

Abstract
Through years of evolutionary selection pressures, organisms have developed potent toxins that coincidentally have marked antineoplastic activity. These natural products have been vital for the development of multiagent treatment regimens currently employed in cancer chemotherapy, and are used in the treatment of a variety of malignancies. Therefore, this review catalogs recent advances in natural product-based drug discovery via the examination of mechanisms of action and available clinical data to highlight the utility of these novel compounds in the burgeoning age of precision medicine. The review also highlights the recent development of antibody-drug conjugates and other immunotoxins, which are capable of delivering highly cytotoxic agents previously deemed too toxic to elicit therapeutic benefit preferentially to neoplastic cells. Finally, the review examines natural products not currently used in the clinic that have novel mechanisms of action, and may serve to supplement current chemotherapeutic protocols.
Key Points
| Natural products have potentiated many novel drug classes employed in cancer chemotherapy, including mechanistic target of rapamycin inhibitors, protein synthesis inhibitors, nucleic acid-directed agents, and microtubule-directed agents |
| Recent advances in immunotherapy have enabled highly cytotoxic natural products to be targeted towards specific tissues |
| There are still many natural products with mechanisms not currently seen in the clinical setting that could be very beneficial to the field of oncology |
Introduction
The diversity of natural products currently used in the clinical setting to treat solid tumors, as well as disseminated cancers is truly extensive. Under the pressure of natural selection, various species produce cytotoxic secondary metabolites to combat potential predators, prey, or competition in the so-called “arms race” of evolution. Remarkably, some of these natural toxins appear to exhibit potent antineoplastic activity, and after years of research, have found their way from the ocean or soil to the highly heterogeneous environment of clinical oncology. The origins of cancer chemotherapy can be traced to human-made compounds, as Goodman, Gilman, and colleagues at Yale University began investigating the potential of nitrogen mustards in 1942 [1], which was shortly followed by Sidney Farber’s use of the antifolate aminopterin to induce remissions among children with leukemia in 1947 [2, 3]. However, the institution of natural products and semisynthetic derivatives of these compounds in the latter part of the 20th century potentiated the idea of concomitant chemotherapy; using a variety of antineoplastic agents with different mechanisms of action to significantly perturb neoplastic development, and in some cases, produce long-term remissions.
Owing to recent advances in molecular biology, investigators have begun unraveling essential oncogenic pathways in carcinogenesis, potentiating an era of chemotherapy in which it is possible to theorize cancer-specific targets. This has launched the introduction of precision medicine in cancer chemotherapy in which clinicians now have the capability of selecting optimal therapies based on the genetic and phenotypic profile of the patient’s malignancy in addition to traditional broad-spanning cytotoxic antineoplastic intervention. Despite these commendable advances in targeted therapy, natural products and their derivatives are still extensively relied upon against malignancies where finding cancer-specific targets has been less successful, and are often used in combination with these targeted approaches to generate more thorough treatment protocols. Further, novel natural product derivatives have shown notably efficacy against previously unresponsive malignancies at the clinical level, suggesting that natural product-based drug discovery still has considerable utility in the burgeoning era of personalized chemotherapy. Finally, natural products have the potential to improve novel immunotherapeutic strategies by conjugating monoclonal antibodies (mABs) or cytokines to highly cytotoxic compounds that have too low of a therapeutic index without an appropriate guidance mechanism.
This review catalogs recent advances in natural product drug discovery that have potentiated promising activity against aggressive malignancies, and have enabled a more precise delivery of highly cytotoxic, natural product-based agents to reduce unintended side effects. Specifically, this review covers the commendable advances in the development of microtubule-directed agents (eribulin and epothilones), mechanistic target of rapamycin (mTOR) inhibitors (everolimus and temsirolimus), protein synthesis inhibitors (omacetaxine mepesuccinate), nucleic acid-directed agents (trabectedin), engineered cytokine proteins (denileukin diftitox), and antibody-drug conjugates (ADCs; brentuximab vedotin, trastuzumab emtansine, calicheamicin conjugated monoclonal antibodies, and exotoxin conjugates). In addition, the review will highlight several novel natural products that act by mechanisms not currently seen in the clinic (cytochalasins and withanolides) to address their potential utility in cancer chemotherapy. Although this review provides an extensive coverage of novel natural product-based antineoplastic agents, additional agents have seen recent success in the clinical setting, and the reader is referred to the following reviews for further information [4–6]. In addition, the diversity of natural product-based antineoplastic agents and their derivatives currently approved by the US Food and Drug Administration (FDA) are highlighted in Table 1. They serve as a reminder of how important nature has been in the treatment of many, if not most types of malignancy.
Table 1.
US Food and Drug Administration (FDA) approved uses of natural products in cancer chemotherapy
| Agent | Drug classification/species of origin | Mechanism of action | FDA approved use |
|---|---|---|---|
| Omacetaxine mepesuccinate (Synribo®) | Alkaloid Cephalotaxus harringtonia |
Inhibits protein synthesis and is independent of direct Bcr-Abl binding | Chronic- or accelerated-phase CML with resistance and/or intolerance to two or more TKIs |
| Daunorubicin (Cerubidine) | Anthracycline Streptomyces peucetius |
Topo II inhibitor, intercalating agent | Remission induction in adult AML or in both children and adults for ALL |
| DaunoXome ® | Citrate liposome formulation | Advanced HIV-associated Kaposi’s sarcoma | |
| Doxorubicin (Adriamycin®) | Anthracycline Streptomyces peucetius |
Topo II inhibitor, intercalating agent | ALL, AML, Wilms tumor, neuroblastoma, soft tissue and bone sarcoma, breast, ovarian, thyroid, bronchiogenic, gastric and transitional cell bladder carcinomas, HL, NHL |
| Doxil ® | Liposome formulation | AIDS-related Kaposi’s sarcoma, ovarian carcinoma, multiple myeloma | |
| Epirubicin (Ellence®) | Anthracycline Streptomyces peucetius |
Topo II inhibitor, intercalating agent | Axillary node-positive breast carcinoma |
| Idarubicin (Idamycin®) | Anthracycline Streptomyces peucetius |
Topo II inhibitor, intercalating agent | Adults with AML classified M1 to M7 (French-American-British system) |
| Valrubicin (Valstar®) | Anthracycline Streptomyces peucetius |
Topo II inhibitor, intercalating agent | Carcinoma in situ of the urinary bladder |
| Mitoxantrone (Novantrone®) |
Anthracycline Streptomyces peucetius |
Topo II inhibitor, intercalating agent | Adult AML, symptomatic hormone-refractory prostate adenocarcinoma |
| Brentuximab vedotin (Adcetris®) | Antibody-drug conjugate, dolastatin, MMAE is derived from peptides found in Dolabella auricularia | MMAE enters cells expressing CD30, potentiating microtubule inhibition in addition to the antineoplastic effects of brentuximab | HL after failure of ASCT or after failure of two prior multiagent chemotherapeutic regimens in those who are not ASCT candidates, sALCL |
| Trastuzumab emtansine (Kadcyla®) | Antibody-drug conjugate, macrolide, DM1 is derived from maytansine, which can be extracted from plants of the genus Maytenus | DM1 enters cells expressing HER2/neu receptor, potentiating microtubule inhibition in addition to the antineoplastic effects of trastuzumab | HER2+ breast carcinoma, metastatic gastric or gastroesophageal adenocarcinoma with HER2 overexpression |
| Mitomycin | Aziridine Streptomyces caespitosus or Streptomyces lavendulae |
Alkylating agent, crosslinks DNA | Disseminated gastric adenocarcinoma, disseminated pancreactic adenocarcinoma |
| Irinotecan (Camptosar®) | Camptothecin Camptotheca acuminata |
Topo I inhibitor | Metastatic colorectal carcinoma |
| Topotecan (Hycamtin®) | Camptothecin Camptotheca acuminata |
Topo I inhibitor | Cervical carcinoma, metastatic ovarian carcinoma, SCLC |
| Denileukin diftitox (Ontak®) |
Engineered cytokine protein Corynebacterium diphtheriae |
Composed of diphtheria toxin fragments linked to IL-2 sequences, interacts with IL-2 cell surface receptors before inhibiting protein synthesis | Persistent or recurrent CTCL in patients who express the CD25 component of the IL-2 receptor |
| Asparaginase (Elspar®, Erwinase®) | Enzyme Elspar: Escherichia coli Erwinase: Erwinia chrysanthemi |
Depletes asparagine, an amino acid required by some leukemias | Component of a multiagent induction regimen for ALL |
| Pegasparagase ® | Pegylated version | Patient has hypersensitivity to asparaginase | |
| Ixabepilone (Ixempra®) |
Epothilone Sorangium cellulosum |
Stabilizes formed microtubules | Metastatic or locally advanced breast carcinoma after failure of an anthracycline and a taxane |
| Bleomycin (Blenoxane®) | Glycopeptide Streptomyces verticillus |
Unresolved, but does induce DNA strand breaks | Squamous cell carcinomas, NHL, testicular cancers, HL, malignant pleural effusions |
| Etoposide (Vepesid®) |
Lignan Podophyllum peltatum |
Topo II inhibitor | Testicular cancers, SCLC |
| Etoposide phosphate (Etophos ® ) | Ester derivative that increases water solubility | Testicular cancer, SCLC | |
| Teniposide (Vumon®) |
Lignan Podophyllum peltatum |
Topo II inhibitor | Refractory childhood ALL |
| Eribulin (Halaven®) | Macrolide Halichondria okadai |
Inhibits the growth phase of microtubules without affecting the shortening phase, sequesters tubulin into nonproductive aggregates | Metastatic breast carcinoma that has received at least two prior chemotherapy regimens for late-stage disease, including both anthracycline- and taxane-based chemotherapies |
| Everolimus (Afinitor®, Afinitor Disperz®) | mTOR inhibitor Streptomyces hygroscopicus |
Inhibits mTOR by binding FKBP-12 | Postmenopausal women with advanced hormone receptor+ and ER− breast carcinoma, PNET, RCC, renal angiomyolipoma, pediatric and adult SEGA |
| Temsirolimus (Torisel®) | mTOR inhibitor Streptomyces hygroscopicus |
Inhibits mTOR by binding FKBP-12 | Advanced RCC |
| Streptozotocin (Zanosar®) | Nitrosourea Streptomyces achromogenes |
Alkylating agent | Metastatic islet cell carcinoma of the pancreas |
| Dactinomycin (Cosmegen®) |
Polypeptide various species of the genus Streptomyces |
Binds DNA at the transcription initiation complex and prevents elongation of RNA chain by RNA polymerase | Wilms tumor, pediatric rhabdomyosarcoma, Ewing’s sarcoma, metastatic and nonseminomatous testicular cancer, gestational trophoblastic neoplasia, locally recurrent or locoregional solid malignancies |
| Paclitaxel (Taxol®) | Taxane Taxus brevifolia |
Stabilizes formed microtubules | Ovarian carcinoma, breast carcinoma, NSCLC, AIDS-related Kaposi’s sarcoma |
| Abraxane ® | Protein bound, conjugated to albumin | Metastatic breast carcinoma, locally advanced or metastatic NSCLC | |
| Docetaxel (Taxotere®) | Taxane Taxus brevifolia |
Stabilizes formed microtubules | NSCLC, breast carcinoma, prostate adenocarcinoma, gastric adenocarcinoma, head and neck carcinomas |
| Cabazitaxel (Jevtana®) |
Taxane Taxus brevifolia |
Stabilizes formed microtubules | Hormone-refractory prostate adenocarcinoma |
| Vincristine (Oncovin®) |
Vinca alkaloid Catharanthus roseus |
Inhibits tubule polymerization | Acute leukemias, HL, NHL, neuroblastoma, Wilms tumor, rhabdomyosarcoma |
| Marqibo ® | Liposome formulation | Philadelphia chromosome+ALL | |
| Vinblastine (Velban®) |
Vinca alkaloid Catharanthus roseus |
Inhibits tubule polymerization | Testicular cancers, HL, NHL, mycosis fungoides, Kaposi’s sarcoma, histiocytic lymphoma, Letterer-Siwe disease (histiocytosis X), breast carcinoma, choriocarcinoma |
| Vinorelbine (Navelbine®) |
Vinca alkaloid Catharanthus roseus |
Inhibits tubulin polymerization | NSCLC |
Underline indicates different formulations of the agent. Bold indicates that the compound is a synthetic or semisynthetic derivative of the original natural product
AIDS acquired human immunodeficiency syndrome, ALL acute lymphoid leukemia, AML acute myeloid leukemia, ASCT autologous stem cell transplant, CD cluster of differentiation, DM1 mertansine, CML chronic myeloid leukemia, CTCL cutaneous T-cell lymphoma, ER estrogen receptor, FKBP12 12 kDa FK506 binding protein, HIV human immunodeficiency virus, HL Hodgkin’s lymphoma, IL-2 interleukin-2, MMAE monomethyl auristatin E, mTOR mechanistic target of rapamycin, NHL non-Hodgkin’s lymphoma, NSCLC non-small-cell lung carcinoma, PNET neuroendocrine tumors of pancreatic origin, RCC renal cell carcinoma, sALCL systemic anaplastic large-cell lymphoma, SEGA subependymal giant-cell astrocytoma, SCLC small-cell lung carcinoma, Topo DNA topoisomerase, TKI tyrosine kinase inhibitor
Microtubule-Disrupting Eribulin
Eribulin is a fully synthetic, macrocyclic ketone analog of the marine sponge natural product halichondrin B (Fig. 1), a potent antimitotic initially isolated in 1986 from Halichondria okadai [7]. Although halicondrin B was designated for preclinical development after it was found to be highly cytotoxic against murine leukemia cells, difficulty in collecting sufficient material for developmental studies slowed its progress, and interest began to fade. However, the discovery that halocondrin B activity resides in the macrocyclic lactone C-1 to C-38 moiety [8] paved the way for development of a simplified synthetic analog, culminating in the design of eribulin.
Fig. 1.

Molecular diversity of antineoplastic agents derived from natural products. AMU atomic mass unit, DM1 mertansine, MW molecular weight, SMCC succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate
As with vinca alkaloids, eribulin exerts its cytotoxic effects by interfering with microtubule dynamics, and inhibiting polymerization [9, 10]. In addition, eribulin also works through an end-poisoning mechanism, resulting in the inhibition of microtubule growth, and even sequesters tubulin into nonfunctional aggregates, promoting G2/M phase arrest and apoptosis [11]. However, the two drug classes contrast in that eribulin does not bind the sides of tubulin polymers, and therefore does not markedly potentiate depolymerization [12, 13]. It does suppress spindle microtubule tension by interfering with centromere dynamics, as seen with some vinca alkaloids (particularly vinorelbine and vinflunine), but does so by inhibiting relaxation rates and the time spent stretching and relaxing, without the corresponding suppressive effects on stretching rates observed with the other agents [14]. Further, eribulin inhibits tubulin polymerization by binding the interdimer interface or the β-tubulin subunit alone contrary to other microtubule-directed agents, including epothilones and taxanes [12]. Interestingly, eribulin demonstrates significant activity against βIII-tubulin, an isotype that is overexpressed in cells resistant to microtubule inhibitors [15, 16], indicative of its unique clinical utility.
In regard to its clinical pharmacologic profile, eribulin demonstrates linear pharmacokinetics with rapid systemic distribution, but has a t1/2 of 40 h and around 49–65 % remains protein bound in circulation [3, 17]. The majority of the agent is eliminated through bile emulsification and fecal excretion. Eribulin is manufactured at 0.5 mg/mL concentrations, and no routine premedication is needed. One of the most common dosing schedules is 1.4 mg/m2 over 2–5 min on days 1 and 8 of a 21-day cycle, either undiluted or diluted in 100 mL of 0.9 % normal saline [17]. Although this schedule is typically well tolerated, eribulin is known to potentiate notable neutropenia (>grade 3 toxicity is ~57 %), elevate transaminases, and induce peripheral neuropathy (~8 % for grade 3 neuropathy and 0.4 % for grade 4 neuropathy).
Owing to the unique mechanisms by which eribulin inhibits microtubule dynamics, the agent is FDA approved for metastatic breast carcinoma refractory or relapsed on at least two prior treatment protocols for late-stage disease, including both anthracycline- and taxane-based chemotherapies [18]. This approval stems in large part from a phase III open-label study (n = 762) in which eribulin improved overall survival [median of 13.1 months, 95 % confidence interval (CI) 11.8–14.3] in comparison to treatments of the physicians’ choice (median of 10.6 months, 9.3–12.5; hazard ratio = 0.81, 95 % CI 0.66–0.99; p = 0.041) [19]. Peripheral neuropathy was the most common adverse event leading to discontinuation from eribulin, occurring in 24 (5 %) of 503 patients. In addition, eribulin has also been investigated for use in a variety of other solid tumors, including non-small-cell lung carcinoma, head and neck carcinoma, pancreatic adenocarcinoma, prostate adenocarcinoma, and various sarcomas. Published data indicate that non-small-cell lung carcinoma patients receive some benefit from eribulin, but the agent does not appear to be effective in the treatment of head and neck or pancreatic malignancies [20]. In addition, eribulin appears to demonstrate activity against metastatic castration-resistant prostate adenocarcinoma and advanced soft-tissue sarcoma with a relatively favorable toxicity profile being observed [21, 22].
