

Abstract
Scope
Exposure of the breast to estrogens and other sex hormones is an important cancer risk factor and estrogen receptor down-regulators are attracting significant clinical interest. Epigallocatechin gallate (EGCG), a polyphenolic compound found in green tea, has gained considerable attention for its antitumor properties. Here we aimed to investigate the molecular mechanisms through which EGCG regulates ERα expression in ER+ PR+ breast cancer cells.
Material and Methods
Western blotting analysis, real time PCR and transient transfections of deletion fragments of the ERα gene promoter show that EGCG down-regulates ERα protein, mRNA and gene promoter activity with a concomitant reduction of ERα genomic and non genomic signal. These events occur through p38MAPK/CK2 activation, causing the release from Hsp90 of PR-B and its consequent nuclear translocation as evidenced by immunofluorescence studies. EMSA and ChIP assay reveal that, upon EGCG treatment, PR-B is recruited at the half PRE site on ERα promoter. This is concomitant with the formation of a corepressor complex containing NCoR and HDAC1 while RNA polymerase II is displaced. The events are crucially mediated by PR-B isoform, since they are abrogated with PR-B siRNA.
Conclusions
Our data provide evidence for a mechanism by which EGCG down-regulates ERα and explain the inhibitory action of EGCG on the proliferation of ER+ PR+ cancer cells tested. We suggest that the EGCG/PR-B signaling should be further exploited for clinical approach.
Keywords: Progesterone receptor, Estrogen receptor, Hsp90, p38MAPK
Introduction
Breast cancer risk factors, such as early age at menarche, late age at menopause, postmenopausal hormone therapy, and high body mass index are thought to affect breast carcinogenesis by increasing the life-time exposure of the breast to estrogens and other sex hormones [1]. Estrogens particularly are mitogenic to breast epithelial cells, and since the effects of estrogens are mediated by both estrogen receptors (ERα and ERβ), the magnitude of their effects may be determined by the individual levels of ERs expressed in the breast.
Up-regulated ERα expression in mice produces ductal hyperplasia, lobular hyperplasia, and ductal carcinoma in situ, demonstrating the consequences of unregulated ERα levels at all stages of breast cancer development [2]; in contrast ERβs acts to regulate the degree of estrogen action by negatively modulating ERα, and the estrogen independent transcriptional activity of ERβ isoforms is inhibited by ERα [3]. Further evidence shows that ERα levels in both hyperplastic lesions and ER-positive tumors are greatly elevated as compared with adjacent normal tissue [4]. Therefore it is not surprising that elevated levels of ERα in benign breast epithelium is, itself, considered a risk factor for progression to invasive breast disease [5].
The strong correlation between ERα expression, breast disease patho-physiology, and therapeutic response have justified the use of estrogen receptor down-regulators as attractive intervention, with significant clinical interest in their use. In this regard, many naturally occurring compounds, commonly present in the diet have gained considerable attention as well [6,7]. In recent years epigallocatechin gallate (EGCG), a polyphenolic compound found in green tea, has demonstrated chemo-preventative and antitumor properties [8].
Epidemiological studies have suggested that green tea consumption is linked to a decrease in the incidence and recurrence of breast cancer [9]. Additionally, treatment with EGCG (50 mg/kg/day, 14 days) reduced the growth of MCF-7 implanted breast tumors in athymic nude mice by 40% [10], and it has been reported that catechin inhibited the proliferation of human breast cancer cells in vitro [11]. This is partly attributable to its effects on modulating the activity of mitogen-activated protein kinases (MAPKs), IGF/IGF-1 receptor, AKT, NF-κB, and CDKs [12,13]. EGCG also inhibits growth factor receptor extracellular signaling, the proteasome, mitochondrial depolarization, and fatty acid synthase [14–16].
Recent studies report that the anti-tumor activity of EGCG is mediated by targeting HSP70 and HSP90 in vitro and in vivo, through a mechanism that involves direct binding of EGCG to the C-terminal region of HSP90 [17], a chaperone protein that is constitutively expressed at high levels in many cancer cells. HSP90 is assembled into heterocomplexes with unliganded steroid receptors, such as GR and PR, thus influencing their cellular distribution and activity [18].
