

Abstract
Lenalidomide, an analog of thalidomide, modified responses of stimulated T cells from healthy young (ages 21–40 years) and old (≥age 65 years) subjects. At 0.03 μM to 1 μM, lenalidomide enhanced generation of IL-2 and IFN-γ by T cell receptor-stimulated T cells of young subjects up to respective maximum increases of 17-fold and three-fold, but at 0.3 μM and 1 μM suppressed IL-17 generation. The same concentrations of lenalidomide enhanced IL-2 and IFN-γ generation by stimulated T cells of old subjects more, with greater respective maximal increases of up to 120-fold and six-fold, without suppressing IL-17 generation. Lenalidomide enhanced proliferation and suppressed apoptosis of stimulated T cells from old subjects, by IL-2-dependent mechanisms, and restored diminished T cell chemotactic responses to CCL21 and sphingosine 1-phosphate. The reversal of T cell abnormalities of immunosenescence by low concentrations of lenalidomide suggest a potential for improvement of immunity in the elderly.
Keywords: Human, T lymphocytes, Cytokines, Proliferation, Apoptosis, Chemotaxis
1. Introduction
The state of human immunosenescence encompasses many changes in immunity and inflammation that develop with aging [1,2]. The numbers and functions of T cells, B cells, macrophages, dendritic cells and NK cells all change during human aging [3–8]. Several profound alterations in T cells, however, constitute the most fundamental basis for diminished adaptive immunity and consequently reduced host defenses in the elderly. There are consistent major reductions in the number of naïve T cells, strikingly diminished diversity of the T cell antigen receptor (TCR) repertoire, increased susceptibility to activation-induced apoptosis, and substantial modifications in profiles of T cell immune cytokine production in elderly humans [9–15]. Additional aspects of dysregulated T cell function regularly observed in immunosenescence include formation of a less effective synapse between antigen-presenting cells and T cells, decreases in some enhancing subsets of T cells, and increases in the number and especially activities of suppressor Treg subsets [16–20]. Other prominent modifications of the human T cell constellation with aging, that have uncertain functional consequences, are increases in some subsets of memory T cells and the CD8+CD28− subset of T cells [21–23].
One universal alteration in elderly human T cell cytokine production is a significant reduction in generation of IL-2, that is vital for TCR-mediated activation of naïve T cells and their differentiation into effector T cells, as well as for many other host defensive activities [15,24,25]. IFN-γ, that like IL-2 is highly protective of human hosts through diverse effects in innate and adaptive immunity, is another T cell cytokine whose generation is altered substantially with human aging [15]. Relative to T cells of concurrently-evaluated healthy young controls, T cells of healthy old women produce more IFN-γ and T cells of healthy old men produce less IFN-γ when stimulated in vitro. Even the T cells of healthy old women that produce more IFN-γ than those of young controls have lesser increments in IFN-γ generation than controls during infections and other inflammatory challenges [15]. It is not clear if any countermeasures that correct dysregulated generation of protective T cell cytokines in the elderly will improve their immune defenses against infections and other challenges. Thus there is great interest in studying welltolerated drugs that are capable of normalizing the immune cytokine alterations of immunosenescence sufficiently to improve elderly human protective immune responses.
Amino-substituted analogs of thalidomide such as lenalidomide and pomalidomide, termed thalidomide-type immunoactive drugs or immunomodulatory drugs (IMDs), have been developed that are much more potent inhibitors of TNF-α production by LPS-stimulated monocytes than thalidomide [26,27]. At concentrations of 0.01 μM to 1 μM, the IMDs also increase the number and ex vivo cytotoxic activity of NK cells from treated subjects and enhance the proliferation and cytokine generation of CD4 and CD8 T cells stimulated by anti-CD3 antibody or phorbol esters plus ionomycin in vitro [28–31]. In contrast to the high potency of IMD stimulation of CD4 and CD8 T cells, inhibition of IL-2-stimulated proliferation and suppressive activity of Treg cells is inhibited more than 50% by IMDs only at concentrations of 10μM or higher [32]. Although the mechanisms of IMD stimulation of T cell activities have not been unequivocally established, three possibilities have been identified. One is a selective increase in AP-1 DNA-binding activity [31]. The second is activation of RhoA and Rac1, but not other Rho GTPases [33]. The third is induction of tyrosine phosphorylation of CD28 and consequently enhanced activation of NF-κB, that reduces dependence on CD28-CD80/CD86 co-stimulation for enhancement of IL-2 generation [34]. In this system, IMDs also reduce the inhibitory effect of CTLA-4-lg on IL-2 generation by minimizing the otherwise requisite role of CD28-CD80/CD86 co-stimulation.
We now show that nM concentrations of the IMD lenalidomide, but not thalidomide or some analogs of thalidomide and lenalidomide, alter cytokine generation by stimulated T cells of old healthy subjects differently than that by T cells of young healthy subjects. In the same range of concentrations, lenalidomide partially reversed aging-related modifications of TCR-stimulated proliferation, activation-induced apoptosis and Chemotaxis of T cells from old subjects. The capacity of low concentrations of lenalidomide to improve several of the T cell abnormalities of immunosenescence, suggest it may be a beneficial therapy for restoration of T cell-dependent immunity in the elderly who have impaired host defenses.
2. Materials and methods
2.1. Selection of study subjects
The healthy old subjects (≥65 years of age) were recruited from the Ambulatory Faculty Practice of UCSF Medical Center and the Jewish Home of San Francisco. Each was evaluated concurrently with a healthy young subject of age 21 to 40 years, who was matched with the old subject for gender, race and national origin. None of the healthy old or young subjects had any history of immune or inflammatory disorders and none was receiving any immunoactive drug or hormone. The study procedures and potential risks were described to every participant, who then signed a consent form that was approved by the UCSF Committee for Human Research. Sixty milliliters of venous blood was taken into a heparinized syringe once from each subject.
2.2. Isolation and stimulation of T cells and monocytes
Heparinized blood diluted 1:1 with Ca++- and Mg++-free Dulbecco’s balanced salt solution was centrifuged on cushions of Ficoll-Paque (GE Healthcare Lifesciences, Pittsburgh, PA) to purify mixed mononuclear leukocytes as described [15]. Monocytes were isolated from mononuclear leukocytes by anti-human CD14 immunomagnetic bead positive selection on columns in a magnetic field (Miltenyi Biotec, Auburn, CA). Twenty milliliter to 30 ml suspensions of 106 monocytes/ml of RPMI-1640 medium with 10% FBS, 100 U/ml of penicillin and 50 μg/ml of streptomycin (complete RPMI) were incubated for 24 h with 104 lU/ml of human recombinant IFN-α 2A (ProSpec-Tany TechnoGene Ltd., Rehovot, Israel) to induce differentiation. T cells were isolated from mononuclear leukocytes by two cycles of immunomagnetic negative selection (human T cell isolation kit II; Miltenyi Biotec, Inc.). T cell purity and the levels of expression of CD4, CD8 and CD28 were delineated by flow cytometry and T cell purity was consistently ≥96% [20].