Microtuble-Stabilizing Epothilones
Epothilones are 16-member macrolide microtubule-stabilizing agents initially isolated in 1987 from the So ce90 strain of the myxobacterium Sorangium cellulosum [23, 24]. Originally investigated for their antimycotic activity, samples of epothilones A and B sent to the National Cancer Institute in 1994 demonstrated potent antineoplastic activity in multiple cancer cell lines that was comparable and sometimes superior to paclitaxel. However, a critical difference between epothilones and other bulky natural products or derivatives is that overexpression of ATP-binding cassette (ABC) transporters does not significantly alter the cytotoxicity of epothilones, as these congeners have minimal substrate affinity for these proteins [25]. Epothilones A and B have marked anti-proliferative activity in neoplastic cells with elevated levels of permeability glycoprotein (P-gp) [26, 27], and tumor samples obtained from patients that respond to ixabepilone have shown significantly elevated levels of ABC transporters P-gp and multidrug resistance-associated protein 1 [27]. It should be noted that multidrug-resistance protein 7 is capable of effluxing a variety of antineoplastic agents, including epothilone B [28]. Although drug resistance to taxanes is often associated with β-III subunit overexpression, epothilones appear to be equally as potent against cells demonstrating this phenotype [29, 30]. Whereas paclitaxel is less effective in suppressing the growth rate and catastrophe frequency of purified α/βIII tubulin, ixabepilone markedly suppresses the dynamic instability of α/βIII dimers [31]. Further, ixabepilone has exhibited substantially higher activity than either taxanes or vinca alkaloids against neoplastic cells in vitro and in vivo in which taxane resistance is associated with βIII overexpression [32–34]. In regards to its structure-activity relationship with tubulin, epothilones appear to bind the taxane pocket of β-tubulin and promote structuring of the M-loop into a short helix, as demonstrated by epothilone A [35]. Consequently, the M-loop establishes lateral tubulin contacts in microtubules, thereby potentiating microtubule assembly and stability.
Modifications to the macrolide ring have been shown to alter both the antineoplastic activity and pharmacologic properties of epothilones. Second- and third-generation congeners have been synthesized that possess higher potency and enhanced water solubility compared with the original natural products [32, 33]. Ixabepilone, a second-generation semisynthetic analog of epothilone B has nitrogen substituted at position 16 of the macrolide ring instead of oxygen, making it a lactam [34] (Fig. 1). This substitution increases water solubility and plasma stability in comparison to epothilones B and D, but also reduces cytotoxicity by one-fold [36]. In addition to ixabepilone, a second semisynthetic derivative of epothilone B, BMS-310705, a congener synthesized by the substitution of a hydroxyl group with an amino group at C-21 of the methylthiazole side chain, is 10-fold more water soluble than epothilone B and is more cytotoxic than epothilone D in multiple human neoplastic cell lines [37, 38]. Several other recent novel epothilone derivatives have gained notable preclinical and clinical interest. 20-Desmethyl-20 methylsulfanyl epothilone B (ABJ-879) is a second-generation derivative that exhibits more cytotoxicity than epothilone B [39]. Sagopilone (ZK-EPO) is the first fully synthetic, third-generation epothilone B derivative that exhibits greater potency in vitro relative to the other epothilones, retains activity in multidrug-resistant malignant cells not observed in other congeners, and even crosses the blood–brain barrier, indicating the potential for penetration into the central nervous system [40]. The preclinical data have been so compelling that sagopilone has been investigated at the clinical level against advanced solid tumors and melanoma, with the agent demonstrating favorable pharmacokinetic data, a feasible toxicity profile, and antitumor activity against melanoma not elicted by other congeners [41, 42]. Finally, a second-generation epothilone D analog, KOS-1584, exhibits 3- to 12-fold increased potency compared with epothilones B and D, enhanced neoplastic tissue penetration, and reduced central nervous system toxicity [43].
Despite the isolation and characterization of epothilones A–F, the only agent of this class to reach FDA approval is ixabepilone. Although ixabepilone has better aqueous solubility than paclitaxel, Kolliphor EL is still typically used as an excipient, and is contraindicated in patients who are hypersensitive to the vehicle [44, 45]. Most clinical investigations of ixabepilone have used a dosing schedule of 32–50 mg/m2 infused over 1 or 3 h on day 1 of a 21-day cycle. Nevertheless, a 3-h infusion time is recommended, as more prominent neurotoxicity is often observed with shorter infusion times, and the FDA-approved dose and schedule is 40 mg/m2 intravenously (i.v.) over 3 h q3w [46]. Multiple phase I/II trials have revealed that ixabepilone exposure is not significantly affected by patient characteristics (age, sex, renal function, body weight, body surface area, race) [44, 47]. As with many mitotic inhibitors, the most common grade 3 or 4 toxicity associated with standard single-agent dosing schedules is neutropenia (grade 3 neutropenia is observed in the range of 10–33 %, while grade 4 neutropenia is observed in 7–32 % of patients, the wide range likely being a reflection of the number and type of prior therapies [46, 48–50].
Similar to eribulin, ixabepilone is currently indicated in metastatic or locally advanced breast carcinoma that is refractory or has relapsed on an anthracycline and a taxane, and is typically administered in combination with capecitabine [48, 50]. Interestingly, ixabepilone has shown notable efficacy against triple-negative breast carcinoma (TNBC), producing a pathologic complete response rate of 26 % in TNBC patients in comparison to 15 % in the non-triple-negative population [51]. Concomitant administration of ixabepilone and capecitabine has resulted in higher overall response rates than monotherapy, and a phase III trial of ixabepilone plus capecitabine produced a median progression-free survival significantly longer for TNBC patients treated with ixabepilone plus capecitabine (4.2 months) in comparison to treatment with capecitabine alone (1.7 months) [51]. In addition to breast carcinoma, ixabepilone has been investigated against non-Hodgkin’s lymphoma and pancreatic adenocarcinoma, both as a stand-alone agent and in concomitant chemotherapy [52–54].
Mechanistic Target of Rapamycin Inhibitors
Although medicinal chemists have made progress in perturbing the PI3K/AKT/mTOR (phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B/mechanistic target of rapamycin) pathway through the development of synthetic compounds, the only mTOR inhibitors currently FDA approved for chemotherapeutic intervention are analogs of the macrolide rapamycin, a natural product initially isolated from Streptomyces hygroscopicus in 1975 within the soils of Easter Island (also referred to as Rapa Nui, giving rise to the name of the agent) [55–57]. Although currently used as an immunosuppressive agent, rapamycin has also demonstrated marked cytostatic activity against several cancer types [58]. Its unique pharmacokinetic profile has prevented rapamycin from being further developed as an antineoplastic agent, but its potent mechanisms of action have inspired the development of temsirolimus, a novel soluble rapamycin derivative that has a favorable toxicity profile in mammalian models, thereby potentiating the use of mTOR inhibitors in chemotherapy. As of now, only temsirolimus and the later developed analog everolimus have received FDA approval as antineoplastic agents.
Rapamycin is a complex 21-member macrolide lactone that contains a pipecolate moiety in the upper left region of the molecule. The agent inhibits mTOR primarily by crosslinking FKBP-12 (12 kDa FK506 binding protein) via its methoxy functional group [59, 60]. The high affinity of rapamycin to FKBP-12 is mediated in part by its pipecolate region, which hydrogen bonds at two different hydrophobic binding pockets, as revealed by X-ray crystallography [61–63]. Once bound, the rapamycin/FKBP-12 complex blocks the binding of the accessory protein RAPTOR (regulatory-associated protein of mTOR) to mTOR, necessary for downstream phosphorylation of S6K1 and 4EBP1. Consequently, S6K1 dephosphorylates, which reduces protein synthesis and decreases cell motility and size [64–66]. In addition, rapamycin induces dephosphorylation of 4EBP1. Such activity potentiates increases in p27, and decreases in cyclin D1 expression, invoking late blockage of G1/S during the cell cycle [66]. Although it is clear that rapamycin induces apoptosis in neoplastic cells, the molecular mechanism of apoptosis has not yet been fully resolved.
Temsirolimus has been an important addition to chemotherapeutic protocols indicated for renal cell carcinoma (RCC) owing to the importance of mTOR in carcinogenesis; in RCC tumors, activated mTOR further exacerbates accumulation of HIF-1α (hypoxia-inducible factor 1-α) by increasing synthesis of this transcription factor and its angiogenic target gene products [67]. As such, rapamycin and its analogs have demonstrated notable antiangiogenic activity [68], indicating the potential of combining these congeners with agents that elicit similar effects on neoplastic vasculature, including bevacizumab, sorafenib, and sunitinib [69, 70]. In addition to its utility in RCC therapy, temsirolimus is being clinically evaluated against other carcinomas known to have elevated mTOR activity, including malignancies of the breast and lung [71, 72]. Everolimus (Fig. 1) is an orally (p.o.) administered 40-O-(2-hydroxyethyl) derivative of rapamycin that is currently indicated for postmenopausal women with advanced hormone receptor positive and estrogen receptor negative breast carcinoma, as well as neuroendocrine tumors of pancreatic origin, RCC, renal angiomyolipoma, and both pediatric and adult subependymal giant cell astrocytoma [18]. The agent has also been clinically evaluated against gastric adenocarcinoma, hepatocellular carcinoma, and in multiple types of lymphoma [73–75].
Protein Synthesis Inhibitor Omacetaxine Mepesuccinate
Omacetaxine mepesuccinate (homoharringtonine) is a natural ester of the alkaloid cephalotaxine, a compound initially isolated and characterized in 1969 from Cephalotaxus harringtonia (Japanese plum yew) [76] (Fig. 1). Although cephalotaxine itself does not exhibit antineoplastic activity, fractionations of extracts obtained from several variants of C. harringtonia produced a series of cephalotaxine esters that demonstrated antineoplastic activity. One of these compounds, homoharringtonine (later renamed omacetaxine mepesuccinate), was shown to influence the progression of acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) in China during the 1970s, with later studies in the United States confirming these findings [77, 78]. However, the clinical development of omacetaxine mepesuccinate was halted after the development of imatinib, which has since shown remarkable activity in patients with Philadelphia chromosome positive CML and acute lymphoid leukemia (ALL). Nevertheless, reemergence in the investigation of omacetaxine mepesuccinate quickly resumed once it was realized that a subset of indicated leukemias are either refractory or develop resistance to imatinib or related tyrosine kinase inhibitors (TKIs), particularly T315I subtypes.
Omacetaxine mepesuccinate elicits its antineoplastic effects through inhibition of protein synthesis. Specifically, the agent prevents aminoacyl-tRNA from binding the ribosomal acceptor site, thereby preventing peptide bond formation at the early stage of protein elongation [79]. In addition, omacetaxine mepesuccinate inhibits the elongation phase of translation by preventing substrate binding to the acceptor site on the 60s ribosome subunit, leading to the blockade of aminoacyl-tRNA binding and peptide bond formation [77, 80]. Further, it has been demonstrated that the agent blocks protein synthesis by competing with the amino acid side chains of incoming aminoacyl-tRNAs for binding at the A-site of formed ribosomes [81]. Interestingly, omacetaxine mepesuccinate has shown notable activity against leukemic stem cells (LSCs) of CML origin with the agent having a similar inhibitory effect on BCR-ABL T315I-expressing LSCs in comparison to non-mutant BCR-ABL-expressing LSCs [82], indicating that the agent may be able to markedly inhibit clonal expansion in select CML patients.
Unlike most antineoplastic agents, omacetaxine mepesuccinate is administered subcutaneously (s.c.). with the standard regimen being 1.25 mg/m2 s.c. b.i.d., days 1–14 every 28 days, with a maintenance treatment of 1.25 mg/m2 s.c. b.i.d. for 7 days every 28 days [79]. Omacetaxine mepesuccinate disappears rapidly from plasma after cessation of infusion, with an observed α-t1/2 of 5 h, a β-t1/2 of 9.3 h, and a mean steady-state terminal t1/2 of 7 h with biexponential decay observed. The agent undergoes a rapid metabolism with urinary excretion representing about 12–15 % of the administered dose. In addition, omacetaxine mepessucinate has a favorable toxicity profile with effects on liver and cardiovascular function being the most prominent [79–81, 83]. Owing to its efficacy in TKI-resistant cells, omacetaxine mepesuccinate is FDA approved for both chronic- and blast-phase CML [18], and clinical trials are currently ongoing to determine optimal agents to use in combination with the protein synthesis inhibitor. Interestingly, not as much interest has been paid towards the potential of omacetaxine mepesuccinate in Philadelphia chromosome-positive ALL that is unresponsive to TKI therapy, and may be an avenue of future clinical interest.
Nucleic Acid-Directed Trabectedin
Trabectedin is a novel antineoplastic agent that was isolated from the sea squirt Ecteinascidia turbinate in 1984 [84, 85], and has a relatively complex structure; three tetrahydroisoquinoline moieties, eight rings including one 10-membered heterocyclic ring containing a cysteine residue, and seven chiral centers (Fig. 1). While the E. turbinate extract that trabectedin is derived from was shown to have antineoplastic activity as early as 1969 [84], separation and characterization of the active molecules was not feasible until the development of sufficiently sensitive techniques. Further delaying its development was the fact that yields from E. turbinate are extremely low; it takes 1000 kg of animals to isolate 1 g of trabectedin. It was not until the development of synthetic methods of preparation that actual clinical investigation was feasible. The current supply of trabectedin is based on a semisynthetic process starting from Safracin B, an antibiotic obtained by fermentation of the bacterium Pseudomonas fluorescens [85].
Trabectedin is extremely potent, requiring only picomolar to low nanomolar concentrations to initiate cell death in various cancer types in vitro [86–88]. This notable cytotoxicity is attributed to at least two separate actions; DNA alkylation and inhibition of transcription. The agent binds the minor groove of DNA, showing a preference for GG- and GC-rich regions, and then alkylates the exocyclic N-2 on guanine [89, 90]. This alkylation step is dependent on the dehydration of the carbinolamine (also referred to as hemiaminal) group found on trabectedin, as this potentiates the formation of an electrophilic iminium intermediate that attracts nucleophilic DNA bases. Two subunits of trabectedin form the primary contacts with DNA, while another subunit protrudes out of the minor groove [91]. It is this subunit that has been associated with the inhibition of transcription, as interaction between the subunit and transcription factors has been observed. Alkylation of DNA produces the standard monoalkylating single-strand breaks that proceed to double-strand breaks when the adduct is recognized by the transcription-coupled nucleotide excision repair (TC-NER) complex [92, 93]. These in vitro observations suggest that patients with BRCA mutations may receive benefit from trabectedin, as BRCA1 locates DNA damage and attracts the TC-NER complex to repair DNA breaks, while BRCA2 mediates homologous recombination by loading other proteins to the double-strand break sites and stalled DNA replication forks [94, 95]. This postulation has been confirmed in select patients with ovarian carcinoma [93], making trabectedin particularly attractive for certain subtypes of breast, lung, and ovarian carcinoma with BRCA mutations.
Separating trabectedin from other alkylating agents is its ability to inhibit the expression of potentially oncogenic transcription factors, including those that code for oncogene products, c-myb, maf, and myc) and cell-cycle related factors (E2F and SRF), and general transcription factors (SCR, NF-Y, SXR, and Sp1) [96]. Its inhibition of P-gp and heat shock protein 70 (Hsp70) expression through NF-Y (as well as SXR, which is also responsible for P-gp transcription) interaction are of particular intrigue [97, 98], as both are integral to neoplastic drug resistance. Further, trabectedin has profound activity on the tumor microenvironment, exerting effects on quiescent tumor-promoting monocytes and macrophages that is atypical of a DNA-directed alkylating agent [96].
Trabectedin has linear pharmacokinetics when administered as a 24-h i.v. infusion as is seen in the standard dosing schedule of 1.5 mg/m2 over 24 h every 3 weeks [99]. In addition, trabectedin is extensively bound to human serum albumin, and the concentration of unbound trabectedin in the plasma at clinically relevant trabectedin plasma concentrations is in the picomolar range. Drug metabolism presides mainly in the liver, with a large number of metabolites being produced by cytochrome P450 isozyme 3A4 and to a lesser extent by other cytochrome P450 isozymes and phase II enzymes [99].