Particularly recent studies on breast cancer models have implicated EGCG in the regulation of ERα. Since the catechin family is structurally similar to isoflavones, it has been shown that catechins, such as EGCG, were co-estrogenic for ERα at lower doses (< 5 μM) but antiestrogenic at higher doses (100–150 μM) and eliciting a concomitant massive cell death at the higher levels [11,19].
In the present study we report that EGCG produces a significant inhibition of ER+ PR+ breast cancer cell proliferation and we define the molecular mechanisms associated with this growth effect. Concomitant with nuclear localization of PR, EGCG treatment causes a down-regulation of ERα protein, mRNA and gene promoter activity. We demonstrate that these effects are crucially mediated by PR-B via its recruitment to the ERα proximal promoter.
Materials and Methods
Materials
Epigallocatechin 3 gallate (EGCG), aprotinin, leupeptin, phenylmethylsulfonyl fluoride (PMFS), and sodium orthovanadate were from Sigma (Milan, Italy). PD 169316 and TBB from Calbiochem (Darmstadt, Germany). ICI 182,780 (ICI, ER antagonist), was purchased from Zeneca Pharmaceuticals (Cheshire, UK). Antibodies used in this study were GAPDH, laminin B, NCoR, RNA Pol II, HDAC1, total p38, phosphorylated p38, ERα, progesterone receptor B (PR-B), EGFR, Raf1, Her2, Hsp90, CK2 from Santa Cruz Biotechnology (Santa Cruz, CA); progesterone receptors (PR-B, PR-A), total MAPK, total AKT, phosphorylated p42/44 MAPK (Thr202/Tyr 204) and pAKT (Ser437), from Cell Signaling Technology (Beverly, MA).
Cell Culture
Cells utilized in the studies were obtained from American Type Culture Collection (Manassas, VA). MCF-7 and T47D, were cultured as described [20]. Ishikawa endometrial cancer cells (ISK) were maintained in DMEM without phenol red supplemented with 10% fetal bovine serum (FBS). SkBr3 cells were cultured in RPMI1640 with 10% FBS. Treatments were performed, after 48h of serum starvation, in 1% dextran charcoal-stripped (CS) FBS to reduce steroid concentration [20].
MTT assay
Cells (3×104cells/mL) were plated in 24 well plates and serum-starved for 48h before the addition of treatment for 4 days (d). The MTT assay was performed as the following: 100 μl of MTT (2mg/mL) (Sigma Aldrich Milan, Italy) were added to each well, and the plates were incubated for 2h at 37 C. Then, 500μl of DMSO were added. The absorbance was measured with the Ultrospec 2100 Prospectrophotometer (Amersham-Biosciences, Milan, Italy) at a test wavelength of 570nm.
Anchorage-independent growth assays
T47D cells (5,000/well) were plated in 4 ml of 0.35% agarose with 5% charcoal-stripped FBS in phenol red-free media, on a 0.7% agarose base in 6-well plates. Two days after plating, media containing control vehicle or hormonal treatments was added to the top layer, and the appropriate media was replaced every 2 days. After 14 d, 150 μl of MTT was added to each well and allowed to incubate at 37°C for 4 h. Plates were then placed in 4°C overnight and colonies ≥50 μm diameter from triplicate assays were counted. Data are the mean colony number of three plates and representative of two independent experiments analyzed for statistical signiӿcance (P<0.05) using a two-tailed student’s Test, performed by Graph Pad Prism 5 (GraphPad Software, Inc., San Diego, CA). Standard deviations are shown.
Plasmids
XETL [21], the wild-type human ERα (HEGO) [22], the full-length PR-B consisting of the full-length PR-B cDNA fused with the SV40 early promoter (a gift from Dr. D. Picard, University of Geneve, Switzerland) [20], the PR DNA-binding mutant C587A (mDBD PR) previously described by Faivre et al. [23] (gift from Dr. C. Lange, University of Minnesota Cancer Center, Minneapolis, MN), the full-length PR-A [24] and the deletion fragments of the ERα gene promoter [25]. The Renilla luciferase expression vector pRL-TK (Promega, Milan, Italy) was used as a transfection standard.
