

Abstract
Syntrophaceticus schinkii strain Sp3 is a mesophilic syntrophic acetate oxidizing bacterium, belonging to the Clostridia class within the phylum Firmicutes, originally isolated from a mesophilic methanogenic digester. It has been shown to oxidize acetate in co-cultivation with hydrogenotrophic methanogens forming methane. The draft genome shows a total size of 3,196,921 bp, encoding 3,688 open reading frames, which includes 3,445 predicted protein-encoding genes and 55 RNA genes. Here, we are presenting assembly and annotation features as well as basic genomic properties of the type strain Sp3.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0092-z) contains supplementary material, which is available to authorized users.
Keywords: Syntrophic acetate oxidizing bacteria, Acetogens, Syntrophy, Methanogens, Hydrogen producer, Methane production
Introduction
During anaerobic degradation of organic material, acetate is formed as a main fermentation product, which is further converted to methane. Two mechanisms for methane formation from acetate have been described: The first one is carried out by aceticlastic methanogens converting acetate to methane and CO2 under low ammonia conditions [1]. The second mechanism, dominating under high ammonia conditions, occurs in two steps, and is performed by acetate-oxidizing bacteria oxidizing acetate to H2 (formate) and CO2 and a methanogenic partner using the hydrogen (formate) to reduce CO2 to methane [2–4]. Most fascinating on this syntrophic relationship is, that the overall reaction operates with a ΔG°´ of -36 kJ x mol−1 close to the thermodynamic equilibrium.
The number of isolated and characterized SAOB is restricted most likely due to their considerable differences in substrate utilization abilities and cultivation requirements. To date three mesophilic SAOB, namely Clostridium ultunense [5], Syntrophaceticus schinkii [6], “Tepidanaerobacter acetatoxydans” [7] and two thermophilic SAOB, namely Thermacetogenium phaeum [2] and Thermotoga lettingae [8] currently renamed to Pseudothermotoga lettingae have been isolated and characterized. Among those, two complete genome sequences of T. phaeum [9], “T. acetatoxydans” [10] and one draft genome sequence of C. ultunense [11] have been published, the later two by this working group. Here, we are presenting the draft genome sequence of the third mesophilic SAOB Syntrophaceticus schinkii strain Sp3. To date, strain Sp3 is the only isolated and characterized representative of the species S. schinkii and was recovered from an up flow anaerobic filter treating wastewater from a fishmeal factory [6]. This process was characterized by high ammonium concentration (6.4 g l−1 NH4+). S. schinkii shows the least narrow substrate spectrum compared to all known SAOB, when growing heterotrophically [6]. The main end product formed is acetate, what allocates the species to the physiological group of acetogens.
Since the recovery of S. schinkii we found it at high abundance in all mesophilic large scale and lab scale biogas producing process we have investigated so far. Genome analysis and comparative genomics might help us to understand general features of syntrophy in particular energy conservation and electron transfer mechanisms during syntrophic acetate oxidation.
The present study summarizes genome sequencing, assembly and annotation as well as general genomic properties of the Syntrophaceticus schinkii strain Sp3 genome.
Organism information
Classification and features
Syntrophaceticus schinkii Sp3 (Fig. 1) is an obligate anaerobic, endospores forming bacterium, whose cells were found to be Gram variable with changing shapes dependent on the growth condition (Table 1, [6]). No flagella have been observed under any condition tested. It can grow up to 0.6 M NH4Cl in pure culture between 25 °C and 40 °C. A more detailed physiological description can be found in Westerholm et al. [6]. Minimum Information about the Genome Sequence (MIGS) of S. schinkii strain Sp3 is provided in Table 1 and Table S1 (Additional file 1).
Fig. 1.
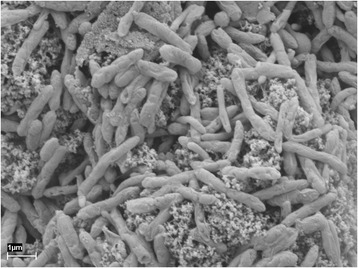
Image. Phase-contrast micrograph of Syntrophaceticus schinkii strain Sp3
Table 1.
