

Abstract
Purpose
We created implantable intraocular devices capable of constant and continuous rapamycin release on the scale of months to years.
Methods
Polycaprolactone (PCL) thin films were used to encapsulate rapamycin to create implantable and biodegradable intraocular devices. Different film devices were studied by modifying the size, thickness, and porosity of the PCL films.
Results
In vitro release of rapamycin was observed to be constant (zero-order) through 14 weeks of study. Release rates were tunable by altering PCL film porosity and thickness. In vivo release of rapamycin was observed out through 16 weeks with concentrations in the retina–choroid in the therapeutic range. Rapamycin concentration in the blood was below the lower limit of quantification. The drug remaining in the device was chemically stable in vitro and in vivo, and was sufficient to last for upwards of 2 years of total release. The mechanism of release is related to the dissolution kinetics of crystalline rapamycin.
Conclusions
Microporous PCL thin film devices demonstrate good ocular compatibility and the ability to release rapamycin locally to the eye over the course of many weeks.
Keywords: intravitreal drug delivery, sirolimus, rapamycin, sustained release, polycaprolactone
Uveitis is a significant cause of blindness characterized by inflammation in the anterior, intermediate, and/or posterior uvea or retina of the eye, with posterior uveitis as the most challenging to treat. Noninfectious uveitis often is treated first with corticosteroids but is followed by immunosuppressants if corticosteroids fail to control inflammation, if inflammation reoccurs despite corticosteroid use, or if systemic side effects occur.1
Rapamycin (also known as sirolimus) is a macrolide drug used currently for immunosuppression in renal transplantation,2 prevention of restenosis by elution from coronary stents,3,4 and for the treatment of chronic uveitis.5,6 However, the oral administration of rapamycin against uveitis can result in severe side effects, so an alternative delivery method is desirable. Currently, an intraocular injection of a liquid formulation of rapamycin is in clinical trials for treatment of uveitis.7 The injection is delivered directly into the vitreous, and treatment requires bimonthly injections for the duration of the chronic disease.8,9 Intraocular administration of rapamycin in a small cohort of patients demonstrated no significant adverse effects, and levels of rapamycin in the blood were well below the concentration where systemic side effects occur.8 Intraocular administration of rapamycin is a potential route for the treatment of posterior uveitis that bypasses the side effects associated with systemic administration. Recent work has explored the use of silicon microparticles for the intravitreal delivery of rapamycin, but release kinetics appear to be limited to approximately 1 month.10,11
Long-term zero-order release behavior is a goal for drug delivery devices because a constant drug elution rate will equilibrate to a steady-state drug concentration, which can be engineered within the therapeutic window of the drug. In the posterior of the eye, zero-order release offers additional potential benefits in treating chronic diseases that otherwise require repeated drug injections into the eye. In addition, zero-order drug release devices can maximize the effect of small drug payloads imposed by the size limits of the eye. Current administration techniques for rapamycin, such as eye drops12 and injections,8 fall short in their ability to achieve sustained drug concentrations in the eye over periods of a month or longer. The importance of long-duration rapamycin delivery is indicated by another mTOR inhibitor, everolimus, which in one study was shown to be effective in refractory uveitis, but uveitis returned in four of eight patients following the end of everolimus therapy.13 Patient compliance also is a major issue for treatments using eye drops, as one study of compliance in glaucoma patients found improper medication delivery (missing a dose or improper administration) in 53.3% of patients.14 Other drug delivery systems for the treatment of uveitis with corticosteroids are available, but none are currently approved for the delivery of rapamycin or similar immunosuppressants.15–17
Zero-order drug delivery devices implanted intravitreally are one way to improve the amount of time an administered drug resides within the therapeutic window at its ocular site of action. One approach in development for sustained drug release in the eye is a device composed of nanoporous, biodegradable polycaprolactone (PCL) thin films which allow zero-order release of therapeutic proteins through the nanopores.18 Polycaprolactone is a polyester with a favorable intraocular biocompatibility profile that can be fabricated into thin films with thicknesses in the range of 10 to 40 μm.18–20 Additionally, PCL thin films can be patterned with nanofeatures or microfeatures by solvent-casting to modify diffusion behavior through the thin films.21 Control of the nano- and microstructure of a PCL thin film allows tuning of drug diffusion behavior across the film. Previous in vivo studies on the safety of these PCL thin films has shown them to be well tolerated in the posterior chamber of rabbit eyes over 6 months, in addition to showing minimal changes in morphology from degradation.19 Based on previous in vivo studies it is estimated that PCL will remain intact for approximately 3 years before losing mechanical integrity.20,22,23
We investigated a drug delivery device for rapamycin that demonstrates zero-order release behavior up to 14 weeks in vitro. The device is composed of a solid rapamycin pellet encapsulated by two PCL thin films, which can be modified to alter the release profile of the device. In vivo results show that the drug is stable in the device, reaches its target tissue at the retina/choroid, and sustains release behavior up to 16 weeks. No systemic adverse effects were observed, and the concentration of rapamycin in the blood was below detection limits.