Reverse transcription and real-time PCR
Cells (6×106) were treated as indicated and processed as described [26]. cDNA diluted were analysed in triplicates by real-time PCR in an iCycler iQ Detection System (Bio-Rad, USA). The primers were: (ERα forward) 5′-AGAGGGCATGGTGGAGATCTT-3′; (ERα reverse) 5′-CAAACTCCTCTCCCTGCAGATT -3′; (pS2 forward) 5′-TTCTATCCTAATACCATCGACG-3′; (pS2 reverse) 5′-TTTGAGTAGTCAAAGTCAGAGC-3′; (IRS1 forward) 5′-AGGATATTTAATTTGCCTCGG-3′; (IRS1 reverse) 5′-AAGCGTTTGTGCATGCTCTTG-3′; (CD1 forward) 5′-TCTAAGATGAAGGAGACCATC-3′; (CD1 reverse) 5′-GCGGTAGTAGGACAGGAAGTTGTT-3′; (18S forward) 5′-GGCGTCCCCCAACTTCTTA-3′ (18S reverse) 5′-GGGCATCACAGACCTGTTATT-3′.
Western blotting and immunoprecipitation
Protein expression or complex formation were assessed as described [27] by Western blotting (WB) or immunoprecipitation (IP) followed by WB, using total protein lysates, cytoplasmic, or nuclear protein lysates, where appropriate. Cells (6×106) were harvested to be analyzed using 500 μl of lysis buffer containing 50 mmol/liter HEPES (pH 7.5), 150 mmol/liter NaCl, 1%b Triton X-100, 1.5 mmol/liter MgCl2, 10 mmol/liter EGTA (pH 7.5), 10% glycerol, and inhibitors (0.1 mmol/liter Na3VO4, 1% phenylmethylsulfonyl fluoride, and 2.0 mg/ml aprotinin) to obtain cytoplasmic proteins. After the collection using a scraper, incubation of 30′ on ice, we lysed the nuclei for 15′ at 4°C using 250 μl of nuclear buffer containing 20 mmol/liter HEPES (pH 8), 0.1mmol/liter EDTA, 5 mmol/liter MgCl2, 0.5 mol/liter NaCl, 20% glycerol, 1% NP-40, and inhibitors(1.7 mg/ml aprotinin, 1 mg/ml leupeptin 200 mmol/liter phenylmethylsulfonylfluoride, 200 mmol/liter sodium orthovanadate, and 100 mmol/liter sodium fluoride). Then lysates were collected and centrifuged at 10.000xg for 10′ at 4°C.
For total protein extracts, 500 μl RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% Na deoxycholate, plus inhibitors1.7 mg/ml aprotinin, 1 mg/ml leupeptin 200 mmol/liter phenylmethylsulfonylfluoride, 200 mmol/liter sodium orthovanadate, and 100 mmol/liter sodium fluoride) was added to the 100 ml cell culture plate for 15′ at 4°C. Then lysates were collected and centrifuged at 10.000xg for 10′ at 4°C. The protein content was determined using Bradford dye reagent (Bio-Rad). For WB, 50 μg of total, cytoplasmic or nuclear lysates were separated on an 11% polyacrylamide denaturing gel (SDS-PAGE) and transferred to nitrocellulose membranes. Proteins of interest were detected with specific Abs, recognized by peroxidase-coupled secondary Abs, and developed using the ECL Plus Western Blotting detection system (Amersham Pharmacia Biotech, United Kingdom). For IP, 500 μg of protein of cytoplasmic or nuclear lysates were precleared for 1h with protein A/G-agarose (Santa Cruz), incubated with primary Abs at 4°C for 18 h in HNTG buffer (20 mmol/liter HEPES, pH 7.5, 150 mmol/liter NaCl, 0.1% Triton X-100, 10% glycerol, and 0.1 mmol/liter Na3VO4), and then the antigen-Ab complexes were precipitated with protein A/G agarose for 2 h in HNTG buffer. The immunoprecipitated proteins were washed three times with HNTG buffer, separated on SDS-PAGE, and processed by WB. The images were acquired by using an Epson Perfection scanner (Epson, Japan) using Photoshop software (Adobe). The optical densities of the spots were analyzed by using ImageJ software (NIH; http://rsb.info.nih.gov/IJ). Images are representative of three different experiments.