Classification and general features of Syntrophaceticus schinkii strain Sp3 according to the “minimum information about a Genome Sequence” (MIGS) specification [22]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [23, 24] | |
| Phylum Firmicutes | TAS [25] | ||
| Class Clostridia | TAS [26, 27] | ||
| Order Thermoanaerobacterales | TAS [26] (p132), [28] | ||
| Family Thermoanaerobacteraceae | TAS [26] (p132), [29] | ||
| Genus Syntrophaceticus | TAS [6, 30] | ||
| Species Syntrophaceticus schinki | TAS [6, 30] | ||
| Strain Sp3 | TAS [6] | ||
| Gram stain | Variable | TAS [6] | |
| Cell shape | Variable b | TAS [6] | |
| Motility | Non motile | TAS [6] | |
| Sporulation | Terminal endospores | TAS [6] | |
| Temperature range | Mesophilic | TAS [6] | |
| Optimum temperature | 37–40 °C | TAS [6] | |
| Carbon source | Heterotroph | TAS [6] | |
| Energy source | Chemoheterotroph | TAS [6] | |
| MIGS-6 | Habitat | Anaerobic sludge | TAS [6] |
| MIGS-6.3 | Salinity | Up to 0.6 M NH4Cl | TAS [6] |
| MIGS-22 | Oxygen | Obligate anaerob | TAS [6] |
| MIGS-15 | Biotic relationship | Syntrophy (beneficial) | TAS [6] |
| MIGS-14 | Pathogenicity | Not reported | NAS |
| MIGS-4 | Geographic location | Spain | NAS |
| MIGS-5 | Sample collection time | 1992 | NAS |
| MIGS-4.1 | Latitude | 42.851329 | NAS |
| MIGS-4.2 | Longitude | −8.475933 | NAS |
| MIGS-4.3 | Depth | Not reported | NAS |
| MIGS-4.4 | Altitude | Not reported | NAS |
aEvidence codes—TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). Evidence codes are from the Gene Ontology project [31]. bShape of cells varies between cocci and straight or slightly curved rods depend on NH4Cl concentration [6]
Phylogentic analysis of the single 16S rRNA gene copy affiliates S. schinkii strain Sp3 to the Clostridia class within the phylum Firmicutes. The RDP Classifier ([12] 2015-08-05) confirmed further the affiliation to Thermoanaerobacteraceae as published by [6] in 2011 (Table 1). The comparison of the 16S rRNA gene sequence with the latest available databases from GenBank (2015-08-05) using NCBI BLAST [13] under default settings identified the thermophilic SAOB T. phaeum(NR_074723.1) as the closest characterized relative sharing 92.12 % identity (Fig. 2). S. schinkii is only distantly related to the characterized mesophilic SAOB C. ultunense ( 82.54 % identity), and “T. acetatoxydans” (84.1 % identity) and the thermophilic P. lettingae (79.64 %). Although S. schinkii has been physiologically affiliated to the group of acetogens, Fig. 2 illustrates a distant relationship to this group, as represented by e.g. the model acetogen Moorella thermoacetica (89.15 % identity).
Fig. 2.
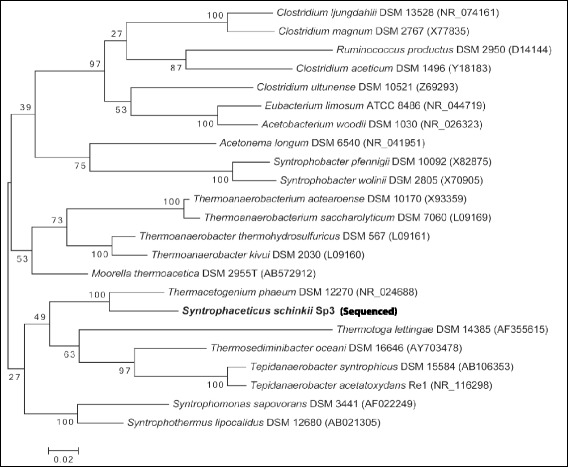
Phylogentic tree. Phylogenetic tree highlighting the relationship of Syntrophaceticus schinkii Sp3 relative to known SAOB, acetogens, and other syntrophic operating bacteria. The 16S rRNA-based alignment was carried out using MUSCLE [32] and the phylogenetic tree was inferred from 1,521 aligned characteristics of the 16S rRNA gene sequence using the maximum-likelihood (ML) algorithm [33] with MEGA 6.06 [34, 35]. Bootstrap analysis [36] with 100 replicates was performed to assess the support of the clusters
Genome sequencing information
Genome project history
Syntrophaceticus schinkii strain Sp3 was sequenced and annotated by the SLU-Global Bioinformatics Centre at the Swedish University of Agricultural Sciences, Uppsala, Sweden. The genome project is deposited in the Genomes OnLine Database [14] with GOLD id Gi0035837 and the working draft genome is deposited in the European Nucleotide Archive database with accession number ERP005192. The SAOB was selected for sequencing on the basis of environmental relevance to issues in global carbon cycling, alternative energy production, and biochemical importance. Table2 contains the summary of project information.