Materials and Methods
Materials
Polycaprolactone (80 kDa Mn), 2,2,2-trifluoroethanol (TFE), and 2 kDa Mn polyethylene glycol (PEG) were obtained from Sigma-Aldrich Corp. (St. Louis, MO, USA). Rapamycin was obtained from Santen, Ltd. (Osaka, Japan). Phosphate-buffered saline (PBS) was obtained from VWR (Radnor, PA, USA), polyoxyl-40-stearate (also known as polyoxyethylene [40] monostearate, Myrj 52) from Spectrum Chemical (New Brunswick, NJ, USA), seco-rapamycin from Santa Cruz Biotech (Dallas, TX, USA), polydimethylsiloxane (PDMS) from Thermo Fisher Scientific (Waltham, MA, USA), and nichrome wire from Consolidated Electronic Wire and Cable (Franklin Park, IL, USA).
Film Fabrication
Polycaprolactone was dissolved in TFE and then spun-cast on a 3-inch silicon wafer to make unfeatured PCL films. Concentrations of PCL in TFE were varied between 100 and 300 mg/mL to change the resulting thickness of the spun-cast film. Microporous PCL films were made by adding polyethylene glycol at a concentration of 150 mg/mL in solutions of PCL and TFE. Films that were rapamycin-loaded in their bulk were prepared by combining solutions of 5 mg/mL rapamycin in TFE with solutions of 150 mg/mL PCL in TFE, or a solution of PCL and PEG at 150 mg/mL in TFE. Polycaprolactone film morphology was investigated by scanning electron microscopy (SEM) performed on a Carl Zeiss Ultra 55 FE-SEM (Carl Zeiss, San Francisco, CA, USA).
Device Fabrication
Rapamycin-loaded devices were prepared by heat-sealing PCL films around a central rapamycin payload (Fig. 1A). Two sets of devices were made, one set 16 mm in diameter and another set 6 mm in diameter. Devices 16 mm in diameter encapsulated 0.5 to 2.0 mg pure loose rapamycin between two PCL films. No excipient was used. These 16-mm devices were made either with two unfeatured PCL films, or one microporous and one unfeatured PCL film. The second set of devices were 6 mm in diameter with a pelleted rapamycin disc of mass 1.0 to 2.5 mg. The drug pellets consisted of pure rapamycin without excipients. Devices 6 mm in diameter were assembled either with two unfeatured PCL films or with one microporous film and one unfeatured PCL film.
Figure 1.

Polycaprolactone devices were sealed by heating PCL thin films inside of a weighted jig (A). Loose rapamycin powder was heat-sealed inside of 16-mm microporous devices (B). Microporous PCL thin films were approximately 10 μm in thickness with micron-sized pores throughout its bulk (C). Rapamycin release behavior from a bulk-loaded film shows nonlinear burst release while drug release from 16-mm microporous and 16-mm unfeatured devices maintains linear kinetics (D).
Devices were assembled by placing the drug pellet between PCL films atop a PDMS slab with an embedded nichrome wire as a heating element. A PDMS annulus and a weight on top of the device held the films together for sealing. Current passed through the wire heat-seals the circumference of the devices.