Aliquots of 5×105 differentiated monocytes in 0.5 ml of complete RPMI-1640 then were preincubated for 60 min at 37°C without and with a thalidomide-type drug, that remained in the suspensions, and stimulated with 100 ng/ml of LPS (E. coli 0111:B4, Sigma Chemicals, St. Louis, MO). Portions of supernatant were removed at 6 h, 24 h and 48 h. Suspensions of 2×106 T cells per ml of complete RPMI were preincubated for 15 min at room temperature without and with 33 μg/ml of anti-human IL-2 receptor-α chain mouse monoclonal antibody (ab62849 at 33 μg/ml; Abcam, Inc., Cambridge, MA). Then suspensions were incubated further for 60 min at 37°C without and with a thalidomide-type drug. Replicate 0.8 ml aliquots of the treated and control T cell suspensions, that still contained the anti-human IL-2 receptor-α chain mouse monoclonal antibody and thalidomide-type drug, were transferred to 24-well plates that had been precoated with 2 μg per well of anti-human CD3 plus anti-human CD28 antibodies (eBioscience, San Diego, CA). One hundred microliter portions of medium from each well were harvested at one, three and five days for cytokine ELISAs.
2.3. Cytokine ELISAs
The concentrations of IL-2, IL-17 and IFN-γ in T cell supernates and TNF-α in monocyte supernates were determined at two dilutions by colorometric ELISAs (eBioscience). Color intensity of each well was determined in an ELISA plate reader (MRX Revelation, Dynex Technologies, Chantilly, VA).
2.4. ELISAs for T cell proliferation, apoptosis and Bcl-XL content
As for cytokine generation, suspensions of 1 × 106 T cells/ml were preincubated without or with 33 μg/ml of anti-human IL-2 receptor-α chain mouse monoclonal antibody and/or a thalidomide-type compound, that remained with the T cells. Aliquots then were transferred to wells of a 96-well plate precoated with either 1 μg each of anti-human CD3 antibody plus anti-CD28 antibody for initiation of proliferation or with 1 μg of anti-human CD3 antibody without anti-human CD28 antibody for induction of apoptosis. After one or two days of proliferation-stimulating incubation, BrdU was added to each well, incubation was continued for one more day, and then the T cells were fixed and their BrdU-labeled DNA was denatured. Peroxidase-conjugated Fab of anti-BrdU antibody and then peroxidase substrate were introduced sequentially. Newly-produced BrdU-labeled DNA, that is a linear indicator of T cell proliferation on day two and three (Cell Proliferation Assay Kit, Roche Biosciences, Indianapolis, IN), was quantified spectroscopically at 450 nm. After two and three days of apoptosis-evoking incubation, T cells were lysed and their nucleosomes in the lysates were trapped by anti-histone antibodies coating each well. The number of antibody-immobilized nucleosomes was quantified by their binding of peroxidase-conjugated anti-DNA antibodies (cell death detection kit, Roche Biosciences, Indianapolis, IN). The consequent rate of conversion of peroxidase substrate to a product with maximal absorption at 405 nm was directly related to the number of nucleosomes and thus the level of apoptosis.
T cells stimulated without and with different treatments for two or three days were washed twice with PBS and solubilized at 107/ml in kit lysis buffer for ELISA quantification of Bcl-XL content using the reagents and procedures of DuoSet IC ELISA system (R&D Systems, Inc., Minneapolis, MN). Values were normalized according to total protein determined with a BCA kit (ThermoFisher-Pierce, Rockford, IL) and the effective range of the ELISA was 200–30,000 pg/ml.
2.5. Quantification of T cell Chemotaxis
To assess Chemotaxis, each upper insert of Transwell permeable supports with a 5 μm diameter pore filter (Corning Life Sciences, Lowell,MA) received 1×106 T cells in 0.1 ml and was placed over 0.6 ml of RPMI-1640 with 10% charcoal- and dextran-extracted fetal bovine serum (lipid-free FBS) (UCSF Cell Culture Facility) without or with a stimulus in the lower compartment. The filters had been preincubated in human type IV collagen for 8 h and then washed and dried overnight, as described [35]. The T cells had been pretreated with 0.01 μM and 0.1 μM lenalidomide for 30 min at 37°C and the stimuli were 10 nM S1P and 100 nM CCL21 and CCL5 (Peprotech, Inc., Rocky Hill, NJ). After 4 h of incubation at 37°C, the number of T cells in each lower compartment was determined by microscopic counting and expressed as a percentage of the initial number added to the upper insert.
2.6. Thalidomide-type compounds
Lenalidomide (Revlimid, CC-5013) was obtained from three sources, each of which was shown in initial studies to have the same effects on T cells. Five milligram pharmaceutical tablets and purified powder from Celgene, Inc. (San Diego, CA) were dissolved in anhydrous dimethyl sulfoxide (DMSO) without further chemical analyses. A third lot of purified powder was synthesized and its structural properties authenticated at NIA (WL, DT and NHG). Thalidomide was obtained from Sigma-Aldrich Co. (St. Louis, MO) and the di-thiol derivatives of thalidomide and lenalidomide were synthesized and their structural properties authenticated at NIA (WL, DT and NHG), as described [36]. All stocks were maintained at −20°C as 10 mM solutions in anhydrous DMSO and diluted in medium just prior to addition to T cell suspensions.
2.7. Data analyses
All groups of data were normally distributed. The statistical significance of each difference between levels of cytokines, proliferation, Chemotaxis and apoptosis was determined by a two-sample t test.
3. Results
3.1. Effects of lenalidomide and other thalidomide-type compounds on the generation of cytokines by T cells of healthy old and young subjects
The capacity of several different thalidomide-type compounds to alter the generation of three distinct immune cytokines by TCR-stimulated T cells of young and old subjects was investigated using concentrations of each compound known to have immune effects [27,37]. In initial studies designed to define the dose-range of thalidomide-type drugs, lenalidomide enhanced generation of IFN-γ by TCR-stimulated T cells of two young and two old healthy subjects significantly at concentrations of 0.03 μM to 3 μM up to a peak of nearly three-fold at 3 μM (Fig. 1A). Significant inhibition of generation of IFN-γ was observed instead for dithiol lenalidomide beginning at 1 μM and di-thiol thalidomide beginning at 3 μM, with only modest inhibition by thalidomide at 100 μM. For stimulated T cell generation of IL-2, again only lenalidomide enhanced the levels significantly at concentrations of 0.03 μM to 30 μM, with mean maximal enhancement of 9.5-fold at 1 μM in this mixed group of young and old subjects (Fig. 1B). Significant inhibition of IL-2 generation was found for di-thiol lenalidomide beginning at 3 μM and for di-thiol thalidomide starting at 30 μM, whereas thalidomide had no detectable effect at up to 100 μM. The distinctively high level of variation of lenalidomide enhancement of IL-2 generation suggested that there may be differences in the effects of lenalidomide on T cells of young and old subjects.
Figure 1.