Table 2.
Genome sequencing project information for the Syntrophaceticus schinkii Sp3 genome
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Draft |
| MIGD-28 | Libraries used | Ion Torrent single end reads |
| MIGS-29 | Sequencing platform | Ion Torrent PGM Systems |
| MIGS-31.2 | Sequencing coverage | 35× |
| MIGS-30 | Assemblers | Newbler 2.8 and MIRA 4.0 |
| MIGS-32 | Gene calling method | PRODIGAL and AMIGene |
| Locus Tag | SSCH | |
| Genbank ID | CDRZ00000000 | |
| GenBank Data of release | March 21, 2014 | |
| GOLD ID | Gi0035837 | |
| BIOPROJECT | PRJNA224116 | |
| MIGS 13 | Source Material Identifier | DSM 21860 |
| Project relevance | Biogas production |
Growth conditions and genomic DNA preparation
Since isolation by our research group, the strain has been kept in liquid cultures and a live culture and medium have been sent to DSMZ, (DSM21860). For DNA isolation batch cultures were grown in basal medium supplemented with 20 mM betaine as described by Westerholm et al. [6]. Cells were grown for 4 weeks at 37 °C without shaking and harvested at 5000 × g. DNA was isolated using the Blood & Tissue Kit from Qiagen (Hilden, Germany) according to the standard protocol recommended by the manufacturer.
Genome sequencing and assembly
The genome of Syntrophaceticus schinkii was sequenced at the SciLifeLab Uppsala, Sweden using Ion torrent PM systems with the mean length of 206 bp, longest read length 392 bp and a total of final library reads of 2,985,963 for single end reads. All general aspects of sequencing performed can be found at Scilifelab website [15]. The FastQC software package [16] was used for reads quality assessment. After preassembly quality checking, the reads were assembled with MIRA 4.0 and Newbler 2.8 assemblers. Possible miss-assemblies were corrected manually by using Tablet, a graphical viewer for visualization of assemblies and read mappings [17]. A comparison of two assemblies obtained from both of the assemblers was used to fill the gaps between contigs. The multiple genome alignment tool Mauve was used for this purpose [18]. The working draft genome sequence of S. schinkii Sp3 contains 3,196,921 bp based on the analysis done with the tools summarized above.
Genome annotation
Automated gene modeling was completed by MaGe [19] a bacterial genome annotation system. Genes were identified using Prodigal [20] and AMIGene [21] as part of MaGe genome annotation pipeline. The predicted CDSs were translated and used to search the NCBI non-redundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases using BLASTP. Predicted coding sequences were subjected to manual analysis using MaGe web-based platform, which also provides functional information of proteins, and which was used to assess and correct genes predicted through the automated pipeline. The predicted functions were also further analyzed by the MaGe annotation system (Fig. 4).
Fig. 4.
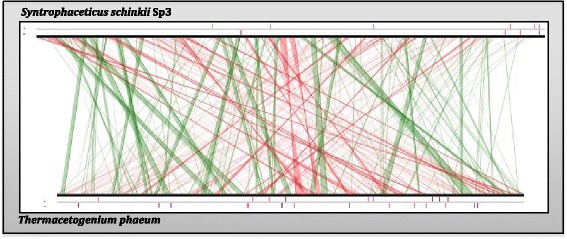
Synteny comparison. Synteny comparison of S. schinkii genome with the closely related genome of T. phaeum. Linear comparison of all predicted gene loci from S. schinkii with T. phaeum was perfomed using built-in tool in MaGe Platform with the synton size of > = 3 genes. The lines indicate syntons between two genomes. Red lines show inversions around the origin of replication. Vertical bars on the boarder line indicate different elements in genomes such as pink: transposases or insertion sequences: blue: rRNA and green: tRNA
Genome properties
The working draft genome comprises 301 contigs in 215 scaffolds with a total size of 3,196,921 bp and a calculated GC content of 46.59 %. The genome shows a protein coding density of 75.21 % with an average intergenic length of 230.2 bp. The genome encodes further 50 tRNA genes and 5 rRNA genes, more precisely three 5S genes, one 16S and one 23S rRNA gene (Table 3, Fig. 3).