Trabectedin is approved in Europe (including Russia) and South Korea for the treatment of advanced soft-tissue sarcoma. This clinical indication has been validated by multicenter phase II trials and a number of noncomparative phase II trials [99, 100]. In addition, the European Commission and the FDA have granted orphan drug status to trabectedin for soft-tissue sarcomas and ovarian carcinoma. The effects of trabectedin against ovarian carcinoma are particularly intriguing; coadministration of trabectedin and pegylated liposomal doxorubicin was associated with a significantly longer (6 weeks; p = 0.019) median progression-free survival than pegylated liposomal doxorubicin monotherapy in patients with recurrent ovarian carcinoma after progression on platinum-based chemotherapy [101]. In addition, concomitant administration of trabectedin and pegylated liposomal doxorubicin was associated with a relative risk reduction (RRR) in disease progression or death of 21 % (HR 0.79; 95 % CI 0.65, 0.96) in comparison to the doxorubicin monotherapy. These findings are encouraging due to the fact that ovarian carcinomas usually portend a poor prognosis (overall 5-year survival rate is 44 %, while stage IV invasive ovarian carcinoma has a 5-year survival rate of 17 %) [102]. The dearth of available treatment options for these patients is apparent, warranting further investigation of trabectedin and other novel therapeutic strategies. The agent has also been investigated in breast carcinoma, lung carcinoma, prostate adenocarcinoma [103–105], and various pediatric sarcomas (Ewing’s sarcoma, and rhabdomyosarcoma) [106].
Finding Novel Utility in Natural Products through Improved Drug Delivery
The natural products discussed so far are potent antineoplastic agents, but are all inherently limited by their untargeted cytotoxic mechanisms. Although malignant cells are preferentially damaged as a result of increased proliferation rates and increased uptake of the given agent, normal tissue is also perturbed, preventing higher concentrations that could elicit more antitumor activity from being administered. Further, there are natural products too potent for clinical use because their activity is not specific enough for neoplastic cells to garner any therapeutic benefit. Finding a delivery system that discriminates between neoplastic and normal cells, and then transports a cytotoxic agent across the plasma membrane of aberrant cells to induce apoptosis would therefore be an ideal situation. As it turns out, the idea of preferentially delivering highly cytotoxic natural products to neoplastic tissue has made commendable progress in recent years owing to advances in immunotherapy. By using epitopes highly expressed on the cell surface of malignant cells, investigators have been able to develop methods capable of transporting a drug payload to the intended target, substantially reducing the toxicity of these agents had they not been conjugated to the delivery protein. This targeted drug delivery can be accomplished through two mechanisms; engineered cytokine proteins and ADCs.
Engineered Cytokine Proteins
Cytokines are small proteins (~5–20 kDa) important for various paracrine and autocrine signaling throughout the body, but are most associated with their role in the immune system. These proteins enable leukocytes to communicate with one another to generate a coordinated, robust, but self-limited response to a target antigen [107, 108]. Cytokines themselves have been used for years in chemotherapy because of their ability to directly stimulate immune effector cells and stromal cells at the tumor site, and enhance neoplastic cell recognition by cytotoxic effector cells [108–110]. Interleukin-2 (IL-2) is particularly appealing in hematological malignancies because the IL-2 receptor (IL-2R) is selectively expressed on activated T lymphocytes, B cells, and natural killer cells [111]. In a normal patient, IL-2R is expressed mostly at a low level in less than 5 % of normal circulating peripheral blood mononuclear cells [112]. However, many transformed leukocytes have a high expression of high or intermediate affinity IL-2R isoforms, particularly in cutaneous T-cell lymphoma (CTCL) in which approximately 50 % of cases express this phenotype as demonstrated by immunohistochemical staining [112, 113]. Therefore, fusing a highly cytotoxic agent to IL-2 may be an effective method by which to target CTCL, while eliciting minimal toxicity to non-hematological tissue.
Denileukin Diftitox
Denileukin diftitox is a fusion protein in which the receptor binding domain of diphtheria toxin is exchanged for that of IL-2 (Fig. 1) [114, 115]. Diphtheria toxin is produced naturally in the pathogen Corynebacterium diphtheriae, and is a single polypeptide chain of 535 amino acids consisting of two subunits linked by disulfide bonds, known as an A–B toxin [116, 117]. The less stable B unit binds the cell surface of a target cell, enabling the more stable A subunit to penetrate the plasma membrane. Once internalized, the diphtheria toxin catalyzes the transfer of NAD+ to a diphthamide residue in eukaryotic elongation factor-2 (eEF2) through ADP-ribosylation, resulting in its deactivation (the same function of endogenous NAD+-diphthamide ADP-ribosyltransferase) [118]. In essence, this action halts translation, as eEF2 facilitates the movement of the peptidyl tRNA-mRNA complex from the A site of the ribosome to the P site during protein synthesis; thereby potently inhibiting the production of new proteins. Consequently, diphtheria toxin is extraordinarily potent with the average human lethal dose being ~0.1 μg/kg [118].
Through human innovation, one of the deadliest toxins in nature can be harnessed for cancer therapy once it is given the appropriate delivery system. Denileukin diftitox contains the full-length sequence of IL-2, as well as protein fragments of diphtheria toxin; 97 amino acids from the native part of the toxin containing the disulfide bond are removed to increase the half-life and affinity of the compound to its target receptor [119, 120]. After diphtheria toxin and IL-2 are fused together, the agent seeks out CD25-bearing cells, as CD25 represents the high-affinity α-subunit of the IL-2 receptor. CTCL cells internalize the agent via receptor-mediated endocytosis after it binds IL-2R, and is subsequently acidified inside the vesicle. This process releases diphtheria toxin fragment A into the cytoplasm, enabling the internalized fragment A to catalyze the transfer of ADP to eEF2. Premature ADP-ribosylation of eEF-2 inhibits further protein synthesis, ultimately potentiating apoptosis [121, 122].
There are limitations to the delivery mechanism by which denileukin diftitox acts. In addition to the expected immunosuppression and potential for bacterial infections, the agent is known to cause acute hypersensitivity-type reactions, asthenia, and nausea/vomiting [123]. Further, the use of IL-2 as a method to selectively transfer diphtheria toxin into CTCL cells is inherently limited. IL-2Rs are classified into three subtypes based on their affinity for IL-2. Each subtype is composed of a combination of subunits: α (CD25 and p 55), β (CD122 and p 75), and γ (CD132 and p 64). Intermediate and high-affinity receptors are composed of IL-2R-β/γ (CD122 and CD132) and IL-2R-α/β/γ (CD25, CD122, and CD133) subunits, respectively, and both perpetuate internalization and signal transduction. By contrast, low-affinity receptors consist of the IL-2R-α/γ (CD25 and CD132) subunits that bind IL-2, but do not cause internalization or activation [112, 124, 125]. Therefore, the presence of the β and γ (CD122 and CD132) subunits is essential for sensitivity and internalization of the fusion toxin. If CTCL cells are lacking appropriate receptors, denileukin diftitox will be ineffective, paving the way for potential drug resistance.
Nevertheless, denileukin diftitox has elicited notable responses in patients, and the agent is currently FDA approved for persistent or recurrent CTCL in patients expressing the critical CD25 component of IL-2R [18]. Because the expression of IL-2R is vital for the T-cell targeting of denileukin diftitox, strategies to upregulate IL-2R expression are of particular clinical interest. Retinoids are a particularly attractive concomitant agent, as they have immunomodulatory function and have been shown to increase IL-2R expression on T-cells [126, 127]. It has been demonstrated that both bexarotene and alitretinoin are modulators of high affinity IL-2R expression, as the agents increase the expression of CD25 and CD75 in CTCL cells. These CTCL cells can be subsequently exposed to denileukin diftitox, resulting in a 50–70 % decrease in protein synthesis [126]. This is particularly exciting because bexarotene is already standard therapy for patients with early- and advanced-stage CTCL. The concomitant use of denileukin diftitox and retinoids is currently being clinically investigated [128, 129], and may potentiate a novel avenue of chemotherapy for CTCL patients.
Antibody-Drug Conjugates
Immunoconjugates are a relatively novel class of chemotherapeutic agents that are gaining increasing interest because of their efficacy and reduced side effects in comparison to some traditional cytotoxic agents. If traditional chemotherapy is equivalated to indiscriminate bombing, immunoconjugates are roughly analogous to modern day missile guidance systems. These agents consist of three main components; the monoclonal antibody (mAb; guidance system), the degradable linker (delivery mechanism), and the cytotoxic agent (warhead) that can either be a toxin or a radioisotope. While not completely specific to neoplastic tissue, immunoconjugates target a given epitope based on the mAb that is chosen, enabling the use of cytotoxic agents that would likely elicit too broad of a toxicity profile in a typical patient. Although the scope of this review limits commentary to ADCs, radioimmunotherapy has shown efficacy in a variety of cancers, and the reader would benefit from the following reviews [130–132].
Brentuximab Vedotin
Brentuximab vedotin is a conjugate of a humanized CD30-targeting mAB and monomethyl auristatin E (MMAE) linked via a cathepsin cleavable linker (valine-citrulline) and a para-aminobenzylcarbamate (PABC) spacer (the name vedotin refers to MMAE plus its linking structure to the mAB) [133–136]. MMAE is a potent mitotic inhibitor derived from the naturally occurring dolastatin 10 that was isolated from Dolabella auricularia (wedge sea hare) in 1987 [137]. Several related molecules have since been produced through total syntheses, establishing the drug class auristatin [138]. As with vinca alkaloids, the auristatins exert antineoplastic activity by inhibiting tubulin polymerization. However. these agents are much more toxic (MMAE is 200 times more potent than vinblastine) [139], substantially limiting their clinical utility. MMAE was subsequently developed with a built-in functionality for stable linker attachment, and still retains high potency in addition to exhibiting water solubility and stability under physiological conditions [133]. The ability to conjugate MMAE to mABs and potentially other proteins has considerably increased the utility of auristatins, enabling these once intolerable agents to be directed towards the intended target, thereby dramatically reducing unintended toxicity.
CD30 is an ideal target for ADCs because the antigen is a tumor necrosis factor receptor that stimulates apoptosis, and is highly expressed in Hodgkin’s lymphoma, systemic anaplastic large cell lymphoma, CTCL, and other selected lymphoid tumors [140, 141], as well as in some non-lymphoid malignancies including germ cell tumors [141, 142]. Further, cross-reactivity of CD30 on normal tissues is very low, with some expression on activated T and B lymphocytes; this expression is not observed on resting T and B lymphocytes [141]. In addition to the CD30-targeting mAB, brentuximab vedotin employs a protease-cleavable linker because protease activity is abundant in lysosomes, where ADCs are often directed [133]. In addition, protease activity is significantly reduced outside of cells owing to secreted protease inhibitors [141]. MMAE is attached to suitable degradable peptides through the built-in N-terminal amine functionality via the self-immolative spacer (PABC). The spacer is required so that the cleavable peptide is situated away from MMAE to allow facile proteolysis. Val-Cit (the clevable peptide) is stable in plasma, but is rapidly hydrolyzed via proteolysis by cathepsin in lysosomes [143]. Upon peptide cleavage, the PABC group rapidly fragments, releasing the highly cytotoxic MMAE into the cytoplasm.
Brentuximab vedotin is currently indicated for Hodgkin’s lymphoma after failure of an autologous stem cell transplant or after failure of two prior multiagent chemotherapeutic regimens in those who are not suitable for autologous stem cell transplantation, as well as systemic anaplastic large cell lymphoma. Due to the prevalence of CD30 in various malignancies, brentuximab vedotin is also being investigated in patients with CTCL, other CD30-positive hematological malignancies, and CD30-positive germ cell tumors [144].
Trastuzumab Emtansine
Trastuzumab emtansine is an ADC consisting of the humanized HER2/Neu-targeting mAB trastuzumab linked to mertansine (DM1) via SMCC (succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate), a heterobifunctional crosslinker (Fig. 1) [145, 146]. While trastuzumab is already effective against HER2-positive breast carcinomas, esophageal carcinomas, and gastric adenocarcinomas [147, 148], conjugating the mAB with the highly cytotoxic DM1 is an effective method to generate activity in refractory or relapsed tumors [149–151]. DM1 refers to mertansine, a derivative of maytansine that has been chemically modified to have a terminal thiol for conjugation. Maytansine is a macrolide of the ansamycin type (contains an aromatic moiety bridged by an aliphatic chain) that was originally isolated in 1972 from the flowering plant Maytenus ovatus [152]. Similar to MMAE, maytansine is a highly potent microtubule inhibitor that by itself lacks the tumor specificity required to elicit therapeutic benefit [153, 154]. It was not until the development of ADCs that the extremely cytotoxic potential of maytansine could be harnessed in the form of conjugate-compatible DM1.
Trastuzumab is a logical choice for ADC enhancement because its target HER2/Neu is preferentially overexpressed in certain subtypes of breast carcinoma, esophageal carcinoma, and gastric adenocarcinoma. In particular, gain- and loss-of-function experiments, as well as immunohistochemistry analyses have indicated HER2 amplification as a driving event in the onset and progression of as much as 25–30 % of breast carcinomas [155, 156]. Further, this subtype of breast carcinoma is often very aggressive and resistant to traditional cytotoxic chemotherapy [157, 158]. Due to its apparent oncogene addiction, targeting HER2 positive breast carcinomas through this vital receptor has become an attractive approach for therapeutic intervention. Each molecule of trastuzumab emtansine consists of a single trastuzumab mAB with several molecules of DM1 attached; trastuzumab may be conjugated with up to 8 DM1 molecules (three to four on average) [159, 160]. SMCC contains two reactive functional groups, a succinimide ester and a maleimide. The succinimide group of SMCC reacts with the free amino group of a lysine residue in the trastuzumab molecule, and the maleimide moiety of SMCC links to the free sulfhydryl group of DM1, forming a covalent bond between the mAB and DM1 [160].
After binding HER2, trastuzumab emtansine gains entry to the cellular interior via receptor-mediated endocytosis [161]. Since the non-reducible SMCC linker is stable in circulation, as well as in the tumor microenvironment, DM1 release occurs only as a result of proteolytic degradation of the mAB part of trastuzumab emtansine in the lysosome. Following release from the lysosome, DM1-containing metabolites potently inhibit microtubule assembly, causing cell-cycle arrest at the G2/M checkpoint, eventually triggering apoptotic mechanisms [162, 163]. Importantly, linkage of DM1 to trastuzumab does not affect the binding affinity of trastuzumab to HER2, nor does it reduce the inherent antineoplastic effects of trastuzumab [162, 164, 165]. Consequently, trastuzumab emtansine benefits from the mechanisms of both trastuzumab and DM1.
Trastuzumab emtansine is currently FDA approved for HER2-positive breast carcinoma, HER2-positive metastatic gastric adenocarcinomas, and gastroesophageal adenocarcinoma with HER2 overexpression [18]. In the USA, trastuzumab emtansine is typically reserved for the treatment of HER2-positive metastatic breast carcinoma in patients who have been treated previously with trastuzumab and a taxane (paclitaxel or docetaxel), and who have already been treated for metastatic breast cancer or developed tumor recurrence within 6 months of adjuvant therapy [166]. This is due in part to the expense of trastuzumab emtansine treatments, as protocols can cost patients US $9800 a month, or US $94,000 for a typical course of treatment. Since trastuzumab and pertuzumab (another HER2/Neu-targeting mAB) bind different epitopes [167, 168], clinical investigation has also been extended to concomitant chemotherapy consisting of both trastuzumab emtansine and pertuzumab against unresectable HER2-positive breast carcinoma that is locally advanced or metastatic [169].
Calicheamicin Conjugated Monoclonal Antibodies
The cytotoxic agents available for mAB conjugation are not limited to highly potent microtubule inhibitors. Perhaps the most notable example is calicheamicin conjugated mABs. Calicheamicins are a class of enediyne nucleic acid-directed agents that were initially characterized in 1989 from the fermentation broth of the bacterium Micromonospora echinospora [170]. Since then, calicheamicin γ1 and the related enediyne esperamicin have become known as two of the most potent antineoplastic agents in existence [171]. As is the case with most extremely cytotoxic agents, their utility would come not as stand-alone agents, but rather as components of epitope-targeting ADCs. The initial attempt to harness the potency of calicheamicins was with gemtuzumab ozogamicin, a humanized CD33-targeting mAB conjugated to a carbohydrate conjugate of N-acetyl-γ calicheamicin dimethyl hydrazide via a bifunctional AcBut linker (4-(4′-acetylphenoxy)butanoic acid) [172, 173]. Gemtuzumab ozogamicin was the first FDA-approved ADC, being approved for the treatment of AML in 2000. Unfortunately, this initial attempt also elicited marked unintended toxicity (most notably the potentiation of sinusoidal obstruction syndrome) [174–176], and was withdrawn from the market in 2010 after a randomized, phase III comparative controlled trial demonstrated that the agent increased patient death and added no benefit over conventional cancer therapies [177]. Nevertheless, an additional calicheamicin conjugated mAb has since been developed (inotuzumab ozogamicin), and calicheamicin congeners are still being investigated for their antineoplastic potential.