In Vitro Release Experiments and Analysis
In vitro release experiments were conducted in 2 mL elution media of PBS (pH 7.4) with 0.1% (wt/vol) polyoxyl-40-stearate to increase rapamycin solubility and stability in the elution media. A 2-mL elution volume was used as it is approximately the same as the vitreous volume in New Zealand White rabbits (∼1.5 mL). Polyoxyl-40-stearate has been shown to improve solubility and stability of rapamycin by approximately a factor of 10.24 Polyoxyl-40-stearate also was added to reduce the tendency of rapamycin to adsorb to the surface of tubes used in elution studies. Devices were incubated in elution media at 37°C with constant shaking on an orbital shaker. Elution media was completely replaced at sampling intervals. Rapamycin concentrations were assayed by ultraviolet spectrophotometry at 278 nm. Rapamycin stability in the devices and dissolution media was assayed by HPLC as described previously.25 Release studies were designed carefully with frequent time points to avoid saturation of the elution media with drug. At all time points the signal from drug loaded devices were blanked to empty controls. Linear regressions were calculated using Microsoft Excel (Microsoft Corp., Redmond, WA, USA).
Adsorption Release and Powder X-Ray Diffraction (PXRD)
Rapamycin was dissolved in methanol and pipetted onto the surface of PCL films. Rapamycin adsorbed to the PCL thin film after methanol evaporation. For release experiments, rapamycin was dissolved to 10 mg/mL and adsorbed by adding varying volumes of the methanol solution to the films. For release experiments, 1, 2.5, and 5 μL solution (10, 25, and 50 μg rapamycin) was adsorbed. For PXRD analysis, rapamycin was dissolved to 200 mg/mL and 30 μL was adsorbed onto PCL thin films.
In Vivo Experiments and Analysis
In vivo release experiments were performed in New Zealand White rabbits for vitreous drug release. Devices 6 mm in diameter with one microporous and one unfeatured PCL film were implanted into the posterior chamber of the right eye under general anesthesia and sterile conditions via a 2-mm scleral incision made circumferentially 2 mm posterior to the limbus using a 20-gauge MVR blade, then closing the incisions with 7-0 vicryl sutures. The left eye was left as an untreated control. In vivo safety and tolerability were evaluated by monitoring IOP, adverse events, and inflammation. Inflammation of the conjunctiva/sclera and surgical insertion site, edema, vitreous leakage, cataract formation, polymer appearance and location, hyphema, retinal detachment, anterior chamber fibrin or cell debris, and vitreous humor change were specifically monitored. Intraocular pressure was taken with a benchtop pneumotonometer (Model 30 Classic; Mentor O&O, Inc., Norwell, MA, USA) for eyes.
The study design had two arms. One arm was designed for six rabbits, with animals euthanized, and whole eye collection and dissection for vitreous and ocular tissue samples, two animals at each time point of 4, 8, and 16 weeks. A second arm was designed for three rabbits as survival studies, to allow longitudinal pharmacokinetic studies with multiple serial vitreous samples taken from each study eye at weeks 1, 2, 4, 8, and 12 by partial microvitrectomy, with a final sample at week 16 after animal euthanization. Microvitrectomy was performed in a sterile fashion under anesthesia using the Intrector (Insight Instruments, Stuart, FL, USA), a 23-gauge sutureless vitreous cutting/aspiration device, entering the sclera 2 mm posterior to the limbus at a site 180° from the thin-film device insertion to collect 100 to 200 μL vitreous at each sampling procedure. As planned, rabbits were killed at 4, 8, and 16 weeks with the exception of one killed at day 3 and one killed at week 3 due to complications from nonocular infections unrelated to the device. The data from the two euthanizations due to complications are included in the results presented.
Rapamycin concentration and stability in the vitreous and the retina–choroid was assayed via LC/MS-MS with positive electrospray ionization in multiple reaction monitoring mode. The lower limit of quantitation for vitreous samples was 0.06 ng/mL, the limit for retina–choroid samples was 1.06 ng/g, and the limit for whole blood samples was 0.5 ng/mL. The ion transition monitored was 931.6 to 864.6 and tacrolimus was used as an internal standard to control for any loss of rapamycin during purification, most notably due to surface adsorption. Owing to the specificity of liquid chromatography/mass spectrometry (LC/MS-MS), the assay is specific to stable rapamycin in the samples tested of the residual drug in the devices and the vitreous.