Table 3.
Genomic statistics for the Syntrophaceticus schinkii strain Sp3 genome
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 3,196,921 | 100.00 |
| DNA Coding (bp) | 2,399,289 | 75.05 |
| DNA G + C content (bp) | 1,489,445 | 46.59 |
| Number of scaffolds | 215 | - |
| Total genes | 3,441 | 100.00 |
| Protein coding genes | 3,281 | 95.35 |
| RNA genes | 55 | 1.59 |
| Pseudo gene | 90 | 2.61 |
| Genes in internal clusters | 2,086 | 60.62 |
| Genes with function prediction | 2,099 | 61.00 |
| Genes assigned to COGs | 2,583 | 75.07 |
| Genes with Pfam domains | 2,749 | 79.88 |
| Genes with signal peptides | 57 | 1.65 |
| CRISPR repeats | 8 | .23 |
Fig. 3.
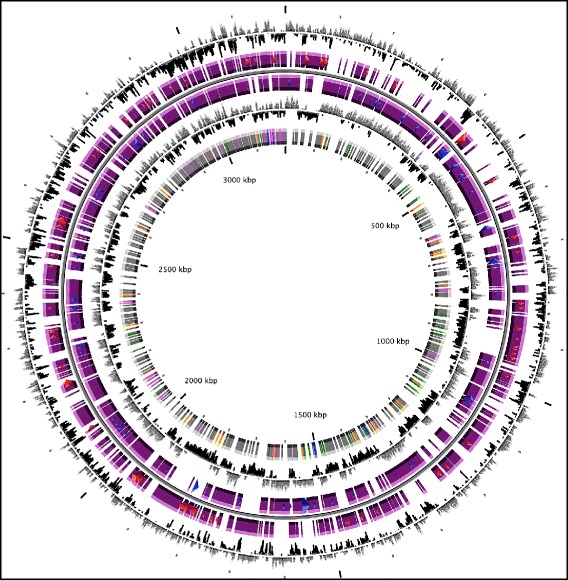
Circular map. Circular map of the Syntrophaceticus schinkii Sp3 genome (from the outside to the center): (1) GC percent deviation (GC window—mean GC) in a 1000-bp window. (2) Predicted CDSs transcribed in the clockwise direction. (3) Predicted CDSs transcribed in the counterclockwise direction. (4) GC skew (G + C/G-C) in a 1000-bp window. (5) rRNA (blue), tRNA (green), misc_RNA (orange), Transposable elements (pink) and pseudogenes (grey)
The genome of S. schinkii genome contains 3,441 predicted protein-encoding genes, of which 2,099 (61 %) have been assigned tentative functions. The remaining 1,346 ORFs are hypothetical / unknown proteins. 2,586 (app. 75 %) of all predicted protein-encoding genes could be allocated to the 22 functional COGs. This is in the same range as described for other acetogenic bacteria such as Acetobacterium woodii WB1 and M. thermoaceticaATCC39073, acetate oxidizing sulfate reducers such as Desulfobacterium autotrophicum HRM2 and Desulfotomaculum kuznetsovii, and the SAOB P. lettingae TMO. Analysis of COGs revealed that ~28 % of all protein-encoding genes fall into four main categories: amino acid transport and metabolism (9.8 %), replication, recombination and repair (6.6 %), energy metabolism (5.9 %), and coenzyme transport and metabolism (4.9 %) (Table 4).
Table 4.