The extreme potency of calicheamicins can be traced back to their unique mechanism of action. All calicheamicins appear to interact with cellular DNA and initiate double-strand cleavage by carbon-centered diradical hydrogen abstraction processes [178, 179]. Calicheamicins bind DNA in the minor groove, wherein they undergo a reaction analogous to a Bergman cyclization to generate a diradical species. This diradical, 1,4-didehydrobenzene, then abstracts hydrogen atoms from the deoxyribose backbone of DNA, ultimately resulting in irreversible strand scission [180]. The affinity of calicheamicin γ1 for the minor groove of DNA is due its aryltetrasaccharide domain [181, 182]. Specifically, calicheamicin γ1 binds the DNA minor groove with its aryltetrasaccharide domain in an extended conformation spanning TCCT/AGGA segments of DNA. Calicheamicin γ1 then inserts itself in an edgewise manner deep into the minor groove with the molecule wedged between the walls of the groove [182]. A range of intermolecular hydrophobic and hydrogen-bonding interactions are also observed, accounting for the sequence-specific recognition in the complex.
In addition to gemtuzumab ozogamicin, another calicheamicin-conjugated humanized mAB, inotuzumab ozogamicin has reached phase III clinical trials. Rather than targeting CD30, inotuzumab ozogamicin consists of a humanized mAb that recognizes the CD22 antigen. Nevertheless, this mAB is still conjugated to N-acetyl-γ-calicheamicin dimethyl hydrazide via the acid labile 4-(4′-acetylphenoxy)butanoic acid linker [183]. Since CD22 is a B-lymphocyte-restricted phosphoglycoprotein [184], inotuzumab ozogamicin has been clinically examined in B-cell leukemias and lymphomas with reversible thrombocytopenia as the main toxicity observed. [185, 186]. It should be noted that CD22 may be expressed in certain lung carcinomas, but this is still inconclusive, as the data have been conflicting [187, 188]. The ADC has shown notable efficacy against pediatric and adult ALL resistant to traditional cytotoxic chemotherapy [186, 189], and is indicative of the potential ADCs have in previously unresponsive cancers.
Exotoxin Conjugates
Antibody/toxin conjugates are not limited to small-molecule cytotoxic agents. In a manner similar to denileukin diftitox, mABs have now been successfully conjugated to large protein bacterial exotoxins, thereby increasing the diversity of available immunotoxins. The two most well-known examples are moxetumomab pasudotox and SS1P.
Moxetumomab pasudotox is a recombinant immunotoxin composed of the Fv fragment of a fully human CD22-targeting mAB fused to a 38-kDa fragment of Pseudomonas exotoxin A [190]. Moxetumomab pasudotox is actually an improved, more active form of a predecessor recombinant immunotoxin, BL22 (also called CAT-3888), which produced complete remission in relapsed/refractory hairy cell leukemia (HCL), but had a <20 % response rate in chronic lymphoid leukemia (CLL) and ALL, which are noted for containing much lower numbers of CD22 [191, 192]. Compared with BL22, moxetumomab pasudotox is up to 50-fold more active against CLL and HCL [193].
Pseudomonas exotoxin is a highly cytotoxic product of the pathogenic bacterium Pseudomonas aeruginosa [194]. With a mechanism very similar to diphtheria toxin, the exotoxin potently inhibits eEF2 through ADP-ribosylation, thereby ceasing further elongation of polypeptides. Crystallographic studies have demonstrated that Pseudomonas exotoxin is made up of three major structural domains; domain Ia is the cell binding domain, domain Ib contains no known function, domain II contains a furin site necessary to release domain III from the cell binding domain, and domain III contains the ADP-ribosylating activity that inactivates eEF2 [194].
In recombinant immunotoxins targeting CD22 (BL22 and moxetumomab pasudotox), domain Ia of PE is removed and replaced by the Fv portion of a mAB reacting with CD22 [190]. Because BL22 was limited by the lower expression of CD22 in CLL and ALL, the mAB portion of the agent was improved. By mutating three residues in the heavy chain of the BL22 Fv (residues 100, 100a, and 100b), investigators were able to increase the affinity of the mAB to HCL by about 15-fold and cytotoxicity toward HCL and CLL cells by up to 50-fold [193]. This improvement in binding affinity and increased cytotoxicity did not interfere with pharmacokinetics or off-target toxicity, indicative of superior antineoplastic potential. Moxetumomab pasudotox has produced complete responses against CD22 positive leukemias in the clinical setting, demonstrating particular promise with relapsed and refractory HCL [195]. Phase III evaluations are currently underway to fully assess the efficacy of moxetumomab pasudotox in comparison to more established therapeutic approaches.
In addition to its potential with CD22-targeting mAB Fvs, the pseudomonas exotoxin has also been fused with an anti-mesothelin Fv to create SS1P. Mesothelin is a 40-kDa cell surface glycoprotein present on some normal mesothelial cells lining the pleura, peritoneum, and pericardium [196, 197]. However, mesothelin is also highly expressed in several malignancies, including epithelial mesotheliomas (~100 % of cases), lung carcinomas (~50 % of cases), ovarian carcinomas (~70 % of cases), and pancreatic/biliary adenocarcinomas (~100 % of cases) [198–201], thereby being an epitope of particular antineoplastic interest. SS1P acts by the same mechanism as moxetumomab pasudotox and is very similar in structure, but is specific for mesothelin-positive cells. This epitomizes the utility of immunoconjugates, as the same highly toxic small molecule or protein can be directed towards different targets once linked to an appropriate effector. Demonstrating potent antitumor activity in preclinical in vivo models [202–204], SS1P has since progressed towards clinical examination. Owing to its response in mesothelioma patients during phase I trials [205–207], SS1P has gained particular attention in the treatment of this malignancy. Nevertheless, SS1P has also been clinically investigated in other cancers with high mesothelin expression, and the agent may gain traction as a multipurpose immunotoxin.
Natural Products with Mechanisms Not Currently Seen in the Clinical Setting
Although the diversity of natural products currently used in the clinical setting is considerably immense, it is not comprehensive. There are in fact many other potential targets inherent to cancer pathology that could be exploited through the implementation of novel natural products. One target of considerable importance is the cytoskeleton. Not only do many malignant cells have a perturbed cytoskeleton owing to the effects of dysplasia and subsequent anaplasia [208, 209], but the cytoskeleton has also been indicated in oncogenic signaling and metastatic progression [210–212]. While microtubule-directed agents have been a mainstay in traditional cytotoxic chemotherapy, they are inherently limited to one component of the cytoskeleton. The other potential targets, intermediate filaments and microfilaments, have remained as elusive clinical prospects.
Cytoskeletal filaments are indeed viable targets to exploit in chemotherapy. Microfilaments are inherently required for cell motility, cytokinesis, and many other processes vital for malignant cell stability [213–216]. Intermediate filaments such as keratins are often overexpressed in carcinomas as a result of the aberrant effects of associated oncogenes [217, 218], and vimentin has been shown to be vital for cell survival in numerous experiments [219–221]. Although there has yet to be a microfilament- or intermediate filament-directed agent approved for clinical use, nature offers potential solutions to these previously elusive targets. Through the course of evolution, a considerable assortment of small molecules has been developed by various organisms that perturb the dynamics of actin polymerization or intermediate filament formation, and have demonstrated profound preclinical antineoplastic activity. A comprehensive review of cytoskeletal filament-directed agents available for further preclinical investigation has already been compiled [222]. Therefore, the potential of exploiting each currently untargeted component of the cytoskeleton will be briefly highlighted by two drug classes; cytochalasins and withanolides.
Cytochalasins
As indicated by both in vitro and in vivo investigation, microfilaments are vital for the progression of many malignancies, and are therefore a suitable target for chemotherapy. One of the most studied drug classes of microfilament-directed agents has been the cytochalasins, which were initially characterized in 1967 as being biological metabolites of the molds Helminthosporium dematiodeum (cytochalasins A and B) and Metarrhizium anisopliae (cytochalasins C and D) [223]. It has since been discovered that these mycotoxins potently inhibit actin polymerization, affecting activities ranging from cell motility and adhesion to cytokinesis. Since their initial discovery, more than 60 different cytochalasins from several species of fungi have been classified into various subgroups based on the size of the macrocyclic ring and the substituent of the perhydroisoindolyl-1-one residue at the C-3 position [224]. While all cytochalasins demonstrate the propensity to bind filamentous (F)-actin and block polymerization, only cytochalasins B and D have been extensively studied for their chemotherapeutic potential. Both congeners bind the fast growing barbed end of microfilaments, essentially fulfilling the role of capping proteins that prevent further actin polymerization once the filament has grown to a sufficient length [222]. However, cytochalasin D is more potent than cytochalasin B, reflected by cytochalasin B being 20-fold less toxic in mice than cytochalasin D [225], a pharmacological property attributed to the affinity cytochalasin D also has for globular (G)-actin.
Although only a handful of laboratories have investigated the potential of cytochalasins as antineoplastic agents, enough studies have been performed to establish mechanisms by which these agents exert their antineoplastic effects (Fig. 2). In preclinical mammalian models of malignancy, cytochalasins B and D exhibit marked antitumor and antimetastatic activity in murine melanomas (B16BL6 and B16F10), lung carcinomas (LA4, Lewis Lung, and M109), leukemias (P388 and P388/ADR), and M5076 sarcoma administered intraperitoneally, intravenously, or subcutaneously [226–239]. In addition, the pharmacokinetics, tissue distribution, and potential toxicities of cytochalasin B and liposome-encapsulated cytochalasin B have been extensively characterized [228–231]. The only major toxicity consistently elicited by cytochalasin B is marked immunosuppression, which has been characterized in multiple murine models [230, 231]. Nevertheless, this in vivo toxicity can be markedly reduced either through the administration of human recombinant IL-2, or liposome encapsulation [228, 229, 231]. Further, liposome encapsulated cytochalasin B shows equal, or even superior antitumor activity in M109 lung carcinoma in vivo when compared with the non-encapsulated compound [228]. It has also been demonstrated by Huang et al. [232] that a pegylated formulation of cytochalasin D is more water soluble, accumulates in tumor tissue more efficiently, and has a much longer t1/2 than natural cytochalasin D (4 h vs. 10 min), while still retaining the antitumor activity of the natural compound.
Fig. 2.
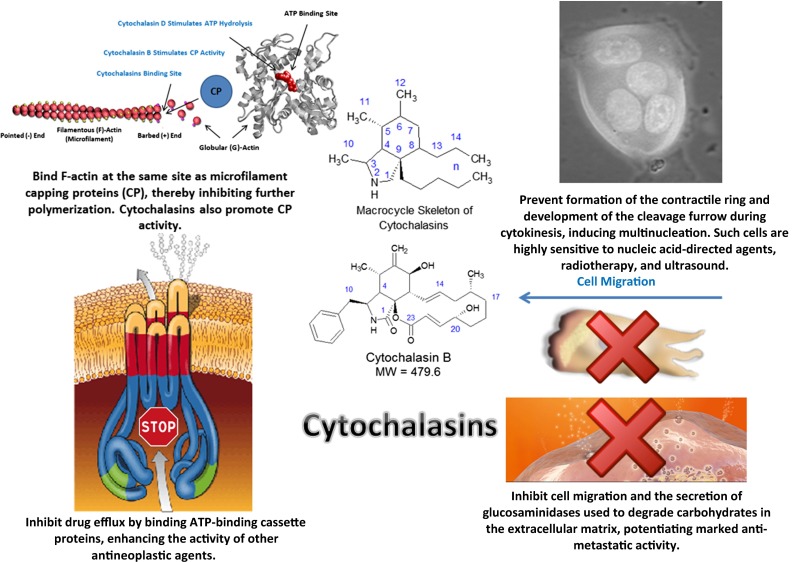
Antineoplastic mechanisms of microfilament-disrupting cytochalasins. MW molecular weight
Withanolides
Along with microfilaments, intermediate filaments are the other component of the cytoskeleton that has yet to be exploited in the clinical setting. It has been repeatedly demonstrated that a variety of malignancies have aberrant or an elevated expression of intermediate filaments keratin (type I or II), nestin (type VI), and vimentin (type III) [233–242]. Although no potential antineoplastic agent has been identified to specifically target aberrant keratin or nestin levels in malignant cells, withanolides have shown promise as a potent type III intermediate filament inhibitor.
The most promising clinical prospect of this drug class is withaferin A, a steroidal lactone that was initially isolated from Withania somnifera (winter cherry) in 1965 [243], and was the first member of the withanolides to be discovered. Withaferin A has been shown to potently inhibit a variety of proteins, with the most notable target being vimentin and other type III intermediate filaments [244–248] (Fig. 3). In addition to its affinity for intermediate filaments, withaferin A has been shown repeatedly to inhibit angiogenesis [247–251], with potent anti-angiogenic activity being exerted at doses as low as 7 µg/kg/day intraperitoneally in C57BL/6J mice [250].
Fig. 3.
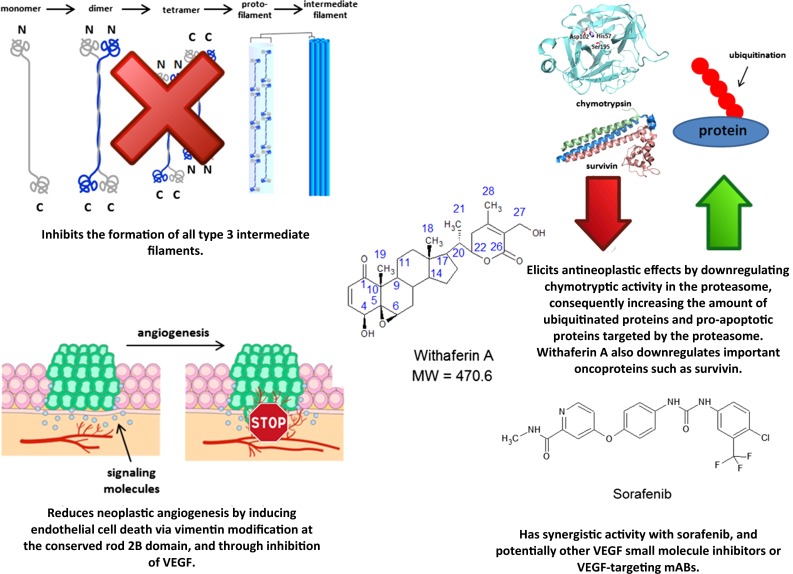
Antineoplastic mechanisms of intermediate filament-disrupting withaferin A. MW molecular weight, VEGF vascular endothelial growth factor
Withaferin A also elicits antineoplastic activity in a considerable variety of cancer cell lines by directly inhibiting neoplastic growth, including carcinomas of the breast, head and neck, ovaries, and thyroid, as well as glioblastoma multiforme and melanoma [251–255]. Owing to its notable anti-angiogenic activity, withaferin A has demonstrated significant synergistic effects with the multikinase inhibitor sorafenib [256], suggesting that withaferin A may be a viable supplement for renal cell carcinomas, and other malignancies notably affected by the inhibition of neoplastic angiogenesis. It should also be noted that withaferin A, withalongolide A (a 19-hydroxy derivative of withaferin A), and several other closely related congeners may influence signaling regulated by the PI3K/AKT/mTOR pathway. These congeners suppress RET and Akt phosphorylation and protein expression, as well as inhibit mTOR activity, the translational activity of 4EBP1, and protein synthesis mediated by p70S6 kinase activation in neoplastic cells [patient-derived medullary thyroid carcinoma cells, as well as human (U87 and U251) and murine (GL26) glioblastoma cells] in vitro [257, 258]. These data indicate a potential synergistic relationship between withanolides and mTOR inhibitors currently used in the clinical setting, and warrant further preclinical investigation.
Conclusion
Beginning with the VAMP (vincristine, amethopterin/methotrexate, 6-mercaptopurine, and prednisone) protocol that potentiated long-term survival in pediatric ALL for the first time in medical history [259], natural products have a long history as antineoplastic agents. They have repeatedly added to the mechanisms of action available to practicing clinicians, as well as inspire semi-synthetic derivations that demonstrate improved clinical utility. Although sometimes archaic by their less than ideal discrimination of neoplastic over normal tissue, these agents are still highly implemented in many forms of chemotherapy. In an age where clinicians are becoming ever more sophisticated in their ability to target solid tumors and disseminated cancers with small-molecule inhibitors and the rapidly expanding field of immunotherapy, we should not lose sight of the importance of natural products for the treatment of cancer. Epitomized by ADCs, natural products previously deemed too potent to elicit therapeutic benefit can now be conjugated to an appropriate protein delivery system, thereby delivering highly cytotoxic and specific treatments to neoplastic tissue. This is an important lesson, as there are currently many cytotoxic mechanisms of action employed in nature that are not currently used in the clinical setting, such as the microfilament-targeting cytochalasins, or the intermediate filament-targeting withanolides.