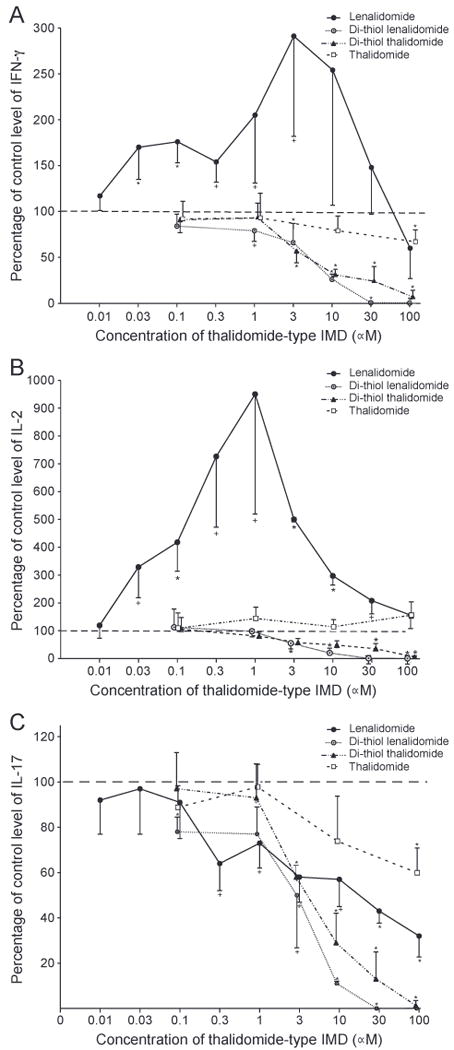
Concentration-dependence of effects of thalidomide-type drugs on human T cell generation of cytokines. Each column and bar depicts the mean ± S.D. of the results of cytokine ELISAs of supernates from T cells of a total of four healthy subjects, of whom two were young and two were old, after three days of incubation with anti-CD3 + anti-CD28 antibodies without (control = 100%) and with a drug. The mean ± S.D. of control (100%) values for IFN-γ, IL-2 and IL-17, respectively, were 30,915 ± 11,206, 5.8 ± 3.4 and 1149 ± 372pg/ml.A= IFN-γ, B = IL-2 and C=IL-17. Statistical significance of differences from the controls without an IMD was calculated by a two-sample t test and indicated by + for pb0.05, * for pb 0.01 and ** for pb 0..001.
No thalidomide-type drug was capable of enhancing TCR-stimulated T cell generation of IL-17. However, there was significant inhibition of generation of IL-17 by di-thiol lenalidomide at 0.1 μM and higher, lenalidomide at 0.3 μM and higher, di-thiol thalidomide at 3 μM and higher, and thalidomide only at 100 μM (Fig. 1C). None of the thalidomide-type drugs altered cytokine generation alone in the absence of TCR-directed stimulation (data not shown).
As lenalidomide enhancement of generation of both IL-2 and IFN-γ were significant in the low concentration range of 0.03 μM to 1 μM for TCR-stimulated T cells from a mixed age group, the time-courses of the effects of these concentrations were delineated for T cells of young and old healthy subjects separately.
For TCR-stimulated T cells of six healthy young subjects (three female and three male), lenalidomide enhancement of generation of IFN-γ was significant at 0.03 μM to 1 μM on days one, three and five, up to maximal levels of two-fold on day three and nearly three-fold on day five (Fig. 2A). Lenalidomide enhancement of IL-2 generation in the same studies was significant at 0.01 μM to 1 μM on all days, was much greater than for IFN-γ, and attained maximal increases of 17-fold on day three and 10-fold on day five (Fig. 2B). In contrast, 0.3 μM and 1 μM lenalidomide suppressed generation of IL-17 on all days, whereas none of the lower concentrations had any significant effects (Fig. 2C). For T cells of six healthy old subjects (three female and three male), 0.01 μM to 1 μM lenalidomide enhanced generation of IFN-γ significantly on all days up to maximal levels of fourfold on day three and six-fold on day five (Fig. 3A). In studies of the same healthy old subjects, generation of IL-2 was enhanced significantly by 0.03 μM to 1 μM lenalidomide on all days with maximal increases of 120-fold and 95-fold at 0.3 μM and 1 μM lenalidomide, respectively (Fig. 3B). The generation of IL-2 by T cells, especially those of old subjects, decreased from a peak on day one to a level approximately 10% of the peak by day five in the absence of lenalidomide. Thus the very striking enhancement of IL-2 generation by T cells of old subjects achieved by lenalidomide effectively prevents this decline and leads to an average level of IL-2 on day five that is ten-fold higher than the level on day one without lenalidomide. Unlike the suppression of IL-17 generation by TCR-stimulated T cells of young subjects by 0.3 μM and 1 μM lenalidomide on days one, three and five, the production of IL-17 by TCR-stimulated T cells of old subjects was not affected significantly by any level of lenalidomide, except for minor suppression of IL-17 at 0.01 μM (Fig. 3C).
Figure 2.
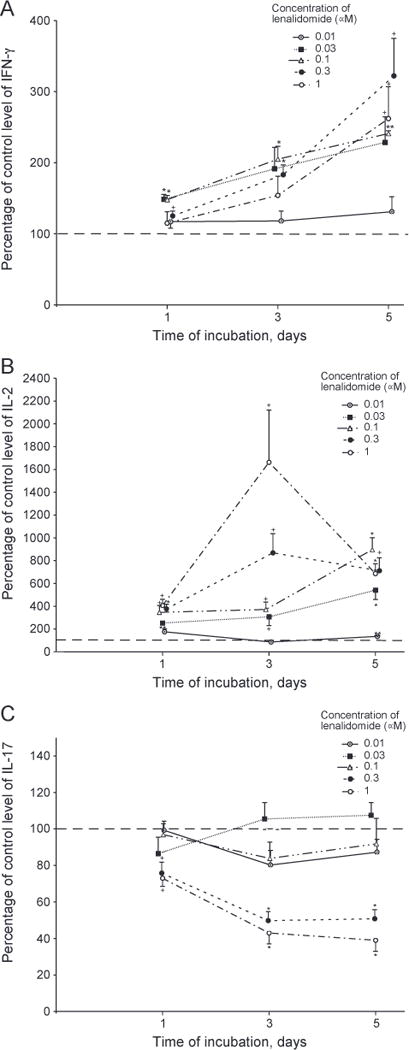
Time-course of effect of lenalidomide on generation of cytokines by T cells of young subjects. Conditions, anti-TCR antibody stimulation, methods of quantification of cytokines, and calculation of statistical significance were the same as in Figure 1. Each column and bar depicts the mean ± S.E.M. of the results of cytokine ELISAs of supernates from T cells of six healthy young subjects (three women and three men). The mean ± S.D. of control (100%) values for IFN-γ, IL-2 and IL-17, respectively, were 3901 ± 1087, 535 ± 85 and 296 ± 92 pg/ml on day 1,33,623 ± 8934, 29 ± 3.4 and 1149 ± 372 pg/ml on day 3, and 49,095 ± 13,501,18 ± 1.8 and 1521 ± 376 pg/ml on day5.A=IFN-γ, B = IL-2 and C = IL-17.
Figure 3.
