd & Wahlestedt C. Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet. 28, 389–396, 10.1016/j.tig.2012.03.013 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K. et al. Natural antisense transcript stabilizes inducible nitric oxide synthase messenger RNA in rat hepatocytes. Hepatology 47, 686–697, 10.1002/hep.22036 (2008). [DOI] [PubMed] [Google Scholar]
- Kimura T. et al. Stabilization of human interferon-alpha1 mRNA by its antisense RNA. Cell. Mol. Life Sci. 70, 1451–1467, 10.1007/s00018-012-1216-x (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongay C. F., Grisafi P. L., Galitski T. & Fink G. R. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell 127, 735–745, 10.1016/j.cell.2006.09.038 (2006). [DOI] [PubMed] [Google Scholar]
- Beiter T., Reich E., Williams R. W. & Simon P. Antisense transcription: a critical look in both directions. Cell. Mol. Life Sci. 66, 94–112, 10.1007/s00018-008-8381-y (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V. & Steinmetz L. M. Gene regulation by antisense transcription. Nat. Rev. Genet. 14, 880–893, 10.1038/nrg3594 (2013). [DOI] [PubMed] [Google Scholar]
- Yla-Herttuala S. & Kaikkonen M. Does mass balance between sense and antisense transcripts fine-tune the outcome of gene expression? EMBO Rep. 15, 125–126, 10.1002/embr.201338030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A. et al. LILRA5 is expressed by synovial tissue macrophages in rheumatoid arthritis, selectively induces pro-inflammatory cytokines and IL-10 and is regulated by TNF-alpha, IL-10 and IFN-gamma. Eur. J. Immunol. 38, 3459–3473, 10.1002/eji.200838415 (2008). [DOI] [PubMed] [Google Scholar]
- Banerjee S. et al. miR-125a-5p regulates differential activation of macrophages and inflammation. J. Biol. Chem. 288, 35428–35436, 10.1074/jbc.M112.426866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingrel J. B. et al. Myeloid-specific Kruppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ. Res. 110, 1294–1302, 10.1161/CIRCRESAHA.112.267310 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesniaux V. et al. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6, 946–959, 10.1016/j.micinf.2004.04.016 (2004). [DOI] [PubMed] [Google Scholar]
- Noss E. H. et al. Toll-like receptor 2-dependent inhibition of macrophage class II MHC expression and antigen processing by 19-kDa lipoprotein of Mycobacterium tuberculosis. J. Immunol. 167, 910–918 (2001). [DOI] [PubMed] [Google Scholar]
- Pai R. K. et al. Prolonged toll-like receptor signaling by Mycobacterium tuberculosis and its 19-kilodalton lipoprotein inhibits gamma interferon-induced regulation of selected genes in macrophages. Infect. Immun. 72, 6603–6614, 10.1128/IAI.72.11.6603-6614.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafuse W. P., Alvarez G. R., Curry H. M. & Zwilling B. S. Mycobacterium tuberculosis and Mycobacterium avium inhibit IFN- gamma -induced gene expression by TLR2-dependent and independent pathways. J. Interferon Cytokine Res. 26, 548–561, 10.1089/jir.2006.26.548 (2006). [DOI] [PubMed] [Google Scholar]
- Loo Y. M. & Gale M. Jr. Immune signaling by RIG-I-like receptors. Immunity 34, 680–692, 10.1016/j.immuni.2011.05.003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T. et al. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 107, 1512–1517, 10.1073/pnas.0912986107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe K. M., McWhirter S. M. & Vance R. E. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5, e1000665, 10.1371/journal.ppat.1000665 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz B. et al. Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 281, 36173–36179, 10.1074/jbc.M604638200 (2006). [DOI] [PubMed] [Google Scholar]
- O’Connell R. M. et al. Immune Activation of Type I IFNs by Listeria monocytogenes Occurs Independently of TLR4, TLR2, and Receptor Interacting Protein 2 but Involves TANK-Binding Kinase 1. J. Immunol. 174, 1602–1607, 10.4049/jimmunol.174.3.1602 (2005). [DOI] [PubMed] [Google Scholar]
- Stanley S. A., Johndrow J. E., Manzanillo P. & Cox J. S. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178, 3143–3152 (2007). [DOI] [PubMed] [Google Scholar]
- Simmons D. P. et al. Mycobacterium tuberculosis and TLR2 agonists inhibit induction of type I IFN and class I MHC antigen cross processing by TLR9. J. Immunol. 185, 2405–2415, 10.4049/jimmunol.0904005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jounai N. et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 104, 14050–14055, 10.1073/pnas.0704014104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Hartman M. & Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med. J. 50, 1–11, 10.3349/ymj.2009.50.1.1 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar S. M. et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 4, 279–287, 10.1038/mi.2011.3 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J., Remold H. G. & Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 164, 2016–2020 (2000). [DOI] [PubMed] [Google Scholar]
- Sly L. M., Hingley-Wilson S. M., Reiner N. E. & McMaster W. R. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J. Immunol. 170, 430–437 (2003). [DOI] [PubMed] [Google Scholar]
- Song W. et al. TFEB regulates lysosomal proteostasis. Hum. Mol. Genet. 22, 1994–2009, 10.1093/hmg/ddt052 (2013). [DOI] [PubMed] [Google Scholar]
- Malik Z. A., Iyer S. S. & Kusner D. J. Mycobacterium tuberculosis phagosomes exhibit altered calmodulin-dependent signal transduction: contribution to inhibition of phagosome-lysosome fusion and intracellular survival in human macrophages. J. Immunol. 166, 3392–3401 (2001). [DOI] [PubMed] [Google Scholar]
- Vergne I. et al. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 102, 4033–4038, 10.1073/pnas.0409716102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surewicz K. et al. The differential interaction of p38 MAP kinase and tumor necrosis factor-alpha in human alveolar macrophages and monocytes induced by Mycobacterium tuberculosis. Cell. Immunol. 228, 34–41, 10.1016/j.cellimm.2004.03.007 (2004). [DOI] [PubMed] [Google Scholar]
- Tomlinson G. S. et al. Adherent human alveolar macrophages exhibit a transient pro-inflammatory profile that confounds responses to innate immune stimulation. PLoS ONE 7, e40348, 10.1371/journal.pone.0040348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.