While precision medicine has captivated the field of oncology in recent years and is gaining an increasing foothold in the clinic, we should also not discourage the continual investigation of natural products as antineoplastic agents. As demonstrated by current protocols, their unique mechanisms can be coupled with synthetic inhibitors or genetically modified proteins to create effective first-line therapies. Further, some derivatives of natural products such as the rapamycin analogs have the potential to be used in select cancers that demonstrate specific aberrant signaling cascades. Nature has already done the hard work through years of evolutionary pressures. Now, modern medicine can harness these complex, but often synthetically reproducible molecules, to develop ever more comprehensive and effective treatment modalities. As demonstrated by the countless lives they have prolonged or even saved, natural products have, are still, and will continue to function as vital components of cancer chemotherapy.
Acknowledgments
The author extends his sincerest gratitude to all of the investigators who have been involved in the development of natural product-based antineoplastic agents, including his mentor, Dr. Thomas Fondy of Syracuse University. In addition, the author thanks the two anonymous reviewers whose suggestions helped improve the quality of the article.
Compliance with Ethical Standards
Funding information
The author thanks Syracuse University for providing funds for research on microfilament-directed agents.
Conflict of interest
The author declares no conflicts of interest.
References
- 1.DeVita VT, Jr, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68(21):8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 2.Miller DR. A tribute to Sidney Farber—the father of modern chemotherapy. Br J Haematol. 2006;134(1):20–26. doi: 10.1111/j.1365-2141.2006.06119.x. [DOI] [PubMed] [Google Scholar]
- 3.Niederhuber JE, Armitage JO, Doroshow JH, MD, Kastan MB, Tepper JE. Abeloff’s clinical oncology. 5th ed. Philadelphia: Elsevier; 2014.
- 4.Gordaliza M. Natural products as leads to anticancer drugs. Clin Transl Oncol. 2007;9(12):767–776. doi: 10.1007/s12094-007-0138-9. [DOI] [PubMed] [Google Scholar]
- 5.Demain AL, Vaishnav P. Natural products for cancer chemotherapy. Microb Biotechnol. 2011;4(6):687–699. doi: 10.1111/j.1751-7915.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75(3):311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirata Y, Uemura D. Halichondrins—antitumor polyether macrolides from a marine sponge. Pure Appl Chem. 1986;58(5):701–710. doi: 10.1351/pac198658050701. [DOI] [Google Scholar]
- 8.Towle MJ, Salvato KA, Budrow J, Wels BF, Kuznetsov G, Aalfs KK, Welsh S, Zheng W, Seletsky BM, Palme MH, Habgood GJ, Singer LA, Dipietro LV, Wang Y, Chen JJ, Quincy DA, Davis A, Yoshimatsu K, Kishi Y, Yu MJ, Littlefield BA. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61(3):1013–1021. [PubMed] [Google Scholar]
- 9.Jain S, Vahdat LT. Eribulin mesylate. Clin Cancer Res. 2011;17(21):6615–6622. doi: 10.1158/1078-0432.CCR-11-1807. [DOI] [PubMed] [Google Scholar]
- 10.Cortes J, Montero AJ, Glück S. Eribulin mesylate, a novel microtubule inhibitor in the treatment of breast cancer. Cancer Treat Rev. 2012;38(2):143–151. doi: 10.1016/j.ctrv.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol Cancer Ther. 2005;4(7):1086–1095. doi: 10.1158/1535-7163.MCT-04-0345. [DOI] [PubMed] [Google Scholar]
- 12.Alday PH, Correia JJ. Macromolecular interaction of halichondrin B analogues eribulin (E7389) and ER-076349 with tubulin by analytical ultracentrifugation. Biochemistry. 2009;48(33):7927–7938. doi: 10.1021/bi900776u. [DOI] [PubMed] [Google Scholar]
- 13.Smith JA, Wilson L, Azarenko O, Zhu X, Lewis BM, Littlefield BA, Jordan MA. Eribulin binds at microtubule ends to a single site on tubulin to suppress dynamic instability. Biochemistry. 2010;49(6):1331–1337. doi: 10.1021/bi901810u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okouneva T, Azarenko O, Wilson L, Littlefield BA, Jordan MA. Inhibition of centromere dynamics by eribulin (E7389) during mitotic metaphase. Mol Cancer Ther. 2008;7(7):2003–2011. doi: 10.1158/1535-7163.MCT-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ortega Vanesa, Cortés Javier. Potential clinical applications of halichondrins in breast cancer and other neoplasms. Breast Cancer (Dove Med Press) 2012;4:9–19. doi: 10.2147/BCTT.S12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swami U, Chaudhary I, Ghalib MH, Goel S. Eribulin: a review of preclinical and clinical studies. Crit Rev Oncol Hematol. 2012;81(2):163–184. doi: 10.1016/j.critrevonc.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shetty N, Gupta S. Eribulin drug review. South Asian J Cancer. 2014;3(1):57–59. doi: 10.4103/2278-330X.126527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abraham J, Gulley JL, Allegra CJ. The Bethesda handbook of clinical oncology, 4th ed. Philadelphia: Lipincott Williams & Wilkins; 2014.
- 19.Cortes J, O’Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, Chollet P, Manikas A, Diéras V, Delozier T, Vladimirov V, Cardoso F, Koh H, Bougnoux P, Dutcus CE, Seegobin S, Mir D, Meneses N, Wanders J, Twelves C. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377(9769):914–923. doi: 10.1016/S0140-6736(11)60070-6. [DOI] [PubMed] [Google Scholar]
- 20.Scarpace SL. Eribulin mesylate (E7389): review of efficacy and tolerability in breast, pancreatic, head and neck, and non-small cell lung cancer. Clin Ther. 2012;34(7):1467–1473. doi: 10.1016/j.clinthera.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 21.de Bono JS, Molife LR, Sonpavde G, Maroto JP, Calvo E, Cartwright TH, Loesch DM, Feit K, Das A, Zang EA, Wanders J, Agoulnik S, Petrylak DP. Phase II study of eribulin mesylate (E7389) in patients with metastatic castration-resistant prostate cancer stratified by prior taxane therapy. Ann Oncol. 2012;23(5):1241–1249. doi: 10.1093/annonc/mdr380. [DOI] [PubMed] [Google Scholar]
- 22.Polastro L, Aftimos PG, Awada A. Eribulin mesylate in the management of metastatic breast cancer and other solid cancers: a drug review. Expert Rev Anticancer Ther. 2014;14(6):649–665. doi: 10.1586/14737140.2014.920693. [DOI] [PubMed] [Google Scholar]
- 23.Reichenbach H, Höfle G. Biologically active secondary metabolites from myxobacteria. Biotechnol Adv. 1993;11(2):219–277. doi: 10.1016/0734-9750(93)90042-L. [DOI] [PubMed] [Google Scholar]
- 24.Reichenbach H, Höfle G. Discovery and development of the epothilones: a novel class of antineoplastic drugs. Drugs R D. 2008;9(1):1–10. doi: 10.2165/00126839-200809010-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fojo T, Menefee M. Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol. 2007;18(Suppl 5):v3–v8. doi: 10.1093/annonc/mdm172. [DOI] [PubMed] [Google Scholar]
- 26.Wolff A, Technau A, Brandner G. Epothilone A induces apoptosis in neuroblastoma cells with multiple mechanisms of drug resistance. Int J Oncol. 1997;11(1):123–126. doi: 10.3892/ijo.11.1.123. [DOI] [PubMed] [Google Scholar]
- 27.Zhuang SH, Hung YE, Hung L, Robey RW, Sackett DL, Linehan WM, Bates SE, Fojo T, Poruchynsky MS. Evidence for microtubule target engagement in tumors of patients receiving ixabepilone. Clin Cancer Res. 2007;13(24):7480–7486. doi: 10.1158/1078-0432.CCR-06-2883. [DOI] [PubMed] [Google Scholar]
- 28.Kathawala RJ, Wang YJ, Ashby CR, Jr, Chen ZS. Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin J Cancer. 2014;33(5):223–230. doi: 10.5732/cjc.013.10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee FY, Borzilleri R, Fairchild CR, Kamath A, Smykla R, Kramer R, Vite G. Preclinical discovery of ixabepilone, a highly active antineoplastic agent. Cancer Chemother Pharmacol. 2008;63(1):157–166. doi: 10.1007/s00280-008-0724-8. [DOI] [PubMed] [Google Scholar]
- 30.Fornier MN. Ixabepilone, first in a new class of antineoplastic agents: the natural epothilones and their analogues. Clin Breast Cancer. 2007;7(10):757–763. doi: 10.3816/CBC.2007.n.036. [DOI] [PubMed] [Google Scholar]
- 31.Dumontet C, Jordan MJ, Lee FFY. Ixabepilone: targeting βIII-tubulin expression in taxane-resistant malignancies. Mol Cancer Ther. 2009;8(1):17–25. doi: 10.1158/1535-7163.MCT-08-0986. [DOI] [PubMed] [Google Scholar]
- 32.Hunt JT. Discovery of ixabepilone. Mol Cancer Ther. 2009;8(2):275–281. doi: 10.1158/1535-7163.MCT-08-0999. [DOI] [PubMed] [Google Scholar]
- 33.Ibrahim NK. Ixabepilone development across the breast cancer continuum: a paradigm shift. Cancer Manag Res. 2010;2:169–179. doi: 10.2147/CMAR.S10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denduluri N, Swain S. Ixabepilone: clinical role in metastatic breast cancer. Clin Breast Cancer. 2011;11(3):139–145. doi: 10.1016/j.clbc.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Prota AE, Bargsten K, Zurwerra D, Field JJ, Díaz JF, Altmann KH, Steinmetz MO. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science. 2013;339(6119):587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 36.Fumoleau P, Coudert B, Isambert N, Ferrant E. Novel tubulin-targeting agents: anticancer activity and pharmacologic profile of epothilones and related analogues. Ann Oncol. 2007;18(Suppl 5):v9–v15. doi: 10.1093/annonc/mdm173. [DOI] [PubMed] [Google Scholar]
- 37.Lee FY, Borzilleri R, Fairchild CR, Kim SH, Long BH, Reventos-Suarez C, Vite GD, Rose WC, Kramer RA. BMS-247550: a novel epothilone analog with a mode of action similar to paclitaxel but possessing superior antitumor efficacy. Clin Cancer Res. 2001;7(5):1429–1437. [PubMed] [Google Scholar]
- 38.Uyar D, Takigawa N, Mekhail T, Grabowski D, Markman M, Lee F, Canetta R, Peck R, Bukowski R, Ganapathi R. Apoptotic pathways of epothilone BMS 310705. Gynecol Oncol. 2003;91(1):173–178. doi: 10.1016/S0090-8258(03)00481-5. [DOI] [PubMed] [Google Scholar]
- 39.Altmann KH. Recent developments in the chemical biology of epothilones. Curr Pharm Des. 2005;11(13):1595–1613. doi: 10.2174/1381612053764715. [DOI] [PubMed] [Google Scholar]
- 40.Nicolaou KC, Ritzén A, Namoto K. Recent developments in the chemistry, biology and medicine of the epothilones. Chem Commun (Camb) 2001;17:1523–1535. doi: 10.1039/b104949f. [DOI] [PubMed] [Google Scholar]
- 41.Schmid P, Kiewe P, Possinger K, Korfel A, Lindemann S, Giurescu M, Reif S, Wiesinger H, Thiel E, Kühnhardt D. Phase I study of the novel, fully synthetic epothilone sagopilone (ZK-EPO) in patients with solid tumors. Ann Oncol. 2010;21(3):633–639. doi: 10.1093/annonc/mdp491. [DOI] [PubMed] [Google Scholar]
- 42.DeConti RC, Algazi AP, Andrews S, Urbas P, Born O, Stoeckigt D, Floren L, Hwang J, Weber J, Sondak VK, Daud AI. Phase II trial of sagopilone, a novel epothilone analog in metastatic melanoma. Br J Cancer. 2010;103(10):1548–1553. doi: 10.1038/sj.bjc.6605931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lam ET, Goel S, Schaaf LJ, Cropp GF, Hannah AL, Zhou Y, McCracken B, Haley BI, Johnson RG, Mani S, Villalona-Calero MA. Phase I dose escalation study of KOS-1584, a novel epothilone, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;69(2):523–531. doi: 10.1007/s00280-011-1724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cobham MV, Donovan D. Ixabepilone: a new treatment option for the management of taxane-resistant metastatic breast cancer. Cancer Manag Res. 2009;1:69–77. [PMC free article] [PubMed] [Google Scholar]
- 45.Pabla N, Sparreboom A. CCR 20th anniversary commentary: BMS-247550-microtubule stabilization as successful targeted therapy. Clin Cancer Res. 2015;21(6):1237–1239. doi: 10.1158/1078-0432.CCR-14-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivera E, Lee J, Davies A. Clinical development of ixabepilone and other epothilones in patients with advanced solid tumors. Oncologist. 2008;13(12):1207–1223. doi: 10.1634/theoncologist.2008-0143. [DOI] [PubMed] [Google Scholar]
- 47.Roché H, Yelle L, Cognetti F, Mauriac L, Bunnell C, Sparano J, Kerbrat P, Delord JP, Vahdat L, Peck R, Lebwohl D, Ezzeddine R, Curé H. Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, as first-line therapy in patients with metastatic breast cancer previously treated with anthracycline chemotherapy. J Clin Oncol. 2007;25(23):3415–3420. doi: 10.1200/JCO.2006.09.7535. [DOI] [PubMed] [Google Scholar]
- 48.Thomas E, Tabernero J, Fornier M, Conté P, Fumoleau P, Lluch A, Vahdat LT, Bunnell CA, Burris HA, Viens P, Baselga J, Rivera E, Guarneri V, Poulart V, Klimovsky J, Lebwohl D, Martin M. Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, in patients with taxane resistant metastatic breast cancer. J Clin Oncol. 2007;25(23):3399–3406. doi: 10.1200/JCO.2006.08.9102. [DOI] [PubMed] [Google Scholar]
- 49.Perez EA, Lerzo G, Pivot X, Thomas E, Vahdat L, Bosserman L, Viens P, Cai C, Mullaney B, Peck R, Hortobagyi GN. Efficacy and safety of ixabepilone (BMS-247550) in a phase II study of patients with advanced breast cancer resistant to an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2007;25(23):3407–3414. doi: 10.1200/JCO.2006.09.3849. [DOI] [PubMed] [Google Scholar]
- 50.Sparano JA, Vrdoljak E, Rixe O, Xu B, Manikhas A, Medina C, Da Costa SC, Ro J, Rubio G, Rondinon M, Perez Manga G, Peck R, Poulart V, Conte P. Randomized phase III trial of ixabepilone plus capecitabine versus capecitabine in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2010;28(20):3256–3263. doi: 10.1200/JCO.2009.24.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez EA, Patel T, Moreno-Aspitia A. Efficacy of ixabepilone in ER/PR/HER2-negative (triple-negative) breast cancer. Breast Cancer Res Treat. 2010;121(2):261–271. doi: 10.1007/s10549-010-0824-0. [DOI] [PubMed] [Google Scholar]
- 52.O’Connor OA, Portlock C, Moskowitz C, Straus D, Hamlin P, Stubblefield M, Dumetrescu O, Colevas AD, Grant B, Zelenetz A. A multicentre phase II clinical experience with the novel aza-epothilone Ixabepilone (BMS247550) in patients with relapsed or refractory indolent non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2008;143(2):201–209. doi: 10.1111/j.1365-2141.2008.07271.x. [DOI] [PubMed] [Google Scholar]
- 53.Churpek JE, Pro B, van Besien K, Kline J, Conner K, Wade JL, 3rd, Hagemeister F, Karrison T, Smith SM. A phase 2 study of epothilone B analog BMS-247550 (NSC 710428) in patients with relapsed aggressive non-Hodgkin lymphomas. Cancer. 2013;119(9):1683–1689. doi: 10.1002/cncr.27917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smaglo BG, Pishvaian MJ. Profile and potential of ixabepilone in the treatment of pancreatic cancer. Drug Des Devel Ther. 2014;8:923–930. doi: 10.2147/DDDT.S52964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic: I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975;28(10):721–6. [DOI] [PubMed]
- 56.Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1(1–4):27–36. doi: 10.1007/s12154-008-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saunders RN, Metcalfe MS, Nicholson ML. Rapamycin in transplantation: a review of the evidence. Kidney Int. 2001;59(1):3–16. doi: 10.1046/j.1523-1755.2001.00460.x. [DOI] [PubMed] [Google Scholar]
- 58.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5(8):671–688. doi: 10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 59.Luengo JI, Yamashita DS, Dunnington D, Beck AK, Rozamus LW, Yen HK, Bossard MJ, Levy MA, Hand A, Newman-Tarr T. Structure-activity studies of rapamycin analogs: evidence that the C-7 methoxy group is part of the effector domain and positioned at the FKBP12-FRAP interface. Chem Biol. 1995;2(7):471–481. doi: 10.1016/1074-5521(95)90264-3. [DOI] [PubMed] [Google Scholar]
- 60.Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16(4):525–537. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 61.Ritacco FV, Graziani EI, Summers MY, Zabriskie TM, Yu K, Bernan VS, Carter GT, Greenstein M. Production of novel rapamycin analogs by precursor-directed biosynthesis. Appl Environ Microbiol. 2005;71(4):1971–1976. doi: 10.1128/AEM.71.4.1971-1976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Graziani EI. Recent advances in the chemistry, biosynthesis and pharmacology of rapamycin analogs. Nat Prod Rep. 2009;26(5):602–609. doi: 10.1039/b804602f. [DOI] [PubMed] [Google Scholar]
- 63.Brachmann S, Fritsch C, Maira SM, García-Echeverría C. PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol. 2009;21(2):194–198. doi: 10.1016/j.ceb.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 64.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci USA. 2008;105(45):17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8(4):567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 66.Gibbons JJ, Abraham RT, Yu K. Mammalian target of rapamycin: discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin Oncol. 2009;36(Suppl 3):S3–S17. doi: 10.1053/j.seminoncol.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 67.Hudes GR, Berkenblit A, Feingold J, Atkins MB, Rini BI, Dutcher J. Clinical trial experience with temsirolimus in patients with advanced renal cell carcinoma. Semin Oncol. 2009;36(Suppl 3):S26–S36. doi: 10.1053/j.seminoncol.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 68.Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51. doi: 10.3389/fnmol.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thompson Coon J, Hoyle M, Green C, Liu Z, Welch K, Moxham T, Stein K. Bevacizumab, sorafenib tosylate, sunitinib and temsirolimus for renal cell carcinoma: a systematic review and economic evaluation. Health Technol Assess. 2010;14(2):1–184. [DOI] [PubMed]