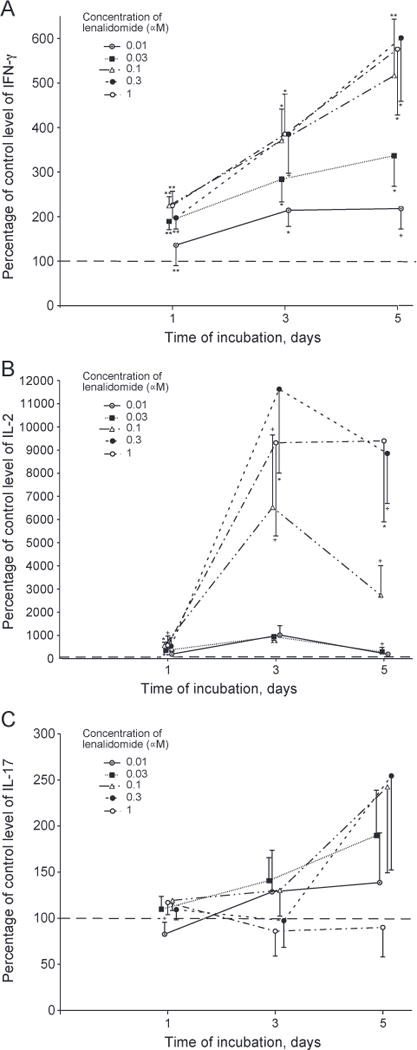
Time-course of effect of lenalidomide on generation of cytokines by T cells of old subjects. Conditions, anti-TCR antibody stimulation, methods of quantification of cytokines, and calculation of statistical significance were the same as in Figure 1. Each column and bar depicts the mean ± S.E.M. of the results of cytokine ELISAs of supernates from T cells of six healthy old subjects (three women and three men). The mean ± S.D. of control (100%) values for IFN-γ, IL-2 and IL-17, respectively, were 1786 ± 1065, 371 ± 247 and 79 ± 57 pg/ml on day 1,26,804 ± 15,216,15 ± 10 and 354 ± 233 pg/ml on day 3, and 115,839 ± 52,211,13 ± 8 and 721 ± 454 pg/ml on day 5.A=IFN-γ, B = IL-2 and C=IL-17.
The observed effects of lenalidomide on cytokine generation were not attributable to alterations in the subset composition of T cells. The mean percentages ± S.D. of CD4 and CD8 T cells in the old subjects (n=4 of 6) were 66 ± 7.4 and 29 ± 5.8, respectively, after two days of TCR stimulation in medium alone and were no different at 69 ± 8.0 and 24 ± 4.6 after two days of TCR stimulation with 1 μM lenalidomide. Similarly, no changes in expression of CD28 were induced by lenalidomide in CD4 and CD8 T cells after 2 days of TCR stimulation with respective mean percentages of 92 and 64 without and 94 and 67 with 1 μM lenalidomide. No NK cells were detected in any suspension as anticipated based on the T cell purification procedure.
3.2. Effects of lenalidomide on T cell generation of IL-2 and IFN-γ: amplification by IL-2 signaling
Lenalidomide significantly enhanced generation of IL-2 by stimulated T cells from two young and three old healthy subjects at concentrations of 0.03 μM to 1 μM on days 1, 3 and 5 and at 0.01 μM on some days (Table 1). In this mixed group of old and young healthy subjects, very significant increases of 21 -fold on day three and 10-fold on day five were observed at 0.1 μM lenalidomide and maximal enhancement of a mean of 32-fold on day three and 37-fold on day five were attained at 1 μM lenalidomide. The presence of a blocking concentration of mouse monoclonal anti-human IL-2 receptor antibody prior to and during exposure to lenalidomide and anti-TCR antibodies (+) significantly decreased all levels of enhancement of IL-2 generation on all days that had been significant in the absence of IL-2 receptor blockade (−) (Table 1). Similarly, the anti-human IL-2 receptor blocking antibody (+) significantly decreased all levels of enhancement of IFN-γ generation on all days that had been significant in the absence of IL-2 receptor blockade (−) (Table 1). The positive feedback enhancement of IL-2 generation and amplification of IFN-γ generation by lenalidomide-evoked increases in the levels of IL-2 thus are mediated by the IL-2 receptor and contribute substantially to the observed steady state levels. In contrast, levels of generation of IL-17 were not affected by the anti-human IL-2 receptor blocking antibody.
Table 1.
IL-2 receptor signaling requirement for optimal lenalidomide enhancement of T cell generation of IL-2 and IFN-γ.
| Day | a-IL-2R Ab | Concentration of lenalidomide (nM) (percentage of control, mean±S.D.) |
||||
|---|---|---|---|---|---|---|
| 10 | 30 | 100 | 300 | 1000 | ||
| IL-2 | ||||||
| 1 | − | 216± 36 | 402± 139 | 669 ± 234 | 724±250 | 677±245 |
| 1 | + | 149±41+ | 175 ± 56* | 264± 161+ | 285± 186+ | 263± 152+ |
| 3 | − | 448± 309 | 903± 160 | 2101 ± 1520 | 2411 ± 1636 | 3239 ± 1147 |
| 3 | + | 234± 188 | 237± 149** | 330 ±242+ | 311 ± 143+ | 368 ± 187* |
| 5 | − | 195± 132 | 398±257 | 1025 ±422 | 2885± 1855 | 3690 ± 1597 |
| 5 | + | 223± 155 | 178±22 | 281 ± 139+ | 206± 115+ | 306 ± 104* |
| IFN-γ | ||||||
| 1 | − | 159 ±64 | 224± 101 | 294± 159 | 239± 100 | 275±95 |
| 1 | + | 101±28 | 104±28+ | 109±38+ | 112 ±36+ | 106±32* |
| 3 | − | 267±61 | 364±92 | 509± 139 | 440± 147 | 463 ± 124 |
| 3 | + | 120±56* | 125±54** | 129±65** | 143 ±77* | 130±62* |
| 5 | − | 307± 110 | 457± 168 | 678 ± 251 | 862 ± 439 | 945 ± 355 |
| 5 | + | 136±55+ | 132±37* | 162 ± 60* | 184 ± 55* | 175 ± 45* |
T cells from two young and three old healthy subjects were incubated with anti-CD3 + anti-CD28 antibodies without (−) or with (+)anti-IL-2R antibody and without (control = 100%) or with lenalidomide. The mean ± S.D. of control (100%) values of IL-2 and IFN-γ, respectively, were 208 ± 138 and 1446 ± 1109 pg/ml on day 1, 32 ± 19 and 6565 ±3686 pg/ml on day 3, and 12 ± 8 and 22,899 ± 16,626 pg/ml on day 5. Statistical significance was calculated to compare values from T cell suspensions that had anti - IL-2R antibody (+) to those with the same concentration of lenalidomide that did not have anti-IL-2R antibody (−).
3.3. Effects of lenalidomide on T cell proliferation and apoptosis: dependence on IL-2 signaling
The proliferation of T cells from healthy old subjects elicited by anti-TCR antibodies was enhanced significantly by 0.03 μM to 1 (μM lenalidomide up to a maximum of approximately two-fold (Fig. 4, left-hand frame). All significant enhancements of proliferation by lenalidomide were eliminated by prior introduction of the anti-human IL-2 receptor blocking antibody (Fig. 4, right-hand frame). The apoptosis of T cells from healthy old subjects induced by TCR-dependent activation was suppressed significantly by 0.01 μM to 1 μM lenalidomide up to a mean maximum of approximately 41% at 0.1 μM lenalidomide (Fig. 5, left-hand frame). As for lenalidomide enhancement of stimulated proliferation, protection of activated T cells from apoptosis by lenalidomide also was lost after inhibition of IL-2 signaling by the anti-human IL-2 receptor blocking antibody (Fig. 5, right-hand frame).
Figure 4.