Data is reported as mean ± SD. Statistics were performed using Student's t-test with a significance threshold at 0.05. ARVO guidelines for the care and use of laboratory animals according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research have been observed.
Results
Rapamycin Release From 16-mm Devices and Bulk Films
Release characteristics of rapamycin devices 16 mm in diameter (Figs. 1A–C) were tested in vitro and compared against the rapamycin release from films that were bulk-loaded with rapamycin. Figure 1D shows the difference in kinetics between rapamycin that has been bulk-loaded throughout a PCL film and rapamycin released from either microporous or unfeatured rapamycin devices. When fit by linear regression, microporous devices released 2.92 μg/d (R2 = 0.999) and unfeatured devices released 2.02 μg/d (R2 = 0.997). Rapamycin release from a bulk-loaded film was nonlinear, though the initial pseudolinear region up to day 6 shows a release of 16.6 μg/d (R2 = 0.822).
Rapamycin Release From 6-mm Devices
A second form factor of device was designed based on enhanced fabrication techniques to result in a 6-mm device containing a pelleted rapamycin payload (Figs. 2A, 2B). In vitro release experiments with 6-mm devices (Fig. 2C) show the release of rapamycin from microporous devices (1.78 mg/d, R2 = 0.993) as nearly identical to the release from rapamycin pellets not contained by a PCL device (1.73 mg/d, R2 = 0.987). Rapamycin released from an unfeatured device shows significantly slower release (0.493 mg/d, R2 = 0.978) while maintaining a zero-order release profile. Release in all device types continued to be zero-order out to 14 weeks, and sufficient payload remained in the devices to release for a further 1.6 years for microporous devices and 4.6 years for unfeatured devices.
Figure 2.

Devices 6 mm in diameter and composed of unfeatured PCL (A) or microporous PCL (B). Surface SEMs also are shown for an unfeatured film (A) and a microporous film (B). Rapamycin release from an unfeatured device is significantly lower than release from either a microporous device or a pellet alone (C). Changing the thickness of the PCL films in an unfeatured device results in a correlated change in the release rate of rapamycin (D). For each condition in (C) and (D), n = 4 and error bars are ± SD.
The ability of PCL thickness to modulate release kinetics was studied using films constructed of two layers of unfeatured 4, 16, or 66 μm thick PCL. Over 5 weeks, devices constructed from 4-μm thick films released 37 mg rapamycin, while devices of 16- and 66-μm thick films released 30 and 22 μg rapamycin, respectively (Fig. 2B). The similar release rates of the three thicknesses of device after week 2 suggest that the impact of PCL film thickness on rapamycin release is focused in the initial 2 weeks of release. The differences in release during the first 2 weeks may be attributed to the amount of time rapamycin takes to reach equilibrium across the films of varying PCL thicknesses.
The stability of solid rapamycin in the device was confirmed in vitro by HPLC and in vivo by LC-MS/MS. In vitro 6-mm unfeatured devices were incubated in an elution experiment for 14 weeks and then dissected to remove the solid rapamycin pellet. High performance liquid chromatography (HPLC) performed on the drug pellets (n = 3) showed peaks identical to pure rapamycin and no presence of the primary degradation product seco-rapamycin, nor any other significant peak indicative of degradation (Supplementary Fig. S1). For in vivo quantification, LC-MS/MS was used to detect the presence of intact rapamycin in the device through 16 weeks of implantation and for in vivo vitreous samples.
Adsorption Release and PXRD
Rapamycin adsorbed to a surface showed an initial burst release followed by slower sustained release out to 3 days (Fig. 3A). Rapamycin pellets in the same in vitro experiment demonstrated zero-order release which continued past 3 days, and demonstrated a much longer potential release in other in vitro experiments. To examine drug crystallinity, PXRD was performed on rapamycin powder and on rapamycin adsorbed to the surface of unfeatured PCL films. The results (Fig. 3B) show characteristic peaks for rapamycin powder, which confirms the crystallinity of the drug. However, PXRD analysis of rapamycin adsorbed to the surface of PCL films showed the loss of peaks and the presence of a halo pattern between zero and 30° for adsorbed rapamycin, indicating the presence of amorphous rapamycin.