- 70.Kelley RK, Nimeiri HS, Munster PN, Vergo MT, Huang Y, Li CM, Hwang J, Mulcahy MF, Yeh BM, Kuhn P, Luttgen MS, Grabowsky JA, Stucky-Marshall L, Korn WM, Ko AH, Bergsland EK, Benson AB, 3rd, Venook AP. Temsirolimus combined with sorafenib in hepatocellular carcinoma: a phase I dose-finding trial with pharmacokinetic and biomarker correlates. Ann Oncol. 2013;24(7):1900–1907. doi: 10.1093/annonc/mdt109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolff AC, Lazar AA, Bondarenko I, Garin AM, Brincat S, Chow L, Sun Y, Neskovic-Konstantinovic Z, Guimaraes RC, Fumoleau P, Chan A, Hachemi S, Strahs A, Cincotta M, Berkenblit A, Krygowski M, Kang LL, Moore L, Hayes DF. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol. 2013;31(2):195–202. doi: 10.1200/JCO.2011.38.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Waqar SN, Robinson C, Bradley J, Goodgame B, Rooney M, Williams K, Gao F, Govindan R. A phase I study of temsirolimus and thoracic radiation in non–small-cell lung cancer. Clin Lung Cancer. 2014;15(2):119–123. doi: 10.1016/j.cllc.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnston PB, Inwards DJ, Colgan JP, Laplant BR, Kabat BF, Habermann TM, Micallef IN, Porrata LF, Ansell SM, Reeder CB, Roy V, Witzig TE. A Phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol. 2010;85(5):320–324. doi: 10.1002/ajh.21664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, Porrata LF, Ansell SM, Colgan JP, Jacobsen ED, Ghobrial IM, Habermann TM. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25(2):341–347. doi: 10.1038/leu.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barnes JA, Jacobsen E, Feng Y, Freedman A, Hochberg EP, LaCasce AS, Armand P, Joyce R, Sohani AR, Rodig SJ, Neuberg D, Fisher DC, Abramson JS. Everolimus in combination with rituximab induces complete responses in heavily pretreated diffuse large B-cell lymphoma. Haematologica. 2013;98(4):615–619. doi: 10.3324/haematol.2012.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Powell RG, Weisleder D, Smith CR, Jr, Wolff IA. Structure of cephalotaxine and related alkaloids. Tetrahedron Lett. 1969;10(46):4081–4084. doi: 10.1016/S0040-4039(01)88620-2. [DOI] [Google Scholar]
- 77.Quintás-Cardama A, Kantarjian H, Cortes J. Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009. Cancer. 2009;115(23):5382–5393. doi: 10.1002/cncr.24601. [DOI] [PubMed] [Google Scholar]
- 78.Chen Y, Li S. Omacetaxine mepesuccinate in the treatment of intractable chronic myeloid leukemia. Onco Targets Ther. 2014;7:177–186. doi: 10.2147/OTT.S41786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heiblig M, Sobh M, Nicolini FE. Subcutaneous omacetaxine mepesuccinate in patients with chronic myeloid leukemia in tyrosine kinase inhibitor-resistant patients: review and perspectives. Leuk Res. 2014;38(10):1145–1153. doi: 10.1016/j.leukres.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 80.Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513(7519):517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- 81.Gürel G, Blaha G, Moore PB, Steitz TA. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009;389(1):146–156. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen Y, Hu Y, Michaels S, Segal D, Brown D, Li S. Inhibitory effects of omacetaxine on leukemic stem cells and BCR-ABL-induced chronic myeloid leukemia and acute lymphoblastic leukemia in mice. Leukemia. 2009;23(8):1446–1454. doi: 10.1038/leu.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim TD, Frick M, le Coutre P. Omacetaxine mepesuccinate for the treatment of leukemia. Expert Opin Pharmacother. 2011;12(15):2381–2392. doi: 10.1517/14656566.2011.613378. [DOI] [PubMed] [Google Scholar]
- 84.Rinehart KL. Antitumor compounds from tunicates. Med Res Rev. 2000;20(1):1–27. doi: 10.1002/(SICI)1098-1128(200001)20:1<1::AID-MED1>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 85.Cuevas C, Francesch A. Development of Yondelis (trabectedin, ET-743): a semisynthetic process solves the supply problem. Nat Prod Rep. 2009;26(3):322–337. doi: 10.1039/b808331m. [DOI] [PubMed] [Google Scholar]
- 86.D’Incalci M, Galmarini CM. A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther. 2010;9(8):2157–2163. doi: 10.1158/1535-7163.MCT-10-0263. [DOI] [PubMed] [Google Scholar]
- 87.Preusser M, Spiegl-Kreinecker S, Lötsch D, Wöhrer A, Schmook M, Dieckmann K, Saringer W, Marosi C, Berger W. Trabectedin has promising antineoplastic activity in high-grade meningioma. Cancer. 2012;118(20):5038–5049. doi: 10.1002/cncr.27460. [DOI] [PubMed] [Google Scholar]
- 88.Grohar PJ, Segars LE, Yeung C, Pommier Y, D’Incalci M, Mendoza A, Helman LJ. Dual targeting of EWS-FLI1 activity and the associated DNA damage response with trabectedin and SN38 synergistically inhibits Ewing sarcoma cell growth. Clin Cancer Res. 2014;20(5):1190–1203. doi: 10.1158/1078-0432.CCR-13-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pommier Y, Kohlhagen G, Bailly C, Waring M, Mazumder A, Kohn KW. DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry. 1996;35(41):13303–13309. doi: 10.1021/bi960306b. [DOI] [PubMed] [Google Scholar]
- 90.Erba E, Cavallaro E, Damia G, Mantovani R, Di Silvio A, Di Francesco AM, Riccardi R, Cuevas C, Faircloth GT, D’Incalci M. The unique biological features of the marine product Yondelis (ET-743, trabectedin) are shared by its analog ET-637, which lacks the C ring. Oncol Res. 2004;14(11–12):579–587. doi: 10.3727/0965040042707925. [DOI] [PubMed] [Google Scholar]
- 91.D’Incalci M, Jimeno J. Preclinical and clinical results with the natural marine product ET-743. Expert Opin Investig Drugs. 2003;12(11):1843–1853. doi: 10.1517/13543784.12.11.1843. [DOI] [PubMed] [Google Scholar]
- 92.Cassier PA, Dufresne A, Blay JY, Fayette J. Trabectedin and its potential in the treatment of soft tissue sarcoma. Ther Clin Risk Manag. 2008;4(1):109–116. doi: 10.2147/tcrm.s1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Monk BJ, Ghatage P, Parekh T, Henitz E, Knoblauch R, Matos-Pita AS, Nieto A, Park YC, Cheng PS, Li W, Favis R, Ricci D, Poveda A. Effect of BRCA1 and XPG mutations on treatment response to trabectedin and pegylated liposomal doxorubicin in patients with advanced ovarian cancer: exploratory analysis of the phase 3 OVA-301 study. Ann Oncol. 2015;26(5):914–920. doi: 10.1093/annonc/mdv071. [DOI] [PubMed] [Google Scholar]
- 94.Venkitaraman AR. Functions of BRCA1 and BRCA2 in the biological response to DNA damage. J Cell Sci. 2001;114(Pt 20):3591–3598. doi: 10.1242/jcs.114.20.3591. [DOI] [PubMed] [Google Scholar]
- 95.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 96.D’Incalci M, Badri N, Galmarini CM, Allavena P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer. 2014;111(4):646–650. doi: 10.1038/bjc.2014.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Uboldi S, Bernasconi S, Romano M, Marchini S, Fuso Nerini I, Damia G, Ganzinelli M, Marangon E, Sala F, Clivio L, Chiorino G, Di Giandomenico S, Rocchi M, Capozzi O, Margison GP, Watson AJ, Caccuri AM, Pastore A, Fossati A, Mantovani R, Grosso F, Tercero JC, Erba E, D’Incalci M. Characterization of a new trabectedin-resistant myxoid liposarcoma cell line that shows collateral sensitivity to methylating agents. Int J Cancer. 2012;131(1):59–69. doi: 10.1002/ijc.26340. [DOI] [PubMed] [Google Scholar]
- 98.Reid A, Martin-Liberal J, Benson C. Trabectedin for advanced soft tissue sarcomas: optimizing use. Ther Clin Risk Manag. 2014;10:1003–1011. doi: 10.2147/TCRM.S49330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carter NJ, Keam SJ. Trabectedin: a review of its use in soft tissue sarcoma and ovarian cancer. Drugs. 2010;70(3):355–376. doi: 10.2165/11202860-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 100.Simpson EL, Rafia R, Stevenson MD, Papaioannou D. Trabectedin for the treatment of advanced metastatic soft tissue sarcoma. Health Technol Assess. 2010;14(Suppl 1):63–67. doi: 10.3310/hta14suppl1/09. [DOI] [PubMed] [Google Scholar]
- 101.Monk BJ, Herzog TJ, Kaye SB, Krasner CN, Vermorken JB, Muggia FM, Pujade-Lauraine E, Lisyanskaya AS, Makhson AN, Rolski J, Gorbounova VA, Ghatage P, Bidzinski M, Shen K, Ngan HY, Vergote IB, Nam JH, Park YC, Lebedinsky CA, Poveda AM. Trabectedin plus pegylated liposomal doxorubicin in recurrent ovarian cancer. J Clin Oncol. 2010;28(19):3107–3114. doi: 10.1200/JCO.2009.25.4037. [DOI] [PubMed] [Google Scholar]
- 102.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 103.Massuti B, Cobo M, Camps C, Dómine M, Provencio M, Alberola V, Viñolas N, Rosell R, Tarón M, Gutiérrez-Calderón V, Lardelli P, Alfaro V, Nieto A, Isla D. Trabectedin in patients with advanced non-small-cell lung cancer (NSCLC) with XPG and/or ERCC1 overexpression and BRCA1 underexpression and pretreated with platinum. Lung Cancer. 2012;76(3):354–361. doi: 10.1016/j.lungcan.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 104.Michaelson MD, Bellmunt J, Hudes GR, Goel S, Lee RJ, Kantoff PW, Stein CA, Lardelli P, Pardos I, Kahatt C, Nieto A, Cullell-Young M, Lewis NL, Smith MR. Multicenter phase II study of trabectedin in patients with metastatic castration-resistant prostate cancer. Ann Oncol. 2012;23(5):1234–1240. doi: 10.1093/annonc/mdr399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Delaloge S, Wolp-Diniz R, Byrski T, Blum JL, Gonçalves A, Campone M, Lardelli P, Kahatt C, Nieto A, Cullell-Young M, Lubinski J. Activity of trabectedin in germline BRCA1/2-mutated metastatic breast cancer: results of an international first-in-class phase II study. Ann Oncol. 2014;25(6):1152–1158. doi: 10.1093/annonc/mdu134. [DOI] [PubMed] [Google Scholar]
- 106.Baruchel S, Pappo A, Krailo M, Baker KS, Wu B, Villaluna D, Lee-Scott M, Adamson PC, Blaney SM. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children’s Oncology Group. Eur J Cancer. 2012;48(4):579–585. doi: 10.1016/j.ejca.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 107.Vilcek J, Feldmann M. Historical review: cytokines as therapeutics and targets of therapeutics. Trends Pharmacol Sci. 2004;25(4):201–209. doi: 10.1016/j.tips.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 108.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4(1):11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 109.Dezfouli S, Hatzinisiriou I, Ralph SJ. Use of cytokines in cancer vaccines/immunotherapy: recent developments improve survival rates for patients with metastatic malignancy. Curr Pharm Des. 2005;11(27):3511–3530. doi: 10.2174/138161205774414574. [DOI] [PubMed] [Google Scholar]
- 110.Kim-Schulze S, Taback B, Kaufman HL. Cytokine therapy for cancer. Surg Oncol Clin N Am. 2007;16(4):793–818. doi: 10.1016/j.soc.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 111.Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010;33(2):153–165. doi: 10.1016/j.immuni.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lansigan F, Stearns DM, Foss F. Role of denileukin diftitox in the treatment of persistent or recurrent cutaneous T-cell lymphoma. Cancer Manag Res. 2010;2:53–59. doi: 10.2147/CMAR.S5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kun L, Hernandez-Ilizaliturri FJ, Reddy NM. Novel therapeutic strategies for cutaneous T-cell lymphoma in advanced stages. Semin Hematol. 2014;51(1):35–41. doi: 10.1053/j.seminhematol.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 114.Wong BY, Gregory SA, Dang NH. Denileukin diftitox as novel targeted therapy for lymphoid malignancies. Cancer Invest. 2007;25(6):495–501. doi: 10.1080/07357900701360096. [DOI] [PubMed] [Google Scholar]
- 115.Manoukian G, Hagemeister F. Denileukin diftitox: a novel immunotoxin. Expert Opin Biol Ther. 2009;9(11):1445–1451. doi: 10.1517/14712590903348135. [DOI] [PubMed] [Google Scholar]
- 116.Wilson BA, Collier RJ. Diphtheria toxin and Pseudomonas aeruginosa exotoxin A: active-site structure and enzymic mechanism. Curr Top Microbiol Immunol. 1992;175:27–41. doi: 10.1007/978-3-642-76966-5_2. [DOI] [PubMed] [Google Scholar]
- 117.Bell CE, Eisenberg D. Crystal structure of diphtheria toxin bound to nicotinamide adenine dinucleotide. Biochemistry. 1996;35(4):1137–1149. doi: 10.1021/bi9520848. [DOI] [PubMed] [Google Scholar]
- 118.Holmes RK. Biology and molecular epidemiology of diphtheria toxin and the tox gene. J Infect Dis. 2000;1(81 Suppl 1):S156–S167. doi: 10.1086/315554. [DOI] [PubMed] [Google Scholar]
- 119.Kaminetzky D, Hymes KB. Denileukin diftitox for the treatment of cutaneous T-cell lymphoma. Biologics. 2008;2(4):717–724. doi: 10.2147/btt.s3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Turturro F. Denileukin diftitox: a biotherapeutic paradigm shift in the treatment of lymphoid-derived disorders. Expert Rev Anticancer Ther. 2007;7(1):11–17. doi: 10.1586/14737140.7.1.11. [DOI] [PubMed] [Google Scholar]
- 121.Thorburn A, Thorburn J, Frankel AE. Induction of apoptosis by tumor cell-targeted toxins. Apoptosis. 2004;9(1):19–25. doi: 10.1023/B:APPT.0000012118.95548.88. [DOI] [PubMed] [Google Scholar]
- 122.Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood. 2007;110(9):3192–3201. doi: 10.1182/blood-2007-06-094615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Foss F. Clinical experience with denileukin diftitox (ONTAK) Semin Oncol. 2006;33(1 Suppl 3):S11–S16. doi: 10.1053/j.seminoncol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 124.Sugamura K, Asao H, Kondo M, Tanaka N, Ishii N, Ohbo K, Nakamura M, Takeshita T. The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu Rev Immunol. 1996;14:179–205. doi: 10.1146/annurev.immunol.14.1.179. [DOI] [PubMed] [Google Scholar]
- 125.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38(1):13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gorgun G, Foss F. Immunomodulatory effects of RXR rexinoids: modulation of high-affinity IL-2R expression enhances susceptibility to denileukin diftitox. Blood. 2002;100(4):1399–1403. doi: 10.1182/blood-2002-01-0300. [DOI] [PubMed] [Google Scholar]
- 127.Foss F. Immunomodulatory effects of rexinoids. Semin Oncol. 2006;33(1 Suppl 3):S21–S25. doi: 10.1053/j.seminoncol.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 128.Foss F, Demierre MF, DiVenuti G. A phase-1 trial of bexarotene and denileukin diftitox in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2005;106(2):454–457. doi: 10.1182/blood-2004-11-4570. [DOI] [PubMed] [Google Scholar]
- 129.Kerl K, Prins C, Cerroni L, French LE. Regression of extranodal natural killer/T-cell lymphoma, nasal type with denileukin diftitox (Ontak) and bexarotene (Targretin): report of a case. Br J Dermatol. 2006;154(5):988–991. doi: 10.1111/j.1365-2133.2006.07151.x. [DOI] [PubMed] [Google Scholar]
- 130.Pasquetto MV, Vecchia L, Covini D, Digilio R, Scotti C. Targeted drug delivery using immunoconjugates: principles and applications. J Immunother. 2011;34(9):611–628. doi: 10.1097/CJI.0b013e318234ecf5. [DOI] [PubMed] [Google Scholar]
- 131.Huang CY, Pourgholami MH, Allen BJ. Optimizing radioimmunoconjugate delivery in the treatment of solid tumor. Cancer Treat Rev. 2012;38(7):854–860. doi: 10.1016/j.ctrv.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 132.Smaglo BG, Aldeghaither D, Weiner LM. The development of immunoconjugates for targeted cancer therapy. Nat Rev Clin Oncol. 2014;11(11):637–648. doi: 10.1038/nrclinonc.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Senter PD, Sievers EL. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat Biotechnol. 2012;30(7):631–637. doi: 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- 134.Minich SS. Brentuximab vedotin: a new age in the treatment of Hodgkin lymphoma and anaplastic large cell lymphoma. Ann Pharmacother. 2012;46(3):377–383. doi: 10.1345/aph.1Q680. [DOI] [PubMed] [Google Scholar]
- 135.Garnock-Jones KP. Brentuximab vedotin: a review of its use in patients with Hodgkin lymphoma and systemic anaplastic large cell lymphoma following previous treatment failure. Drugs. 2013;73(4):371–381. doi: 10.1007/s40265-013-0031-5. [DOI] [PubMed] [Google Scholar]
- 136.Newland AM, Li JX, Wasco LE, Aziz MT, Lowe DK. Brentuximab vedotin: a CD30-directed antibody-cytotoxic drug conjugate. Pharmacotherapy. 2013;33(1):93–104. doi: 10.1002/phar.1170. [DOI] [PubMed] [Google Scholar]
- 137.Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB, Bontems RJ. The isolation and structure of a remarkable marine animal antineoplastic constituent: dolastatin 10. J Am Chem Soc. 1987;109(22):6883–6885. doi: 10.1021/ja00256a070. [DOI] [Google Scholar]
- 138.Alley SC, Zhang X, Okeley NM, Anderson M, Law CL, Senter PD, Benjamin DR. The pharmacologic basis for antibody-auristatin conjugate activity. J Pharmacol Exp Ther. 2009;330(3):932–938. doi: 10.1124/jpet.109.155549. [DOI] [PubMed] [Google Scholar]
- 139.Chabner BA, Longo DL. Cancer chemotherapy and biotherapy: principles and practice. 5th ed. Philadelphia: Lipincott Williams & Wilkins; 2011.