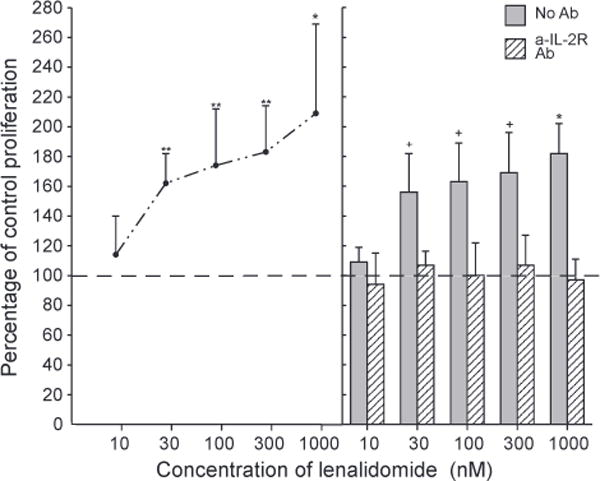
Enhancement of activation-induced T cell proliferation by lenalidomide: dependence on IL-2 signaling. Each point and bar in the left-hand frame and each column and bar in the right-hand frame depict the mean±S.D. of the results of proliferation assays of activated T cells from the same five old subjects as in Figure 5. Incubation with anti-CD3 + anti-CD28 antibodies was for two days without (control = 100%) or with lenalidomide prior to addition of BrdU. The mean±S.D. of control values was 0.468±0.166 AU (450 nm–690 nm). Statistical significance was calculated and depicted as in Figure 1, with comparisons between values with lenalidomide and the 100% control for points in the left-hand frame and between values without and with a-IL-2 R monoclonal antibody for each set of two columns in the right-hand frame.
Figure 5.
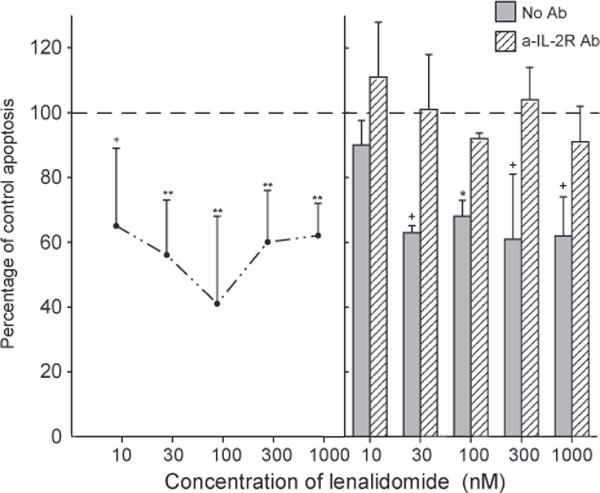
Suppression of activation-induced T cell apoptosis by lenalidomide: dependence on IL-2 signaling. Each point and bar in the left-hand frame and each column and bar in the right-hand frame depict the mean ± S. D. of the results of apoptosis assays of activated T cells from the same five old subjects as in Figure 4. Incubation with anti-CD3 antibody was for two days (right-hand frame) or three days (left-hand frame) without (control = 100%) or with lenalidomide. The mean ± S.D. of control values was 0.599±0.121 AU (405 nm–490 nm). Statistical significance was calculated and depicted as in Figure 1, with comparisons between values with lenalidomide and the 100% control for points in the left-hand frame and between values without and with a-IL-2 R monoclonal antibody for each set of two columns in the right-hand frame.
Initial investigations of possible mechanisms by which lenalidomide suppressed apoptosis of activated T cells from old subjects showed significant increases in T cell content of Bcl-XL. After two and three days of incubation with anti-CD3 antibody, the mean content trange of Bcl-XL was 8290 ± 1048 pg/106 T cells and 6270 ± 691 pg/106 T cells, respectively, and the values for the two times were increased to respective means of 133, 151 and 158% and 123, 164 and 193% of controls by 0.01 μM, 0.1 μM and 1 μM lenalidomide (n=two, pb0.05 for all except 1 μ.M lenalidomide on day 3 = pb0.001 and 0.1 μM lenalidomide on day 2 = not significant).
3.4. Effects of Lenalidomide on T cell Chemotaxis
The chemotactic responses of T cells from healthy young subjects to S1P and CCL21 were significantly higher and that to CCL5 was significantly lower than the responses of T cells from healthy old subjects (Table 2). The chemotactic response of T cells from healthy old subjects to CCL21 was augmented significantly by 0.01 μM lenalidomide to a level indistinguishable from that of T cells from healthy young subjects. Similarly, the chemotactic responses of T cells from healthy old subjects to CCL21 and S1P were enhanced significantly by 0.1 μM lenalidomide and the restored levels were indistinguishable from those of T cells from healthy young subjects. Lenalidomide had no effect on the chemotactic responses of T cells from healthy young subjects.
Table 2.
Effects of lenalidomide on T cell Chemotaxis.
| Concentration of lenalidomide (nM) | Response to each chemotactic stimulus (net percentage of initial T cells, mean ± S.D.)
|
||
|---|---|---|---|
| S1P | CCL5 | CCL21 | |
| Young subjects | |||
| 0 | 2.77±0.70++ | 4.13 ±0.45++ | 37.4±2.7** |
| 10 | 2.47 ±0.76 | 3.93 ±0.31 | 32.3 ± 1.9 |
| 100 | 2.27±0.67 | 3.60±0.56 | 35.8±4.3 |
| Old subjects | |||
| 0 | 1.70 ±0.36 | 5.30±0.40 | 27.8 ±2.9 |
| 10 | 2.10±0.26 | 5.00±0.87 | 33.8±2.7+ |
| 100 | 2.40±0.10+ | 5.07±0.71 | 33.2±2.9+ |
Each value is the mean ± S.D. of the results for T cells from three healthy young subjects and three healthy old subjects responding chemotactically to 10 nMS1 P or 100 nM CCL5 or CCL21. The levels of statistical significance of differences in baseline values between T cells of young and old subjects without lenalidomide are depicted by ++ = pb0.05 or **=pb0.01. The level of statistical significance of differences between baseline values and those with lenalidomide for T cells of old subjects is depicted by +=pb0.05.
3.5. Effect of lenalidomide on the generation of TNF-α by differentiated monocytes
The lowest concentrations of lenalidomide that altered diverse T cell functions were examined for their capacity to inhibit generation of TNF-α by differentiated human blood monocytes, which was the immune effect of thalidomide-type IMDs observed initially [26]. The generation of TNF-α by LPS-stimulated differentiated monocytes from a mixed group of healthy young and old subjects was inhibited significantly by 0.1, 0.3 and 1 μM lenalidomide at all times, by 0.03 μM lenalidomide at 24 and 48 h, and by 0.01 μM lenalidomide at 24 h (Table 3). Thus a macrophage-dependent anti-inflammatory effect of lenalidomide accompanies its T cell-mediated immunostimulatory activities.
Table 3.
Lenalidomide effects on LPS-stimulated macrophage generation of TNF-α.
| Concentration of lenalidomide (nM) (percentage of control, mean±S.D.) | |||||
|---|---|---|---|---|---|
| 10 | 30 | 100 | 300 | 1000 | |
| Incubation time (h) | |||||
| 6 | 91 ±14 | 78±13 | 67±7.8* | 65±4.5** | 46 ±14* |
| 24 | 69±11* | 54±17* | 35 ±4.0** | 33±1.5** | 20±4.5** |
| 48 | 73 ±12 | 57±2.5** | 48±3.2** | 36±4.6** | 36 ±7.6** |
Each value is the mean +S.D. of the results from studies of two adults of ages 21–45 and two adults of ages N65 years. Production of TNF-α evoked by 100 ng/ml of LPS in the absence of lenalidomide (100% control) ranged from 4439 to 6039 pg/ml, 1843 to 2038 pg/ml, and 222 to 308 pg/ml after 6, 24 and 48 h of incubation. The significance of decreases in TNF-α generation induced by lenalidomide relative to the control (100%) value were calculated by a two-sample t test;
=pb0.05,
=pb0.01 and
=pb0.001.