Figure 3.

Elution experiments (A) of adsorbed rapamycin show nonlinear burst release, while rapamycin pellets maintain linear release. Mass given in the legend is mass attempted to adsorb onto the film. For each condition, n = 4 and error bars are ± SD. Powder x-ray diffraction analysis (B) of rapamycin (1), rapamycin adsorbed onto PCL films (2), and blank PCL films (3). Adsorbed rapamycin on a PCL film (2) shows the gentle hill of a halo effect between zero and 30°. The presence of the halo and sharp peaks for adsorbed rapamycin indicates the presence of amorphous and crystalline rapamycin.
In Vivo Rapamycin Pharmacokinetics
Microporous devices 6 mm in diameter were used for in vivo trials and show constant release kinetics up to 16 weeks (Fig. 4; Supplementary Fig. S2). Vitreous concentrations at week 16 were 5.2 ng/mL with a standard deviation of 3.1 (n = 5). Retina–choroid concentrations at week 16 were on average 206 ng/g with a standard deviation of 74 (n = 5) and were relatively constant over the course of the study. Drug concentrations at 4- and 8-week time points in the vitreous averaged 3.6 ± 1.5 ng/mL (n = 4) and in the retina–choroid averaged 407 ± 387 ng/g (n = 4). The concentration of rapamycin in whole blood and the control eye was below the lower limit of quantitation for the assay (0.5 ng/mL) throughout the study. The outlying data point for vitreous concentration at day 3 may imply a burst release immediately following device implantation before equilibrium is reached throughout the eye. Further in vivo trials may investigate early release behavior of these devices.
Figure 4.

In vivo rapamycin release maintained a constant rapamycin concentration through 16 weeks as detected in the vitreous (A) and in the retina–choroid (B). Average of the data from weeks 4 to 16 is shown as a dashed line. Average ± SD is shown as a dotted line. Despite the failings of microvitrectomy as a sampling method (squares), sampling of microvitrectomy-arm animals at euthanization yielded results consistent with the acute sampling-only arm (circles).
Following implantation, eyes were monitored for adverse events and device migration. No evidence of device migration was seen and no evidence of fibrosis or other cellular debris was evident on the devices after explantation (Fig. 5A). Differences in IOP between pre- and postimplantation eyes were not statistically significant for device and control eyes (Fig. 5B). The lone adverse event observed was a cataract attributed to surgical trauma during device implantation (Fig. 5C). No intraocular inflammation was observed during the course of the in vivo study.
Figure 5.
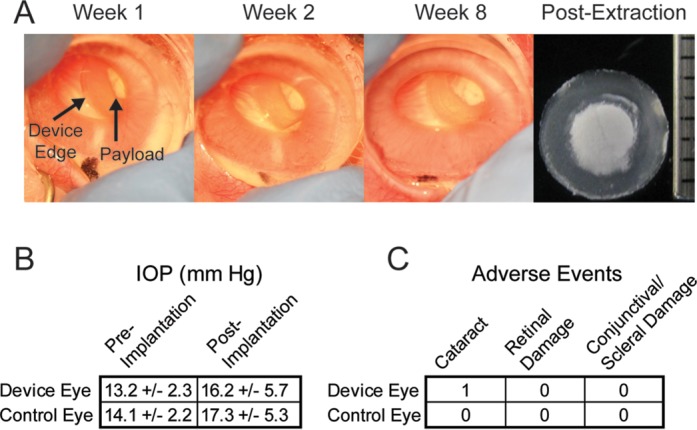
Devices maintained consistent position in the eye over the course of observation and no fibrosis was found after device removal (A). No statistically significant difference in IOP was found between pre- and postimplantation eyes (B). Only one adverse event was observed and it was attributed to contact between the lens and the device during implantation (C).
Discussion
The release behavior of rapamycin is dependent upon device structure and rapamycin crystalline properties. The use of PCL thin films to encapsulate crystalline rapamycin creates a device capable of sustained, zero-order release in vitro and in vivo. More traditional bulk-loaded films show a characteristic release curve with an initial burst release phase that decays into a slower rate of release. Microporous PCL devices show a zero-order release curve for rapamycin with a constant amount of drug released per day. Unfeatured rapamycin devices also released drug in a zero-order curve, except with a lower slope owing to the absence of micropores. Due to the small percentage of rapamycin releasing from the devices over the studied period of time, much of the drug mass still was intact. The percentage of drug mass remaining in each in vivo device by week 16 is 84% ± 15% (Supplementary Fig. S3). Future work will further examine the long-term behavior of the devices, including mass balance and release behavior as the devices near exhaustion of their drug payloads.