- 140.Dosio F, Brusa P, Cattel L. Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components. Toxins (Basel) 2011;3(7):848–883. doi: 10.3390/toxins3070848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Muta H, Podack ER. CD30: from basic research to cancer therapy. Immunol Res. 2013;57(1–3):151–158. doi: 10.1007/s12026-013-8464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Leroy X, Augusto D, Leteurtre E, Gosselin B. CD30 and CD117 (c-kit) used in combination are useful for distinguishing embryonal carcinoma from seminoma. J Histochem Cytochem. 2002;50(2):283–285. doi: 10.1177/002215540205000216. [DOI] [PubMed] [Google Scholar]
- 143.Sanderson RJ, Hering MA, James SF, Sun MM, Doronina SO, Siadak AW, Senter PD, Wahl AF. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res. 2005;11(2 Pt 1):843–852. [PubMed] [Google Scholar]
- 144.Ansell SM. Brentuximab vedotin. Blood. 2014;124(22):3197–3200. doi: 10.1182/blood-2014-06-537514. [DOI] [PubMed] [Google Scholar]
- 145.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res 2011;17(20):6437–47. [DOI] [PubMed]
- 146.Boyraz B, Sendur MA, Aksoy S, Babacan T, Roach EC, Kizilarslanoglu MC, Petekkaya I, Altundag K. Trastuzumab emtansine (T-DM1) for HER2-positive breast cancer. Curr Med Res Opin. 2013;29(4):405–414. doi: 10.1185/03007995.2013.775113. [DOI] [PubMed] [Google Scholar]
- 147.Kümler I, Tuxen MK, Nielsen DL. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat Rev. 2014;40(2):259–270. doi: 10.1016/j.ctrv.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 148.Boku N. HER2-positive gastric cancer. Gastric Cancer. 2014;17(1):1–12. doi: 10.1007/s10120-013-0252-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Oostra DR, Macrae ER. Role of trastuzumab emtansine in the treatment of HER2-positive breast cancer. Breast Cancer (Dove Med Press) 2014;6:103–113. doi: 10.2147/BCTT.S67297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Singh JC, Jhaveri K, Esteva FJ. HER2-positive advanced breast cancer: optimizing patient outcomes and opportunities for drug development. Br J Cancer. 2014;111(10):1888–1898. doi: 10.1038/bjc.2014.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Corrigan PA, Cicci TA, Auten JJ, Lowe DK. Ado-trastuzumab emtansine: a HER2-positive targeted antibody-drug conjugate. Ann Pharmacother. 2014;48(11):1484–1493. doi: 10.1177/1060028014545354. [DOI] [PubMed] [Google Scholar]
- 152.Kupchan SM, Komoda Y, Court WA, Thomas GJ, Smith RM, Karim A, Gilmore CJ, Haltiwanger RC, Bryan RF. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J Am Chem Soc. 1972;94(4):1354–1356. doi: 10.1021/ja00759a054. [DOI] [PubMed] [Google Scholar]
- 153.Edmonson JH, Rubin J, Kvols LK, O’Connell MJ, Frytak S, Green SJ. Phase II study of maytansine in advanced breast cancer. Cancer Treat Rep. 1981;65(5–6):536–537. [PubMed]
- 154.Ravry MJ, Omura GA, Birch R. Phase II evaluation of maytansine (NSC 153858) in advanced cancer. a Southeastern Cancer Study Group trial. Am J Clin Oncol. 1985;8(2):148–150. doi: 10.1097/00000421-198504000-00007. [DOI] [PubMed] [Google Scholar]
- 155.Latta EK, Tjan S, Parkes RK, O’Malley FP. The role of HER2/neu overexpression/amplification in the progression of ductal carcinoma in situ to invasive carcinoma of the breast. Mod Pathol. 2002;15(12):1318–1325. doi: 10.1097/01.MP.0000038462.62634.B1. [DOI] [PubMed] [Google Scholar]
- 156.Chung SS, Giehl N, Wu Y, Vadgama JV. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int J Oncol. 2014;44(2):403–411. doi: 10.3892/ijo.2013.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Colomer R, Montero S, Lluch A, Ojeda B, Barnadas A, Casado A, Massutí B, Cortés-Funes H, Lloveras B. Circulating HER2 extracellular domain and resistance to chemotherapy in advanced breast cancer. Clin Cancer Res. 2000;6(6):2356–2362. [PubMed] [Google Scholar]
- 158.Marquette C, Nabell L. Chemotherapy-resistant metastatic breast cancer. Curr Treat Options Oncol. 2012;13(2):263–275. doi: 10.1007/s11864-012-0184-6. [DOI] [PubMed] [Google Scholar]
- 159.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 160.Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, Burris Iii HA, LoRusso PM, Yi JH, Saad O, Tong B, Chu YW, Holden S, Joshi A. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69(5):1229–1240. doi: 10.1007/s00280-011-1817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Barok M, Joensuu H, Isola J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014;16(2):209. doi: 10.1186/bcr3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Barok M, Tanner M, Köninki K, Isola J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Res. 2011;13(2):R46. doi: 10.1186/bcr2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cretella D, Saccani F, Quaini F, Frati C, Lagrasta C, Bonelli M, Caffarra C, Cavazzoni A, Fumarola C, Galetti M, La Monica S, Ampollini L, Tiseo M, Ardizzoni A, Petronini PG, Alfieri RR. Trastuzumab emtansine is active on HER-2 overexpressing NSCLC cell lines and overcomes gefitinib resistance. Mol Cancer. 2014;13:143. doi: 10.1186/1476-4598-13-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347–356. doi: 10.1007/s10549-010-1090-x. [DOI] [PubMed] [Google Scholar]
- 165.Barok M, Tanner M, Köninki K, Isola J. Trastuzumab-DM1 is highly effective in preclinical models of HER2-positive gastric cancer. Cancer Lett. 2011;306(2):171–179. doi: 10.1016/j.canlet.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 166.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Diéras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–1791. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Fuentes G, Scaltriti M, Baselga J, Verma CS. Synergy between trastuzumab and pertuzumab for human epidermal growth factor 2 (Her2) from colocalization: an in silico based mechanism. Breast Cancer Res. 2011;13(3):R54. doi: 10.1186/bcr2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McCormack PL. Pertuzumab: a review of its use for first-line combination treatment of HER2-positive metastatic breast cancer. Drugs. 2013;73(13):1491–1502. doi: 10.1007/s40265-013-0109-0. [DOI] [PubMed] [Google Scholar]
- 169.Miller KD, Diéras V, Harbeck N, Andre F, Mahtani RL, Gianni L, Albain KS, Crivellari D, Fang L, Michelson G, de Haas SL, Burris HA. Phase IIa trial of trastuzumab emtansine with pertuzumab for patients with human epidermal growth factor receptor 2-positive, locally advanced, or metastatic breast cancer. J Clin Oncol. 2014;32(14):1437–1444. doi: 10.1200/JCO.2013.52.6590. [DOI] [PubMed] [Google Scholar]
- 170.Maiese WM, Lechevalier MP, Lechevalier HA, Korshalla J, Kuck N, Fantini A, Wildey MJ, Thomas J, Greenstein M. Calicheamicins, a novel family of antitumor antibiotics: taxonomy, fermentation and biological properties. J Antibiot (Tokyo) 1989;42(4):558–563. doi: 10.7164/antibiotics.42.558. [DOI] [PubMed] [Google Scholar]
- 171.Lambert JM. Drug-conjugated antibodies for the treatment of cancer. Br J Clin Pharmacol. 2013;76(2):248–262. doi: 10.1111/bcp.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hamann PR, Hinman LM, Hollander I, Beyer CF, Lindh D, Holcomb R, Hallett W, Tsou HR, Upeslacis J, Shochat D, Mountain A, Flowers DA, Bernstein I. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug Chem. 2002;13(1):47–58. doi: 10.1021/bc010021y. [DOI] [PubMed] [Google Scholar]
- 173.Fenton C, Perry CM. Gemtuzumab ozogamicin: a review of its use in acute myeloid leukaemia. Drugs. 2005;65(16):2405–2427. doi: 10.2165/00003495-200565160-00014. [DOI] [PubMed] [Google Scholar]
- 174.Giles FJ, Kantarjian HM, Kornblau SM, Thomas DA, Garcia-Manero G, Waddelow TA, David CL, Phan AT, Colburn DE, Rashid A, Estey EH. Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer. 2001;92(2):406–413. doi: 10.1002/1097-0142(20010715)92:2<406::AID-CNCR1336>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 175.Wadleigh M, Richardson PG, Zahrieh D, Lee SJ, Cutler C, Ho V, Alyea EP, Antin JH, Stone RM, Soiffer RJ, DeAngelo DJ. Prior gemtuzumab ozogamicin exposure significantly increases the risk of veno-occlusive disease in patients who undergo myeloablative allogeneic stem cell transplantation. Blood. 2003;102(5):1578–1582. doi: 10.1182/blood-2003-01-0255. [DOI] [PubMed] [Google Scholar]
- 176.Chevallier P, Prebet T, Turlure P, Hunault M, Vigouroux S, Harousseau JL, Blaise D, Ifrah N, Milpied N, Mohty M. Prior treatment with gemtuzumab ozogamicin and the risk of veno-occlusive disease after allogeneic haematopoietic stem cell transplantation. Bone Marrow Transpl. 2010;45(1):165–170. doi: 10.1038/bmt.2009.153. [DOI] [PubMed] [Google Scholar]
- 177.Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, Larson RA, Erba HP, Stiff PJ, Stuart RK, Walter RB, Tallman MS, Stenke L, Appelbaum FR. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24):4854–4860. doi: 10.1182/blood-2013-01-466706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Walker S, Landovitz R, Ding WD, Ellestad GA, Kahne D. Cleavage behavior of calicheamicin gamma 1 and calicheamicin T. Proc Natl Acad Sci USA. 1992;89(10):4608–4612. doi: 10.1073/pnas.89.10.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nicolaou KC, Smith AL, Yue EW. Chemistry and biology of natural and designed enediynes. Proc Natl Acad Sci USA. 1993;90(13):5881–5888. doi: 10.1073/pnas.90.13.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kraka E, Tuttle T, Cremer D. The reactivity of calicheamicin gamma(1)(I) in the minor groove of DNA: the decisive role of the environment. Chemistry. 2007;13(33):9256–9269. doi: 10.1002/chem.200700504. [DOI] [PubMed] [Google Scholar]
- 181.Kumar RA, Ikemoto N, Patel DJ. Solution structure of the calicheamicin gamma 1I-DNA complex. J Mol Biol. 1997;265(2):187–201. doi: 10.1006/jmbi.1996.0718. [DOI] [PubMed] [Google Scholar]
- 182.Ellestad GA. Structural and conformational features relevant to the anti-tumor activity of calicheamicin γ 1I. Chirality. 2011;23(8):660–671. doi: 10.1002/chir.20990. [DOI] [PubMed] [Google Scholar]
- 183.Kantarjian H, Thomas D, Wayne AS, O’Brien S. Monoclonal antibody-based therapies: a new dawn in the treatment of acute lymphoblastic leukemia. J Clin Oncol. 2012;30(31):3876–3883. doi: 10.1200/JCO.2012.41.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tu X, LaVallee T, Lechleider R. CD22 as a target for cancer therapy. J Exp Ther Oncol. 2011;9(3):241–248. [PubMed] [Google Scholar]
- 185.Ogura M, Hatake K, Ando K, Tobinai K, Tokushige K, Ono C, Ishibashi T, Vandendries E. Phase I study of anti-CD22 immunoconjugate inotuzumab ozogamicin plus rituximab in relapsed/refractory B-cell non-Hodgkin lymphoma. Cancer Sci. 2012;103(5):933–938. doi: 10.1111/j.1349-7006.2012.02241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kantarjian H, Thomas D, Jorgensen J, Kebriaei P, Jabbour E, Rytting M, York S, Ravandi F, Garris R, Kwari M, Faderl S, Cortes J, Champlin R, O’Brien S. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119(15):2728–2736. doi: 10.1002/cncr.28136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tuscano JM, Kato J, Pearson D, Xiong C, Newell L, Ma Y, Gandara DR, O’Donnell RT. CD22 antigen is broadly expressed on lung cancer cells and is a target for antibody-based therapy. Cancer Res. 2012;72(21):5556–5565. doi: 10.1158/0008-5472.CAN-12-0173. [DOI] [PubMed] [Google Scholar]
- 188.Pop LM, Barman S, Shao C, Poe JC, Venturi GM, Shelton JM, Pop IV, Gerber DE, Girard L, Liu XY, Behrens C, Rodriguez-Canales J, Liu H, Wistuba II, Richardson JA, Minna JD, Tedder TF, Vitetta ES. A reevaluation of CD22 expression in human lung cancer. Cancer Res. 2014;74(1):263–271. doi: 10.1158/0008-5472.CAN-13-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rytting M, Triche L, Thomas D, O’Brien S, Kantarjian H. Initial experience with CMC-544 (inotuzumab ozogamicin) in pediatric patients with relapsed B-cell acute lymphoblastic leukemia. Pediatr Blood Cancer. 2014;61(2):369–372. doi: 10.1002/pbc.24721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17(20):6398–6405. doi: 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJ, Wilson WH, Pastan I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23(27):6719–6729. doi: 10.1200/JCO.2005.11.437. [DOI] [PubMed] [Google Scholar]
- 192.Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, Stetler-Stevenson M, Fitzgerald DJ, Pastan I. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res. 2010;16(6):1894–1903. doi: 10.1158/1078-0432.CCR-09-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.CAT-8015: a second-generation pseudomonas exotoxin A-based immunotherapy targeting CD22-expressing hematologic malignancies. Clin Cancer Res. 2009;15(3):832–9. [DOI] [PMC free article] [PubMed]
- 194.Weldon JE, Pastan I. A guide to taming a toxin: recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011;278(23):4683–4700. doi: 10.1111/j.1742-4658.2011.08182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, Fitzgerald DJ, Lechleider R, Pastan I. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30(15):1822–1828. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kelly RJ, Sharon E, Pastan I, Hassan R. Mesothelin-targeted agents in clinical trials and in preclinical development. Mol Cancer Ther. 2012;11(3):517–525. doi: 10.1158/1535-7163.MCT-11-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pastan I, Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014;74(11):2907–2912. doi: 10.1158/0008-5472.CAN-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, Murugesan SR, Leach SD, Jaffee E, Yeo CJ, Cameron JL, Kern SE, Hruban RH. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE) Clin Cancer Res. 2001;7(12):3862–3868. [PubMed] [Google Scholar]
- 199.Ordóñez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27(11):1418–1428. doi: 10.1097/00000478-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 200.Ordóñez NG. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod Pathol. 2003;16(3):192–197. doi: 10.1097/01.MP.0000056981.16578.C3. [DOI] [PubMed] [Google Scholar]
- 201.Hassan R, Kreitman RJ, Pastan I, Willingham MC. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005;13(3):243–247. doi: 10.1097/01.pai.00000141545.36485.d6. [DOI] [PubMed] [Google Scholar]
- 202.Zhang Y, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, Le N, Ho M, Pastan I. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin Cancer Res. 2006;12(15):4695–4701. doi: 10.1158/1078-0432.CCR-06-0346. [DOI] [PubMed] [Google Scholar]