Number of genes associated with the general COG functional categories
| Code | Value | % age | Description |
|---|---|---|---|
| J | 156 | 4.53 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.00 | RNA processing and modification |
| K | 211 | 6.12 | Transcription |
| L | 230 | 6.68 | Replication, recombination and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 59 | 1.71 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.00 | Nuclear structure |
| V | 117 | 3.39 | Defense mechanisms |
| T | 136 | 3.95 | Signal transduction mechanisms |
| M | 169 | 4.90 | Cell wall/membrane/envelope biogenesis |
| N | 37 | 1.07 | Cell motility |
| Z | 1 | 0.02 | Cytoskeleton |
| W | 1 | 0.03 | Extracellular structures |
| U | 61 | 1.77 | Intracellular trafficking, secretion, and vesicular transport |
| O | 101 | 2.93 | Posttranslational modification, protein turnover, chaperones |
| C | 204 | 5.92 | Energy production and conversion |
| G | 138 | 4.00 | Carbohydrate transport and metabolism |
| E | 339 | 9.84 | Amino acid transport and metabolism |
| F | 70 | 2.03 | Nucleotide transport and metabolism |
| H | 172 | 4.99 | Coenzyme transport and metabolism |
| I | 52 | 1.51 | Lipid transport and metabolism |
| P | 206 | 5.98 | Inorganic ion transport and metabolism |
| Q | 54 | 1.57 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 369 | 10.71 | General function prediction only |
| S | 219 | 6.36 | Function unknown |
| 342 | 9.93 | Not in COGs |
Insights from the genome sequence
Synteny-based analyses with all bacterial genomes present in the NCBI Reference Sequence database confirmed again that T. phaeum is the closest relative of S. schinkii having approximately 50 % of the total genome size in synteny (Fig. 4). A comparison of all inferred proteins of S. schinkii with all proteins collected in the NCBI RefSeq database revealed the highest number of orthologous (1788: 51.90 %) with T. phaeum. Both S. schinkii and T. phaeum, are known as syntrophic acetate oxidizing bacteria able to oxidize acetate in co-culture with a hydrogenotrophic methanogenic partner, but differ clearly in their substrate utilization patterns [2, 6] Moreover, in contrast to the thermophilic T. phaeum, S. schinkii possess mesophilic characteristics and cannot switch to a chemolithoautotrophic lifestyle.
The genome has been analyzed regarding general phenotypic features such as sporulation, oxygen tolerance, secreted and selenocystein-containing proteins and motility. The genome contains the master regulator Spo0A (SSCH_630004) needed for sporulation but lacks genes encoding the phosphorelays Spo0F and Spo0B as it has been observed in other clostridia. All the sporulation-specific sigma factors SigE (SSCH_460001), SigG (SSCH_1070017), and SigK (SSCH_700028) were predicted except for SigF. Two putative manganese containing catalases (SSCH_1760003, SSCH_2560004) and two putative rubrerythrin encoding genes (SSCH_590006, SSCH_180042) identified within the genome give reasons to believe, that this organism posses the ability to tolerate small amounts of oxygen. According to the observed immobility S. schinkii does not harbor any flagellum related genes including hook-associated proteins (FlgE, FlgK, FlgL), basal and hook proteins (FlgE), capping proteins (FliD), biosynthesis secretory proteins (FlhA, FlhB, FliF, FliH and FliI), flagella formation proteins, motor proteins (FliG and FliM) and the basal proteins (FlgC and FlgB).
Genes encoding key components of the selenocysteine-decoding (SelA, SelB, SelC, SelD) machinery are widely distributed in bacterial genomes. Also S. schinkii appears to have the ability to express selenocysteine proteins: The genome contains a single copy of the L-selenocysteinyl-tRNASec transferase (selA: SSCH_110005/6), monoselenophosphate synthase (selD: SSCH_970007), the selenocysteinyl-tRNA specific elongation factor (selB: SSCH_110004) and potential selenocysteine-specific tRNASec (selC: SSCH_tRNA31). We found two potential selenocysteine containing glycine/sarcosine/betaine reductase complexes encoded by the genome (SSCH_440002-8, SSCH_960012-15) consisting of selenoprotein subunit A, the substrate specific selenoprotein subunit B and acetyl phosphate forming subunit C. Since S. schinkii can only grow on betaine but not on glycine or sarcosine [6], this reductase complex might be specifically involved in betaine utilization. 57 CDSs were predicted to encode surface associated or secreted proteins identified by putative N-terminal signal peptides (signal peptide I and II).
Conclusions
Acetate oxidation under anoxic conditions is thermodynamically unfavorable and requires the metabolic cooperation of a partner organism in order to make endergonic reactions more exergonic through the efficient removal of the products. S. schinkii oxidizes acetate to hydrogen and/or formate, which is directly used by a hydrogenotrophic methanogen. Since the methanogenic partner has been isolated and sequenced S. schinkii appears to have great potential to serve as a model organism for studying methane producing syntrophic relationships. The working draft genome sequence presented here will open the door for understanding the preferred habitats, the metabolism behind different life styles, and the mechanisms initiating syntrophy. This knowledge will help us to trigger SAOB towards an efficient and stable hydrogen/biogas production in engineered anaerobic digestion processes suffering high ammonia release.