The zero-order release behavior of unfeatured devices, microporous devices, and unencapsulated rapamycin pellets was explored by comparing solid crystalline rapamycin to amorphous rapamycin adsorbed onto PCL films. The PXRD results and the in vitro release results support crystallinity as the quality driving slow rapamycin dissolution, which results in linear release rates from these types of encapsulation devices. As the crystal structure of rapamycin also is lost through its dissolution for the formation of rapamycin bulk-loaded films, the release behavior of bulk-loaded films is not dependent on rapamycin crystallinity and instead follows traditional burst release kinetics.26
The differences in behavior between rapamycin releasing devices may be attributed to the microporosity of the film, the thickness of the film, and the surface area of crystallized rapamycin available for dissolution. The influence of porosity is seen in the contrast between the release rates of the unfeatured devices when compared to the microporous devices. Drug diffusion through the films is hindered in unfeatured devices due to a lack of pores in their PCL films, while the microporous films have porous networks through their bulk. Varying the thickness of 6-mm unfeatured devices also resulted in differences in cumulative release in the first 5 weeks, as thicker PCL films decreased the release of rapamycin from the devices.
In vivo performance of the microporous 6-mm device demonstrated zero-order release behavior by showing relatively constant vitreous concentrations over time. The concentration of rapamycin in the retina–choroid averaging 206 ± 74 ng/g at 16 weeks demonstrates significantly longer sustained delivery than other rapamycin delivery technologies. A study using repeated injection of solubilized rapamycin in rabbit vitreous in vivo results in a dose-dependent concentration between 109 and 1050 ng/g at 4 weeks after injections, followed by a decline to approximately 20 ng/g by 8 weeks.8 A study using a nanomicellar eye drop formulation achieved 362 ± 56 (SEM), but was only measured at 1 hour after a single dose.12
For our in vivo study, there was a large variability in the concentration of rapamycin measured in the retina–choroid for one device measured at week 4. For this device, the concentration of the drug in the vitreous and the blood did not show a similar increase. Two possible reasons for the increase seen only in the retina–choroid could be the implantation of the device extremely near to the retina, or contamination of the retina–choroid sample during dissection or analysis.
The risk of systemic side effects from this type of device is potentially much lower than other delivery methods. The effective concentration for systemic immunosuppression by rapamycin is measured typically in whole blood at 5 to 15 ng/mL.8 Concentrations above this range are at risk of side effects. The concentration of rapamycin in whole blood was below the lower limit of quantitation for the assay (0.5 ng/mL) which suggests that local administration of rapamycin in these devices minimizes systemic exposure to the drug and avoids the associated side effects. Of note, the concentration of rapamycin in retina–choroid is much higher than the 5 to 15 mg/mL threshold value, which is desirable when targeting the choroid as a site of uveitis. The above-threshold concentrations of rapamycin suggest that an effective dose of the drug exists in the retina–choroid.
The risk of obstructing the visual axis also must be considered; however, throughout the course of this study no change in the location of the device was observed. Given the location of the device outside of the visual axis, it is unlikely the device or its soluble degradation products will result in a visual obstruction over the complete lifetime of the device.
An attempt was made with this study to use microvitrectomy to reduce the number of animals required for studying pharmacokinetics in the vitreous. Samples taken from the partial microvitrectomy arm consistently yielded rapamycin concentrations approximately a factor of 5 lower than samples from the nonmicrovitrectomy arm. However, samples taken from the animals in the microvitrectomy arm at time of death yielded drug concentrations on par with the nonmicrovitrectomy arm. Thus, for rapamycin it seems that partial microvitrectomy may not yield representative drug concentrations in the vitreous. Potential reasons for this failure may be related to inhomogenous distribution of rapamycin within the vitreous due to altered vitreous structure after repeated partial vitrectomy, the hydrophobic nature of rapamycin limiting diffusion in the vitreous, or the tendency of dissolved rapamycin to form a drug depot in the vitreous.8 Further work may be necessary to find out if and how this result extends to other drugs administered intravitreally and if the behavior can be predicted by drug hydrophobicity.