- 203.Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. Anti-mesothelin immunotoxin SS1P in combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 2007;13(23):7166–7171. doi: 10.1158/1078-0432.CCR-07-1592. [DOI] [PubMed] [Google Scholar]
- 204.Zhang Y, Xiang L, Hassan R, Pastan I. Immunotoxin and taxol synergy results from a decrease in shed mesothelin levels in the extracellular space of tumors. Proc Natl Acad Sci USA. 2007;104(43):17099–17104. doi: 10.1073/pnas.0708101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, Pastan I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13(17):5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 206.Kreitman RJ, Hassan R, Fitzgerald DJ, Pastan I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15(16):5274–5279. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hassan R, Sharon E, Thomas A, Zhang J, Ling A, Miettinen M, Kreitman RJ, Steinberg SM, Hollevoet K, Pastan I. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer. 2014;120(21):3311–3319. doi: 10.1002/cncr.28875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Trendowski M. Using cytochalasins to improve current chemotherapeutic approaches. Anticancer Agents Med Chem. 2015;15(3):327–335. doi: 10.2174/1871520614666141016164335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Trendowski M. The inherent metastasis of leukaemia and its exploitation by sonodynamic therapy. Crit Rev Oncol Hematol. 2015;94(2):149–163. doi: 10.1016/j.critrevonc.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 210.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172(7):973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773(5):642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ivaska J, Pallari HM, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp Cell Res. 2007;313(10):2050–2062. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- 213.Kato A, Kurita S, Hayashi A, Kaji N, Ohashi K, Mizuno K. Critical roles of actin-interacting protein 1 in cytokinesis and chemotactic migration of mammalian cells. Biochem J. 2008;414(2):261–270. doi: 10.1042/BJ20071655. [DOI] [PubMed] [Google Scholar]
- 214.Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105(4):1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yahara I, Harada F, Sekita S, Yoshihira Y, Natori S. Correlation between effects of 24 different cytochalasins on cellular structures and cellular events and those on actin in vitro. J Cell Biol. 1982;92(1):69–78. doi: 10.1083/jcb.92.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.MacLean-Fletcher S, Pollard TD. Mechanism of action of cytochalasin B on actin. Cell. 1980;20(2):329–341. doi: 10.1016/0092-8674(80)90619-4. [DOI] [PubMed] [Google Scholar]
- 217.Bisgaard HC, Ton PT, Nagy P, Thorgeirsson SS. Phenotypic modulation of keratins, vimentin, and alpha-fetoprotein in cultured rat liver epithelial cells after chemical, oncogene, and spontaneous transformation. J Cell Physiol. 1994;159(3):485–494. doi: 10.1002/jcp.1041590313. [DOI] [PubMed] [Google Scholar]
- 218.Oshima RG, Baribault H, Caulín C. Oncogenic regulation and function of keratins 8 and 18. Cancer Metastasis Rev. 1996;15(4):445–471. doi: 10.1007/BF00054012. [DOI] [PubMed] [Google Scholar]
- 219.Ulirsch J, Fan C, Knafl G, Wu MJ, Coleman B, Perou CM, Swift-Scanlan T. Vimentin DNA methylation predicts survival in breast cancer. Breast Cancer Res Treat 2013; 137(2):383–396. [DOI] [PMC free article] [PubMed]
- 220.Thomas PA, Kirschmann DA, Cerhan JR, Folberg R, Seftor EA, Sellers TA, Hendrix MJ. Association between keratin and vimentin expression, malignant phenotype, and survival in postmenopausal breast cancer patients. Clin Cancer Res. 1999;5(10):2698–2703. [PubMed] [Google Scholar]
- 221.Byun Y, Chen F, Chang R, Trivedi M, Green KJ, Cryns VL. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 2001;8(5):443–450. doi: 10.1038/sj.cdd.4400840. [DOI] [PubMed] [Google Scholar]
- 222.Trendowski M. Exploiting the cytoskeletal filaments of neoplastic cells to potentiate a novel therapeutic approach. Biochim Biophys Acta Rev Cancer. 2014;1846(2):599–616. doi: 10.1016/j.bbcan.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 223.Aldridge DC, Armstrong JJ, Speake RN, Turner WB. The cytochalasins, a new class of biologically active mould metabolites. Chem Commun (London) 1967;1:26–27. doi: 10.1039/c19670000026. [DOI] [Google Scholar]
- 224.Van Goietsenoven G, Mathieu V, Andolfi A, Cimmino A, Lefranc F, Kiss R, Evidente A. In vitro growth inhibitory effects of cytochalasins and derivatives in cancer cells. Planta Med. 2011;77(7):711–717. doi: 10.1055/s-0030-1250523. [DOI] [PubMed] [Google Scholar]
- 225.Trendowski M, Wong V, Wellington K, Hatfield S, Fondy TP. Tolerated doses in zebrafish of cytochalasins and jasplakinolide for comparison with tolerated doses in mice in the evaluation of pre-clinical activity of microfilament-directed agents in tumor model systems in vivo. Vivo. 2014;28(6):1021–1031. [PubMed] [Google Scholar]
- 226.Trendowski M, Mitchell JM, Corsette CM, Acquafondata C, Fondy TP. Chemotherapy with cytochalasin congeners in vitro and in vivo against murine models. Invest New Drugs. 2015;33(2):290–299. doi: 10.1007/s10637-014-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bousquet PF, Paulsen LA, Fondy C, Lipski KM, Loucy KJ, Fondy TP. Effects of cytochalasin B in culture and in vivo on murine Madison 109 lung carcinoma and on B16 melanoma. Cancer Res. 1990;50(5):1431–1439. [PubMed] [Google Scholar]
- 228.Trendowski M, Mitchell JM, Corsette CM, Acquafondata C, Fondy TP. Chemotherapy in vivo against murine M109 lung carcinoma with cytochalasin B by localized, systemic, and liposomal administration. Invest New Drugs. 2015;33(2):280–289. doi: 10.1007/s10637-014-0202-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Trendowski M, Zoino JN, Christen TD, Acquafondata C, Fondy TP. Preparation, in vivo administration, dose-limiting toxicities, and antineoplastic activity of cytochalasin B. Transl Oncol. 2015;8(4):308–317. doi: 10.1016/j.tranon.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lipski KM, McQuiggan JD, Loucy KJ, Fondy TP. Cytochalasin B: preparation, analysis in tissue extracts, and pharmacokinetics after intraperitoneal bolus administration in mice. Anal Biochem. 1987;161(2):332–340. doi: 10.1016/0003-2697(87)90459-3. [DOI] [PubMed] [Google Scholar]
- 231.Bogyo D, Fondy SR, Finster L, Fondy C, Patil S, Fondy TP. Cytochalasin-B-induced immunosuppression of murine allogeneic anti-tumor response and the effect of recombinant human interleukin-2. Cancer Immunol Immunother. 1991;32(6):400–405. doi: 10.1007/BF01741335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Huang FY, Mei WL, Li YN, Tan GH, Dai HF, Guo JL, Wang H, Huang YH, Zhao HG, Zhou SL, Li L, Lin YY. The antitumour activities induced by pegylated liposomal cytochalasin D in murine models. Eur J Cancer. 2012;48(14):2260–2269. doi: 10.1016/j.ejca.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 233.Karantza V. Keratins in health and cancer: more than mere epithelial cell markers. Oncogene. 2011;30(2):127–138. doi: 10.1038/onc.2010.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bauman PA, Dalton WS, Anderson JM, Cress AE. Expression of cytokeratin confers multiple drug resistance. Proc Natl Acad Sci USA. 1994;91:5311–5314. doi: 10.1073/pnas.91.12.5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Anderson JM, Heindl LM, Bauman PA, Ludi CW, Dalton WS, Cress AE. Cytokeratin expression results in a drug-resistant phenotype to six different chemotherapeutic agents. Clin Cancer Res. 1996;2(1):97–105. [PubMed] [Google Scholar]
- 236.Daly N, Meleady P, Walsh D, Clynes M. Regulation of keratin and integrin gene expression in cancer and drug resistance. Cytotechnology. 1998;27(1–3):321–344. doi: 10.1023/A:1008066216490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Liu F, Fan D, Qi J, Zhu H, Zhou Y, Yang C, Zhu Z, Xiong D. Co-expression of cytokeratin 8 and breast cancer resistant protein indicates a multifactorial drug-resistant phenotype in human breast cancer cell line. Life Sci. 2008;83(13–14):496–501. doi: 10.1016/j.lfs.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 238.Kleeberger W, Bova GS, Nielsen ME, Herawi M, Chuang AY, Epstein JI, Berman DM. Roles for the stem cell associated intermediate filament Nestin in prostate cancer migration and metastasis. Cancer Res. 2007;67(19):9199–9206. doi: 10.1158/0008-5472.CAN-07-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Teranishi N, Naito Z, Ishiwata T, Tanaka N, Furukawa K, Seya T, Shinji S, Tajiri T. Identification of neovasculature using nestin in colorectal cancer. Int J Oncol. 2007;30(3):593–603. [PubMed] [Google Scholar]
- 240.Krupkova O, Jr, Loja T, Zambo I, Veselska R. Nestin expression in human tumors and tumor cell lines. Neoplasma. 2010;57(4):291–298. doi: 10.4149/neo_2010_04_291. [DOI] [PubMed] [Google Scholar]
- 241.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68(18):3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Takemura K, Hirayama R, Hirokawa K, Inagaki M, Tsujimura K, Esaki Y, Mishima Y. Expression of vimentin in gastric cancer: a possible indicator for prognosis. Pathobiology. 1994;62(3):149–154. doi: 10.1159/000163895. [DOI] [PubMed] [Google Scholar]
- 243.Lavie D, Glotter E, Shvo Y. Constituents of withania somnifera dun: III. The side chain of withaferin A. J Org Chem. 1965;30(6):1774–1778. doi: 10.1021/jo01017a015. [DOI] [Google Scholar]
- 244.Vanden Berghe W, Sabbe L, Kaileh M, Haegeman G, Heyninck K. Molecular insight in the multifunctional activities of Withaferin A. Biochem Pharmacol. 2012;84(10):1282–91. [DOI] [PubMed]
- 245.Bargagna-Mohan P, Hamza A, Kim YE, Khuan Abby Ho Y, Mor-Vaknin N, Wendschlag N, Liu J, Evans RM, Markovitz DM, Zhan CG, Kim KB, Mohan R. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem Biol. 2007;14(6):623–634. doi: 10.1016/j.chembiol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Bargagna-Mohan P, Paranthan RR, Hamza A, Dimova N, Trucchi B, Srinivasan C, Elliott GI, Zhan CG, Lau DL, Zhu H, Kasahara K, Inagaki M, Cambi F, Mohan R. Withaferin A targets intermediate filaments glial fibrillary acidic protein and vimentin in a model of retinal gliosis. J Biol Chem. 2010;285(10):7657–7669. doi: 10.1074/jbc.M109.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Bargagna-Mohan P, Paranthan RR, Hamza A, Zhan CG, Lee DM, Kim KB, Lau DL, Srinivasan C, Nakayama K, Nakayama KI, Herrmann H, Mohan R. Corneal antifibrotic switch identified in genetic and pharmacological deficiency of vimentin. J Biol Chem. 2012;287(2):989–1006. doi: 10.1074/jbc.M111.297150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Grin B, Mahammad S, Wedig T, Cleland MM, Tsai L, Herrmann H, Goldman RD. Withaferin a alters intermediate filament organization, cell shape and behavior. PLoS One. 2012;7(6):e39065. doi: 10.1371/journal.pone.0039065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yokota Y, Bargagna-Mohan P, Ravindranath PP, Kim KB, Mohan R. Development of withaferin A analogs as probes of angiogenesis. Bioorg Med Chem Lett. 2006;16:2603–2607. doi: 10.1016/j.bmcl.2006.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mohan R, Hammers HJ, Bargagna-Mohan P, Zhan XH, Herbstritt CJ, Ruiz A, Zhang L, Hanson AD, Conner BP, Rougas J, Pribluda VS. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis. 2004;7(2):115–122. doi: 10.1007/s10456-004-1026-3. [DOI] [PubMed] [Google Scholar]
- 251.Bargagna-Mohan P, Ravindranath PP, Mohan R. Small molecule anti-angiogenic probes of the ubiquitin proteasome pathway: potential application to choroidal neovascularization. Invest Ophthalmol Vis Sci. 2006;47:4138–4145. doi: 10.1167/iovs.05-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Samadi AK, Tong X, Mukerji R, Zhang H, Timmermann BN, Cohen MS. Withaferin A, a cytotoxic steroid from Vassobia breviflora, induces apoptosis in human head and neck squamous cell carcinoma. J Nat Prod. 2010;73(9):1476–1481. doi: 10.1021/np100112p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Zhang X, Mukerji R, Samadi AK, Cohen MS. Down-regulation of estrogen receptor-alpha and rearranged during transfection tyrosine kinase is associated with withaferin a-induced apoptosis in MCF-7 breast cancer cells. BMC Complement Altern Med. 2011;11:84. doi: 10.1186/1472-6882-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zhang X, Samadi AK, Roby KF, Timmermann B, Cohen MS. Inhibition of cell growth and induction of apoptosis in ovarian carcinoma cell lines CaOV3 and SKOV3 by natural withanolide Withaferin A. Gynecol Oncol. 2012;124(3):606–612. doi: 10.1016/j.ygyno.2011.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Samadi AK, Cohen SM, Mukerji R, Chaguturu V, Zhang X, Timmermann BN, Cohen MS, Person EA. Natural withanolide withaferin A induces apoptosis in uveal melanoma cells by suppression of Akt and c-MET activation. Tumour Biol. 2012;33(4):1179–1189. doi: 10.1007/s13277-012-0363-x. [DOI] [PubMed] [Google Scholar]
- 256.Cohen SM, Mukerji R, Timmermann BN, Samadi AK, Cohen MS. A novel combination of withaferin A and sorafenib shows synergistic efficacy against both papillary and anaplastic thyroid cancers. Am J Surg 2012;204(6):895–900 (discussion 900–1). [DOI] [PubMed]
- 257.Grogan PT, Sleder KD, Samadi AK, Zhang H, Timmermann BN, Cohen MS. Cytotoxicity of withaferin A in glioblastomas involves induction of an oxidative stress-mediated heat shock response while altering Akt/mTOR and MAPK signaling pathways. Invest New Drugs. 2013;31(3):545–557. doi: 10.1007/s10637-012-9888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Samadi AK, Bazzill J, Zhang X, Gallagher R, Zhang H, Gollapudi R, Kindscher K, Timmermann B, Cohen MS. Novel withanolides target medullary thyroid cancer through inhibition of both RET phosphorylation and the mammalian target of rapamycin pathway. Surgery. 2012;152(6):1238–1247. doi: 10.1016/j.surg.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Freireich EJ. The history of leukemia therapy:a personal journey. Clin Lymphoma Myeloma Leuk. 2012;12(6):386–392. doi: 10.1016/j.clml.2012.09.014. [DOI] [PubMed] [Google Scholar]