Acknowledgements
This work was supported by the Higher Education Commission, Pakistan, University of the Punjab, Lahore, Pakistan. Uppsala Genome Center performed sequencing supported by Science for Life Laboratory (Uppsala), the Swedish Bioinformatics Infrastructure for the Life Sciences supporting the SGBC bioinformatics platform at SLU, University of the Punjab, Lahore, Pakistan and Uppsala Multidisciplinary Center for Advanced Computational Science, Uppsala, Sweden. The contribution of SM and EB-R was supported by EU-COST action BM1006-SeqAhead. EB-R was also partially supported by EU FP7 ALLBIO project, grant number 289452, The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We wish to thank Maria Westerholm, who isolated and characterized Syntrophaceticus schinkii strain Sp3 for providing the micrograph.
Abbreviations
- SAOB
Syntrophic acetate-oxidizing bacteria
- DSMZ
Deutsche Sammlung für Mikroorganismen und Zellkulturen
- MIRA
Mimicking Intelligent Read Assembly
- MaGe
Magnifying Genomes
- BLASTP
Basic local alignment search tool for proteins
- NCBI
National Center for Biotechnology Information
Additional file
Table S1. Associated MIGS record for Syntrophaceticus schinkii strain Sp3. (DOCX 111 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SM, BM and AS contributed to the conception and design of this project. SM was involved in the acquisition and initial analysis of the data. SM and BM were involved in the interpretation of the data. SM prepared the first draft of the manuscript. EBR and AS provided financial support. All authors were involved in the critical revision of the manuscript and have given final approval of the version to be published and agree to be accountable for all aspects of the work.
References
- 1.Ferry JG. Fermentation of Acetate. In: Ferry DJG, ed. Methanogenesis. Chapman & Hall Microbiology Series. Springer US; 1993:304–334. Available at: http://link.springer.com/chapter/10.1007/978-1-4615-2391-8_7. Accessed March 3, 2014.
- 2.Hattori S, Kamagata Y, Hanada S, Shoun H. Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate-oxidizing bacterium. Int. J. Syst. Evol. Microbiol. 2000;50 Pt 4:1601–1609. doi: 10.1099/00207713-50-4-1601. [DOI] [PubMed] [Google Scholar]
- 3.Petersen SP, Ahring BK. Acetate oxidation in a thermophilic anaerobic sewage-sludge digestor: the importance of non-aceticlastic methanogenesis from acetate. FEMS Microbiol. Lett. 1991;86:149–152. doi: 10.1111/j.1574-6968.1991.tb04804.x. [DOI] [Google Scholar]
- 4.Schnürer A, Svensson BH, Schink B. Enzyme activities in and energetics of acetate metabolism by the mesophilic syntrophically acetate-oxidizing anaerobe Clostridium ultunense. FEMS Microbiol. Lett. 1997;154:331–336. doi: 10.1016/S0378-1097(97)00350-9. [DOI] [Google Scholar]
- 5.Anna Schnürer BS. Clostridium ultunense sp. nov., a Mesophilic Bacterium Oxidizing Acetate in Syntrophic Association with a Hydrogenotrophic Methanogenic Bacterium. First Publ Int. J. Syst. Bacteriol. 1996;46(4):1145–1152. doi: 10.1099/00207713-46-4-1145. [DOI] [PubMed] [Google Scholar]
- 6.Westerholm M, Roos S, Schnürer A. Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol. Lett. 2010;309:100–104. doi: 10.1111/j.1574-6968.2010.02023.x. [DOI] [PubMed] [Google Scholar]
- 7.Westerholm M, Roos S, Schnürer A. Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate-oxidizing bacterium isolated from two ammonium-enriched mesophilic methanogenic processes. Syst. Appl. Microbiol. 2011;34:260–266. doi: 10.1016/j.syapm.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 8.Balk M, Weijma J, Stams AJM. Thermotoga lettingae sp. nov., a novel thermophilic, methanol-degrading bacterium isolated from a thermophilic anaerobic reactor. Int. J. Syst. Evol. Microbiol. 2002;52:1361–1368. doi: 10.1099/00207713-52-4-1361. [DOI] [PubMed] [Google Scholar]
- 9.Oehler D, Poehlein A, Leimbach A, et al. Genome-guided analysis of physiological and morphological traits of the fermentative acetate oxidizer Thermacetogenium phaeum. BMC Genomics. 2012;13:723. doi: 10.1186/1471-2164-13-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller B, Manzoor S, Niazi A, Bongcam-Rudloff E, Schnürer A. Genome-guided analysis of physiological capacities of Tepidanaerobacter acetatoxydans provides insights into environmental adaptations and syntrophic acetate oxidation. PloS One. 2015;10 doi: 10.1371/journal.pone.0121237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manzoor S, Müller B, Niazi A, Bongcam-Rudloff E, Schnürer A. Draft Genome Sequence of Clostridium ultunense Strain Esp, a Syntrophic Acetate-Oxidizing Bacterium. Genome Announc. 2013;1:e00107–13. doi: 10.1128/genomeA.00107-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anon. Classifier. Available at: https://rdp.cme.msu.edu/classifier/classifier.jsp. Accessed November 2, 2015.