The ability to release rapamycin in the vitreous was demonstrated over 16 weeks in vivo, with a drug payload capable of lasting on the order of years, which is significantly longer than the 2 months between the necessary dosing of rapamycin by injection.8 Rapamycin concentrations in the vitreous and retina–choroid were relatively unchanging over the course of the study, demonstrating the potential for a continuous therapeutically effective drug concentration over the lifetime of the drug payload.
Conclusions
A drug delivery system composed of PCL thin films surrounding rapamycin is an effective means to deliver the drug up to 16 weeks, with the potential for an additional 2 to 5 years of release. Release behavior of rapamycin is zero-order in in vitro and in vivo experiments. Drug concentration is high in the retina–choroid site of action after intravitreal implantation, and systemic concentrations are well below the limit for immunosuppression. The release behavior observed in this study may be attributed to the dissolution properties associated with crystalline rapamycin and further research on this property is merited.
Supplementary Material
Acknowledgments
The authors thank San Francisco State University for the use of the Carl Zeiss Ultra 55 FE-SEM and supporting equipment, Research to Prevent Blindness for support of the microsurgery suite used for animal studies, and Elliot Chan for his kind assistance.
Supported by National Institutes of Health (NIH; Bethesda, MD, USA) Grant #R01EY021574, and Santen Pharmaceutical Co., and UC Proof of Concept Grant 247211 from the UC Office of the President. The FE-SEM and supporting facilities were obtained under NSF-MRI Award #0821619 and NSF-EAR Award #0949176, respectively.
Disclosure: K.D. Lance, P; S.D. Good, None; T.S. Mendes, None; M. Ishikiriyama, None; P. Chew, None; L.S. Estes, None; K. Yamada, Santen Pharmaceutical Co. (E); S. Mudumba, Santen, Inc. (E); R.B. Bhisitkul, Santen Pharmaceutical Co. (F, C), P; T.A. Desai, Santen Pharmaceutical Co. (F, C), P
References
- 1. Pato E,, Muñoz-Fernández S,, Francisco F,, et al. Systematic review on the effectiveness of immunosuppressants and biological therapies in the treatment of autoimmune posterior uveitis. Semin Arthritis Rheum. 2011; 40: 314–323. [DOI] [PubMed] [Google Scholar]
- 2. Morath C,, Arns W,, Schwenger V,, et al. Sirolimus in renal transplantation. Nephrol Dial Transplant. 2007; 22 (suppl 8): viii61–viii65. [DOI] [PubMed] [Google Scholar]
- 3. Puranik AS,, Dawson ER,, Peppas N. Recent advances in drug eluting stents. Int J Pharm. 2013; 441: 665–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dasari TW,, Patel B,, Saucedo JF. Systematic review of effectiveness of oral sirolimus after bare-metal stenting of coronary arteries for prevention of in-stent restenosis. Am J Cardiol. 2013; 112: 1322–1327. [DOI] [PubMed] [Google Scholar]
- 5. Shanmuganathan VA,, Casely EM,, Raj D,, et al. The efficacy of sirolimus in the treatment of patients with refractory uveitis. Br J Ophthalmol. 2005; 89: 666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Phillips BN,, Wroblewski KJ. A retrospective review of oral low-dose sirolimus (rapamycin) for the treatment of active uveitis. J Ophthalmic Inflamm Infect. 2011; 1: 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nguyen QD,, Ibrahim MA,, Watters A,, et al. Ocular tolerability and efficacy of intravitreal and subconjunctival injections of sirolimus in patients with non-infectious uveitis: primary 6-month results of the SAVE Study. J Ophthalmic Inflamm Infect. 2013; 3: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mudumba S,, Bezwada P,, Takanaga H,, et al. Tolerability and pharmacokinetics of intravitreal sirolimus. J Ocul Pharmacol Ther. 2012; 28: 507–514. [DOI] [PubMed] [Google Scholar]
- 9. Study Assessing Double-masked Uveitis Treatment (SAKURA). Available at: http://clinicaltrials.gov/show/NCT01358266. Accessed July 16, 2015.