4. Discussion
Potent thalidomide-type drugs, such as lenalidomide (Revlimid), are in regular use at oral doses of 5 mg to 20 mg per day for treatment of some forms of leprosy, del 5q myelodys-plastic syndromes, chronic lymphocytic leukemia and multiple myeloma. Pharmacokinetic studies of lenalidomide in humans have documented mean plasma levels of 320 to 440 nM at 1 h and of over 30 nM for up to 10 h after a single 5 mg oral dose [38,39]. Deductions from these data predict that a lower oral dose of 1 mg to 3 mg would result in plasma levels of 76 nM to 228 nM at 1 h and of 6 nM to 18 nM for up to 10 h. At the plasma concentrations of approximately 0.01 nM to 0.1 μM attained by such a low-dose, lenalidomide enhanced T cell generation of IL-2 and IFN-γ in young and old subjects for at least five days (Figs. 2 and 3). The enhancement of T cell generation of IL-2 and IFN-γ continued to be observed in both age groups with 0.3 μM to 1 μM lenalidomide, in which higher concentration range there also was significant inhibition of generation of IL-17 by stimulated T cells of young but not old subjects (Figs. 2 and 3). In the same plasma concentration range of 0.01 μM to 0.1 μM lenalidomide, achieved by a low-dose, TNF-α generation by LPS-stimulated differentiated monocytes was inhibited significantly for at least 48 h (Table 3). Low-dose lenalidomide thus concurrently enhances T cell generation of protective immune cytokines and inhibits macrophage generation of inflammatory cytokines.
In the concentration range of 0.03 μM to 1 μM, lenalidomide augmented significantly anti-TCR antibody-initiated proliferation of T cells from healthy old subjects by an IL-2 receptor signaling-dependent mechanism (Fig. 4). Anti-TCR antibody activation-induced apoptosis, to which the T cells of old subjects are especially susceptible, also was suppressed by 0.01 μM to 1 μM lenalidomide in T cells of healthy old subjects by an IL-2 receptor signaling-dependent mechanism (Fig. 5). An IL-2 positive-feedback mechanism enhanced generation of IL-2 and IFN-γ by stimulated T cells through a mechanism that was inhibited by the same concentration of anti-IL-2 receptor-α chain blocking antibody which prevented IL-2-mediated lenalidomide enhancement of proliferation and survival (Table 1). Promotion of increased proliferation and survival of T cells by 0.01 μM to 1 μM lenalidomide through IL-2 receptor signaling leads to more cytokine-generating T cells and thereby may contribute to the sustained enhancement of generation of IL-2 and IFN-γ by low concentrations of lenalidomide. It has long been known that differentiation of naïve CD4 T cells into effector CD4 T cells and subsequently into memory CD4 T cells by TCR-plus co-receptor-stimulation requires concurrently-generated IL-2 [40,41]. TCR-mediated activation of the diminished number of naïve T cells in old mice and humans generates fewer effector T cells and lower levels of IL-2 than for naïve T cells of young subjects [24,42,43]. The deficiency of effector T cells in older mice may be corrected by addition of exogenous IL-2 but not other cytokines of the cluster whose receptors utilize the common γ chain-binding unit [40]. Lenalidomide enhancement of generation of endogenous IL-2 appears to mimic addition of exogenous IL-2 and consequently reverses the deficient proliferation and survival of activated naïve T cells in old subjects. This could be a central mechanism through which lenalidomide restores the capacity of immunosenescent activated naïve T cells to differentiate efficiently into longer-lived effector T cells.
The many roles of IL-2 in T cell development, subset differentiation, proliferation, activation and the balance between apoptosis and survival are complex and only partially understood. Although IL-2 itself may have immunosuppressive effects by directly promoting activation-induced T cell death and by indirectly fostering differentiation of Treg cells, the mechanisms of IL-2 signaling in these processes and in the establishment of tolerance in vivo are controversial [44,45]. In contrast, apoptosis evoked by the state of IL-2 deprivation that ensues rapidly after transient activation of CD4 and CD8 T cells involves well-defined transcriptional programs that result in appearance of pro-apoptotic factors and concurrent repression of anti-apoptotic principles [46,47]. Our experimental design does not include restimulation of the T cells at a time of IL-2 sufficiency, so that elicitation of apoptosis on day two and especially day three after primary TCR-dependent stimulation is most likely attributable to inadequate levels of IL-2 and possibly other cytokine growth factors. The persistent promotion of adequately protective levels of IL-2 by lenalidomide may account for the observed reduction in apoptosis (Fig. 5). The administration of blocking doses of anti-IL-2α receptor antibody to patients with autoimmune diseases similarly leads to decreases in T cell survival and reduced proliferative responses of CD4 and CD8 T cells to their cognate antigens [48,49].
The capacity of lenalidomide to augment the diminished chemotactic responses of T cells from healthy old subjects to S1P and CCL21 resulted in levels of migration that were the same as those of T cells from healthy young subjects (Table 2). As this enhancement is similar for two different chemotactic factor systems, that share only decreased activity for T cells of healthy old subjects compared to healthy young subjects, lenalidomide appears to be acting on a common signal transduction or T cell intrinsic mobility pathway rather than at the level of the chemotactic factor receptors.
The principal functionally-relevant difference between responses to lenalidomide of T cells from old subjects and those from young subjects is the magnitude of enhancement of IL-2 generation (Figs. 2 and 3). For the maximal enhancing effects of nanomolar concentrations of lenalidomide on days three and five, increments in IL-2 levels were six- to twelve-fold higher for stimulated T cells of old than young subjects. Although partially attributable to lower baseline levels of IL-2 from stimulated T cells of old than young subjects, lenalidomide nonetheless effectively raised IL-2 generation by stimulated T cells of old subjects to a ten-fold higher actual mean peak level on day three than that on day one in the absence of lenalidomide. These data also reveal the differential cytokine specificity of lenalidomide, as there were no significant effects on IL-17 generation by T cells of old subjects and only modest decreases for those of young subjects. Thus a low-dose regimen of lenalidomide in old subjects also would not be expected to increase the risk of bacterial infections by impairing recruitment of neutrophils nor increase the risk of autoimmunity by mobilization and activation of Th17 cells and related subsets of effector T cells. Additionally, low-dose lenalidomide may improve T cell-dependent immunity by augmenting Chemotaxis, proliferation and post-activation survival.
In considering the possible beneficial effects of low-dose lenalidomide in immunosenescence, two other effects may prove to be important. First is the capacity of lenalidomide to increase the number and activity of NK cells, that is attained at 1 μM or lower concentrations [28,29]. Second is the capability of lenalidomide at 1 μM or lower concentrations to suppress stimulated mononuclear phagocyte generation of a range of inflammatory mediators, including IL-6, TNF-α and IL-1β, as well as to increase their production of immunosuppressive IL-10 (Table 3) [26]. Both effects of lenalidomide will add to its therapeutic value in immunosenescence by reducing excessive inflammation while correcting diminished innate and adaptive immunity. Future testing of low-dose lenalidomide IMD will focus on its potential roles in improving diverse aspects of immunity in the elderly population ranging from the effectiveness of vaccines to host defenses against microbial infections to resistance to cancers.