- 13.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008;36:D475–D479. doi: 10.1093/nar/gkm884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anon. SciLifeLab. Available at: https://www.scilifelab.se/. Accessed November 2, 2015.
- 16.Andrews S. FastQC A Quality Control tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 17.Milne I, Stephen G, Bayer M, et al. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013;14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 18.Darling AE, Mau B, Perna NT. ProgressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE; 2010;5:e11147. [DOI] [PMC free article] [PubMed]
- 19.Vallenet D, Labarre L, Rouy Z, et al. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006;34:53–65. doi: 10.1093/nar/gkj406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bocs S, Cruveiller S, Vallenet D, Nuel G, Medigue C. AMIGene: Annotation of MIcrobial Genes. Nucleic Acids Res. 2003;31:3723–3726. doi: 10.1093/nar/gkg590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Field D, Garrity G, Gray T, et al. The minimum information about a genome sequence (MIGS) specification. Nat. Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludwig W, Schleifer K-H, Whitman WB. Revised road map to the phylumFirmicutes. In: Vos PD, Garrity GM, Jones D, et al., eds. Bergey’s Manual® of Systematic Bacteriology. Springer New York; 2009:1–13. Available at: http://link.springer.com/chapter/10.1007/978-0-387-68489-5_1. Accessed March 3, 2014.
- 24.Wolf M, Müller T, Dandekar T, Pollack JD. Phylogeny of Firmicutes with special reference to Mycoplasma (Mollicutes) as inferred from phosphoglycerate kinase amino acid sequence data. Int. J. Syst. Evol. Microbiol. 2004;54:871–875. doi: 10.1099/ijs.0.02868-0. [DOI] [PubMed] [Google Scholar]
- 25.Gibbons NE, Murray RGE. Proposals Concerning the Higher Taxa of Bacteria. Int. J. Syst. Bacteriol. 1978;28:1–6. doi: 10.1099/00207713-28-1-1. [DOI] [Google Scholar]
- 26.Editor L. List of new names and new combinations previously effectively, but not validly, published. List no. 132. Int. J. Syst. Evol. Microbiol. 2010;60:469–472. doi: 10.1099/ijs.0.024562-0. [DOI] [PubMed] [Google Scholar]
- 27.Rainey FA, Class II. Clostridia class nov. 2nd ed. (Vos PD, Garrity G, Jones D, et al., eds.) New York: Springer-Verlag; 2009. [Google Scholar]
- 28.Wiegel J, Order III. Thermoanaerobacterales ord. nov. 2nd ed. (Vos PD, Garrity G, Jones D, et al., eds.) New York: Springer-Verlag; 2009. [Google Scholar]
- 29.Wiegel J, Family I. Thermoanaerobacteraceae fam. nov. 2nd ed. (Vos PD, Garrity G, Jones D, et al., eds.) New York: Springer-Verlag; 2009. [Google Scholar]
- 30.Editor L. List of new names and new combinations previously effectively, but not validly, published. Int. J. Syst. Evol. Microbiol. 2011;61:1011–1013. doi: 10.1099/ijs.0.033498-0. [DOI] [Google Scholar]
- 31.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Felsenstein J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 34.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 35.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felsenstein J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution. 1985;39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]