- 10. Nieto A,, Hou H,, Moon SW,, Sailor MJ,, Freeman WR, Cheng L. surface engineering of porous silicon microparticles for intravitreal sustained delivery of rapamycin. Invest Ophthalmol Vis Sci. 2015; 56: 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hou H,, Nieto A,, Belghith A,, et al. A sustained intravitreal drug delivery system with remote real time monitoring capability. Acta Biomater. 2015; 24: 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cholkar K,, Gunda S,, Earla R,, Pal D,, Mitra AK. Nanomicellar topical aqueous drop formulation of rapamycin for back-of-the-eye delivery. AAPS PharmSciTech. 2014; 16: 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heiligenhaus A,, Zurek-Imhoff B,, Roesel M,, Hennig M,, Rammrath D,, Heinz C. Everolimus for the treatment of uveitis refractory to cyclosporine A: a pilot study. Graefes Arch Clin Exp Ophthalmol. 2013; 251: 143–152. [DOI] [PubMed] [Google Scholar]
- 14. Kholdebarin R,, Campbell RJ,, Jin Y-P,, Buys YM. Multicenter study of compliance and drop administration in glaucoma. Can J Ophthalmol. 2008; 43: 454–461. [DOI] [PubMed] [Google Scholar]
- 15. Hazirolan D,, Pleyer U. Think global - act local: intravitreal drug delivery systems in chronic noninfectious uveitis. Ophthalmic Res. 2012; 49: 59–65. [DOI] [PubMed] [Google Scholar]
- 16. Edelhauser HF,, Rowe-Rendleman CL,, Robinson MR,, et al. Ophthalmic drug delivery systems for the treatment of retinal diseases: basic research to clinical applications. Invest Ophthalmol Vis Sci. 2010; 51: 5403–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tempest-Roe S,, Joshi L,, Dick AD,, Taylor SR. Local therapies for inflammatory eye disease in translation: past present and future. BMC Ophthalmol. 2013; 13: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bernards DA,, Lance KD,, Ciaccio NA,, Desai TA. Nanostructured thin film polymer devices for constant-rate protein delivery. Nano Lett. 2012; 12: 5355–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bernards DA,, Bhisitkul RB,, Wynn P,, et al. Ocular biocompatibility and structural integrity of micro- and nanostructured poly(caprolactone) films. J Ocul Pharmacol Ther. 2013; 29: 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Silva-Cunha A,, Fialho SL,, Naud M-C,, Behar-Cohen F. Poly-epsilon-caprolactone intravitreous devices: an in vivo study. Invest Ophthalmol Vis Sci. 2009; 50: 2312–2318. [DOI] [PubMed] [Google Scholar]
- 21. Bernards DA,, Desai TA. Nanotemplating of biodegradable polymer membranes for constant-rate drug delivery. Adv Mater. 2010; 22: 2358–2362. [DOI] [PubMed] [Google Scholar]
- 22. Sun H,, Mei L,, Song C,, Cui X,, Wang P. The in vivo degradation absorption and excretion of PCL-based implant. Biomaterials. 2006; 27: 1735–1740. [DOI] [PubMed] [Google Scholar]
- 23. Natu MV,, de Sousa HC,, Gil MH. Influence of polymer processing technique on long term degradation of poly(ε-caprolactone) constructs. Polym Degrad Stab. 2013; 98: 44–51. [Google Scholar]
- 24. Kim M-S,, Kim J-S,, Park HJ,, Cho WK,, Cha K-H,, Hwang S-J, Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int J Nanomedicine. 2011; 6: 2997–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Il'ichev Y,, Alquier L,, Maryanoff C. Degradation of rapamycin and its ring-opened isomer: role of base catalysis. ARKIVOC. 2007; xii: 110–131. [Google Scholar]
- 26. Carvalho SR,, Watts AB,, Peters JI,, et al. Characterization and pharmacokinetic analysis of crystalline versus amorphous rapamycin dry powder via pulmonary administration in rats. Eur J Pharm Biopharm. 2014; 88: 136–147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.