Acknowledgments
This research was supported by a grant from the Kenneth Rainin Foundation and by the Intramural Research Program of the National Institute on Aging. The authors are grateful to Judith H. Goetzl for preparation of graphics and for textual editing.
Abbreviations
- IMD
immunomodulatory drug
- DMSO
dimethyl sulfoxide
Footnotes
The authors declare no conflicts of interest.
Authors’ contributions: N.H.G., D.L.L., J.B.S. and E.J.G. designed research; L.F., W.B.E., J.B.S. and E.J.G. identified and evaluated subjects; D.T., W.L. and N.H.G. synthesized and analyzed compounds: M.-C. H. and E.J.G. performed research and analyzed data; E.J.G., L.F. and J.B.S. wrote the paper.
References
- 1.Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9:57–62. doi: 10.1038/nri2471. [DOI] [PubMed] [Google Scholar]
- 2.Caruso C, Buffa S, Candore G, Colonna-Romano G, Dunn-Walters D, Kipling D, Pawelec G. Mechanisms of immunosenescence. Immun Ageing. 2009;6:10. doi: 10.1186/1742-4933-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson KM, Owen K, Witte PL. Aging and developmental transitions in the B cell lineage. Int Immunol. 2002;14:1313–1323. doi: 10.1093/intimm/dxf092. [DOI] [PubMed] [Google Scholar]
- 4.Johnson SA, Cambier JC. Ageing, autoimmunity and arthritis: senescence of the B cell compartment – implications for humoral immunity. Arthritis Res Ther. 2004;6:131–139. doi: 10.1186/ar1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agrawal A, Agrawal S, Cao JN, Su H, Osann K, Gupta S. Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signaling pathway. J Immunol. 2007;178:6912–6922. doi: 10.4049/jimmunol.178.11.6912. [DOI] [PubMed] [Google Scholar]
- 6.Agrawal A, Agrawal S, Tay J, Gupta S. bBiology of dendritic cells in aging. J Clin Immunol. 2008;28:14–20. doi: 10.1007/s10875-007-9127-6. [DOI] [PubMed] [Google Scholar]
- 7.Gomez CR, Nomellini V, Faunce DE, Kovacs EJ. Innate immunity and aging. Exp Gerontol. 2008;43:718–728. doi: 10.1016/j.exger.2008.05.0168.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agius E, Lacy KE, Vukmanovic-Stejic M, Jagger AL, Papageorgiou AP, Hall S, Reed JR, Curnow SJ, Fuentes-Duculan J, Buckley CD, Salmon M, Taams LS, Krueger J, Greenwood J, Klein N, Rustin MH, Akbar AN. Decreased TNF-alpha synthesis by macrophages restricts cutaneous immuno-surveillance by memory CD4+ T cells during aging. J Exp Med. 2009;206:1929–1940. doi: 10.1084/jem.20090896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wack A, Cossarizza A, Heltai S, Barbieri D, D’Addato S, Fransceschi C, Dellabona P, Casorati G. Age-related modifications of the human alphabeta T cell repertoire due to different clonal expansions in the CD4+ and CD8+ subsets. Int Immunol. 1998;10:1281–1288. doi: 10.1093/intimm/10.9.1281. [DOI] [PubMed] [Google Scholar]
- 10.Fagnoni FF, Vescovini R, Passeri G, Bologna G, Pedrazzoni M, Lavagetto G, Casti A, Franceschi C, Passeri M, Sansoni P. Shortage of circulating naive CD8(+) T cells provides new insights on immunodeficiency in aging. Blood. 2000;95:2860–2868. [PubMed] [Google Scholar]
- 11.Gupta S, Bi R, Su K, Yel L, Chiplunkar S, Gollapudi S. Characterization of naive, memory and effector CD8+ T cells: effect of age. Exp Gerontol. 2004;39:545–550. doi: 10.1016/j.exger.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Naylor K, Li G, Vallejo AN, Lee WW, Koetz K, Bryl E, Witkowski J, Fulbright J, Weyand CM, Goronzy JJ. The influence of age on T cell generation and TCR diversity. J Immunol. 2005;174:7446–7452. doi: 10.4049/jimmunol.174.11.7446. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Agrawal A, Agrawal S, Su H, Gollapudi S. A paradox of immunodeficiency and inflammation in human aging: lessons learned from apoptosis. Immun Ageing. 2006;3:5. doi: 10.1186/1742-4933-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilpatrick RD, Rickabaugh T, Hultin LE, Hultin P, Hausner MA, Detels R, Phair J, Jamieson BD. Homeostasis of the naive CD4+ T cell compartment during aging. J Immunol. 2008;180:1499–1507. doi: 10.4049/jimmunol.180.3.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goetzl EJ, Huang MC, Kon J, Patel K, Schwartz JB, Fast K, Ferrucci L, Madara K, Taub DD, Longo DL. Gender specificity of altered human immune cytokine profiles in aging. FASEB J. 2010;24:3580–3589. doi: 10.1096/fj.10-160911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gregg R, Smith CM, Clark FJ, Dunnion D, Khan N, Chakraverty R, Nayak L, Moss PA. The number of human peripheral blood CD4+ CD25 high regulatory T cells increases with age. Clin Exp Immunol. 2005;140:540–546. doi: 10.1111/j.1365-2249.2005.02798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 18.Marko MG, Ahmed T, Bunnell SC, Wu D, Chung H, Huber BT, Meydani SN. Age-associated decline in effective immune synapse formation of CD4(+) T cells is reversed by vitamin E supplementation. J Immunol. 2007;178:1443–1449. doi: 10.4049/jimmunol.178.3.1443. [DOI] [PubMed] [Google Scholar]
- 19.Lages CS, Suffia I, Velilla PA, Huang B, Warshaw G, Hildeman DA, Belkaid Y, Chougnet C. Functional regulatory T cells accumulate in aged hosts and promote chronic infectious disease reactivation. J Immunol. 2008;181:1835–1848. doi: 10.4049/jimmunol.181.3.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang MC, Patel K, Taub DD, Longo DL, Goetzl EJ. Human CD4-8- T cells are a distinctive immunoregulatory subset. FASEB J. 2010;24:2558–2566. doi: 10.1096/fj.09-153148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monteiro J, Batliwalla F, Ostrer H, Gregersen PK. Shortened telomeres in clonally expanded CD28–CD8+ T cells imply a replicative history that is distinct from their CD28+CD8+ counterparts. J Immunol. 1996;156:3587–3590. [PubMed] [Google Scholar]
- 22.Effros RB, Dagarag M, Spaulding C, Man J. The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005;205:147–157. doi: 10.1111/j.0105-2896.2005.00259.x. [DOI] [PubMed] [Google Scholar]
- 23.Gupta S, Gollapudi S. TNF-alpha-induced apoptosis in human naive and memory CD8+ T cells in aged humans. Exp Gerontol. 2006;41:69–77. doi: 10.1016/j.exger.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Rea IM, Stewart M, Campbell P, Alexander HD, Crockard AD, Morris TC. Changes in lymphocyte subsets, inter-leukin 2, and soluble interleukin 2 receptor in old and very old age. Gerontology. 1996;42:69–78. doi: 10.1159/000213775. [DOI] [PubMed] [Google Scholar]
- 25.Caruso C, Candore G, Cigna D, DiLorenzo G, Sireci G, Dieli F, Salerno A. Cytokine production pathway in the elderly. Immunol Res. 1996;15:84–90. doi: 10.1007/BF02918286. [DOI] [PubMed] [Google Scholar]
- 26.Corral LG, Muller GW, Moreira AL, Chen Y, Wu M, Stirling D, Kaplan G. Selection of novel analogs of thalidomide with enhanced tumor necrosis factor alpha inhibitory activity. Mol Med. 1996;2:506–515. [PMC free article] [PubMed] [Google Scholar]
- 27.Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, Patterson RT, Stirling DI, Kaplan G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163:380–386. [PubMed] [Google Scholar]
- 28.Hayashi T, Hideshima T, Akiyama M, Podar K, Yasui H, Raje N, Kumar S, Chauhan D, Treon SP, Richardson P, Anderson KC. Molecular mechanisms whereby immunomodulatory drugs activate natural killer cells: clinical application. Br J Haematol. 2005;128:192–203. doi: 10.1111/j.1365-2141.2004.05286.x. [DOI] [PubMed] [Google Scholar]
- 29.Reddy N, Hernandez-llizaliturri FJ, Deeb G, Roth M, Vaughn M, Knight J, Wallace P, Czuczman MS. Immunomodulatory drugs stimulate natural killer-cell function, alter cytokine production by dendritic cells, and inhibit angiogenesis enhancing the anti-tumour activity of rituximab in vivo. Br J Haematol. 2008;140:36–45. doi: 10.1111/j.1365-2141.2007.06841.x. [DOI] [PubMed] [Google Scholar]
- 30.Schäfer PH, Gandhi AK, Loveland MA, Chen RS, Man HW, Schnetkamp PP, Wolbring G, Govinda S, Corral LG, Payvandi F, Muller GW, Stirling DI. Enhancement of cytokine production and AP-1 transcriptional activity in T cells by thalidomide-related immunomodulatory drugs. J Pharmacol Exp Ther. 2003;305:1222–1232. doi: 10.1124/jpet.102.048496. [DOI] [PubMed] [Google Scholar]
- 31.Payvandi F, Wu L, Naziruddin SD, Haley M, Parton A, Schafer PH, Chen RS, Muller GW, Hughes CC, Stirling DI. Immunomodulatory drugs (IMiDs) increase the production of IL-2 from stimulated T cells by increasing PKC-theta activation and enhancing the DNA-binding activity of AP-1 but not NF-kappaB, OCT-1, or NF-AT. J Interferon Cytokine Res. 2005;25:604–616. doi: 10.1089/jir.2005.25.604. [DOI] [PubMed] [Google Scholar]
- 32.Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, Todryk S, Chen R, Muller G, Stirling D, Schafer P, Bartlett JB, Dalgleish AG. The anti-cancer agents lenalido-mide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother. 2009;58:1033–1045. doi: 10.1007/s00262-008-0620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Y, Li J, Ferguson GD, Mercurio F, Khambatta G, Morrison L, Lopez-Girona A, Corral LG, Webb DR, Bennett BL, Xie W. Immunomodulatory drugs reorganize cyto-skeleton by modulating Rho GTPases. Blood. 2009;114:338–345. doi: 10.1182/blood-2009-02-200543. [DOI] [PubMed] [Google Scholar]
- 34.LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, Mitsiades N, Mitsiades C, Cheema P, Chauhan D, Richardson PG, Anderson KC, Munshi NC. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood. 2004;103:1787–1790. doi: 10.1182/blood-2003-02-0361. [DOI] [PubMed] [Google Scholar]
- 35.Graler MH, Huang MC, Watson S, Goetzl EJ. Immunological effects of transgenic constitutive expression of the type 1 sphingosine 1-phosphate receptor by mouse lymphocytes. J Immunol. 2005;174:1997–2003. doi: 10.4049/jimmunol.174.4.1997. [DOI] [PubMed] [Google Scholar]
- 36.Zhu X, Giordano T, Yu QS, Holloway HW, Perry TA, Lahiri DK, Brossi A, Greig NH. Thiothalidomides: novel isosteric analogues of thalidomide with enhanced TNF-alpha inhibitory activity. J Med Chem. 2003;46:5222–5229. doi: 10.1021/jm030152f. [DOI] [PubMed] [Google Scholar]
- 37.Haslett PA, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med. 1998;187:1885–1892. doi: 10.1084/jem.187.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tohnya TM, Hwang K, Lepper ER, Fine HA, Dahut WL, Venitz J, Sparreboom A, Figg WD. Determination of CC-5013, an analogue of thalidomide, in human plasma by liquid chromatography-mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci. 2004;811:135–141. doi: 10.1016/j.jchromb.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 39.Dahut WL, Aragon-Ching JB, Woo S, Tohnya TM, Gulley JL, Arlen PM, Wright JJ, Ventiz J, Figg WD. Phase I study of oral lenalidomide in patients with refractory metastatic cancer. J Clin Pharmacol. 2009;49:650–660. doi: 10.1177/0091270009335001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haynes L, Linton PJ, Eaton SM, Tonkonogy SL, Swain SL. Interleukin 2, but not other common gamma chain-binding cytokines, can reverse the defect in generation of CD4 effector T cells from naive T cells of aged mice. J Exp Med. 1999;190:1013–1024. doi: 10.1084/jem.190.7.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchell DM, Ravkov EV, Williams MA. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J Immunol. 2010;184:6719–6730. doi: 10.4049/jimmunol.0904089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gillis S, Kozak R, Durante M, Weksler ME. Immunological studies of aging. Decreased production of and response to T cell growth factor by lymphocytes from aged humans. J Clin Invest. 1981;67:937–942. doi: 10.1172/JCI110143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swain S, Clise-Dwyer K, Haynes L. Homeostasis and the age-associated defect of CD4 T cells. Semin Immunol. 2005;17:370–377. doi: 10.1016/j.smim.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol. 2004;172:3983–3988. doi: 10.4049/jimmunol.172.7.3983. [DOI] [PubMed] [Google Scholar]
- 45.Brenner D, Krammer PH, Arnold R. Concepts of activated T cell death. Crit Rev Oncol Hematol. 2008;66:52–64. doi: 10.1016/j.critrevonc.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Eastman A. Apoptosis in an interleukin-2-dependent cytotoxic T lymphocyte cell line is associated with intracellular acidification. Role of the Na(+)/H(+)-antiport. J Biol Chem. 1995;270:3203–3211. doi: 10.1074/jbc.270.7.3203. [DOI] [PubMed] [Google Scholar]
- 47.Devireddy LR, Green MR. Transcriptional program of apoptosis induction following interleukin 2 deprivation: identification of RC3, a calcium/calmodulin binding protein, as a novel proapoptotic factor. Mol Cell Biol. 2003;23:4532–4541. doi: 10.1128/MCB.23.13.4532-4541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nussenblatt RB, Fortin E, Schiffman R, Rizzo L, Smith J, Van Veldhuisen P, Sran P, Yaffe A, Goldman CK, Waldmann TA, Whitcup SM. Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: a phase l/ll clinical trial. Proc Natl Acad Sci USA. 1999;96:7462–7466. doi: 10.1073/pnas.96.13.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bielekova B, Richert N, Howard T, Blevins G, Markovic-Plese S, McCartin J, Frank JA, Wurfel J, Ohayon J, Waldmann TA, McFarland HF, Martin R. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc Natl Acad Sci USA. 2004;101:8705–8708. doi: 10.1073/pnas.0402653101. [DOI] [PMC free article] [PubMed] [Google Scholar]