Immunofluorescence
T47D cells seeded on glass cover-lips were treated with 40 μM EGCG for 12h, washed with PBS, and then fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Next, cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min, blocked with 5% bovine serum albumin for 30 min, and incubated with anti-PR-B antibody (1:50) in PBS overnight at 4°C. The day after the cells were washed three times with PBS and incubated with the secondary antibody anti-rabbit IgG-fluorescein isothiocyanate (FITC) (1:200) for 1h at room temperature. To check the specificity of immunolabeling the primary antibody was replaced by normal rabbit serum (negative control). The blue-fluorescent DAPI was used for nuclear stain. Immunofluorescence analysis was carried out on a OLYMPUS BX51 microscope using a ×40 objective. Images are representative of three different experiments.
Transfections and luciferase assays
Transfections were done as described [20] using Fugene 6 reagent (Roche Diagnostics, Milan, Italy). Luciferase activity was measured with the Dual Luciferase kit (Promega, Milan, Italy). Results represent mean of luciferase activities observed in three independent experiments done in triplicate.
Site directed mutagenesis
Mutagenesis was performed on Fragment D of the ERα promoter using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) following the manufacturer’s instructions. The sequence for the sense primer was: 5′-AGCAGGGAGATGAGGATTGCTGAAGTCCATGGGGGTATGT-3′. The plasmids were then sequenced to confirm the mutation of the desired site.
Lipid-Mediated Transfection of siRNA Duplexes
Custom-synthesized siRNA (Invitrogen)-annealed duplexes (25-bp double-stranded RNA [dsRNA]) were used for effective depletion of PR-B. A non specific siRNA (NS) (Invitrogen) that lacked identity with known gene targets was used as a control for non-sequence-specific effects. Cells were transfected using Lipofectamine 2000 reagent (Invitrogen, Paisley, UK) according to the manufacturer’s instructions and then treated as indicated.
Electrophoretic mobility shift assay (EMSA)
EMSA was carried out as previously described [25], with a few modifications. Cells were treated for 6h before harvesting for the assay. The sequence of ERα-half PRE oligonucleotide used as probe or the unlabelled competitor was 5′- AGGATTGTGTTCTCCATGGG -3′, mutated 5′- AGGATTGTTAAGTCCATGGG-3′. To test specific binding nuclear extracts were pre-incubated with rabbit polyclonal PR antibody or normal rabbit IgG. The reactions were separated on 6% polyacrylamide gel in 0.25x Tris borate–EDTA for 3h at 150V. Images are representative of three different experiments.
Chromatin immunoprecipitation (ChIP) assays
Cells were treated for 12h before harvesting for the assay performed as described [28]. ERα promoter primers used for PCR: forward, 5′-ACGTTCTTGATCCAGCAGGGTA-3′ and reverse, 5′-ACCTGCCAAATTATATGCAAATGGCAG-3′ containing the half-PRE site; and forward, 5′-GTGGCCATTGTTGACCTACAG- 3′ and reverse, 5′-CTGTAGGTCAACAATGGCCAC-3′ upstream the half-PRE site. Images are representative of three different experiments.
Statistical Analysis
Each datum point represents the mean ± S.D. of three different experiments. Data were analyzed by Student’s t test using the GraphPad Prism 4 software program. P<0.05 was considered as statistically significant.
Results
EGCG treatment decreases ER+ PR+ cancer cell proliferation
We first investigated whether EGCG affects cancer cell proliferation following extended treatments with low doses of the catechin. We tested ER+ PR+ breast cancer cell lines, including T47D cells, which are known to express ERα and elevated levels of endogenous PRs [20], MCF-7 cells which express ERα and low levels of PR, and a well-differentiated ER+ PR+ human endometrial adenocarcinoma cell line (Ishikawa cells). Proliferating cells were exposed to different nontoxic concentrations of EGCG (10μM; 20μM; 40μM) and then analyzed in MTT growth assays (Fig.1A). After 4 d treatment, 10μM EGCG inhibited basal cell proliferation of both T47D (50%), and MCF-7 (20%). With increasing doses of EGCG (20 and 40μM), T47D cell number was further reduced by 52% to 67%, respectively. MCF-7 (25% to 50%) and ISK (55% to 68%) cells growth was also significantly decreased. EGCG inhibitory action was still present after pre-treatment with 1μM ICI 182,780, a potent and specific antagonist with excellent growth-inhibitory effects in several cell and animal models of human breast cancer, which induces ERα degradation through ubiquitin-mediated mechanism [29]. This suggests that growth inhibition was not mediated by binding of EGCG to ER.
Figure 1. EGCG inhibits ER+ PR+ cancer cell proliferation.
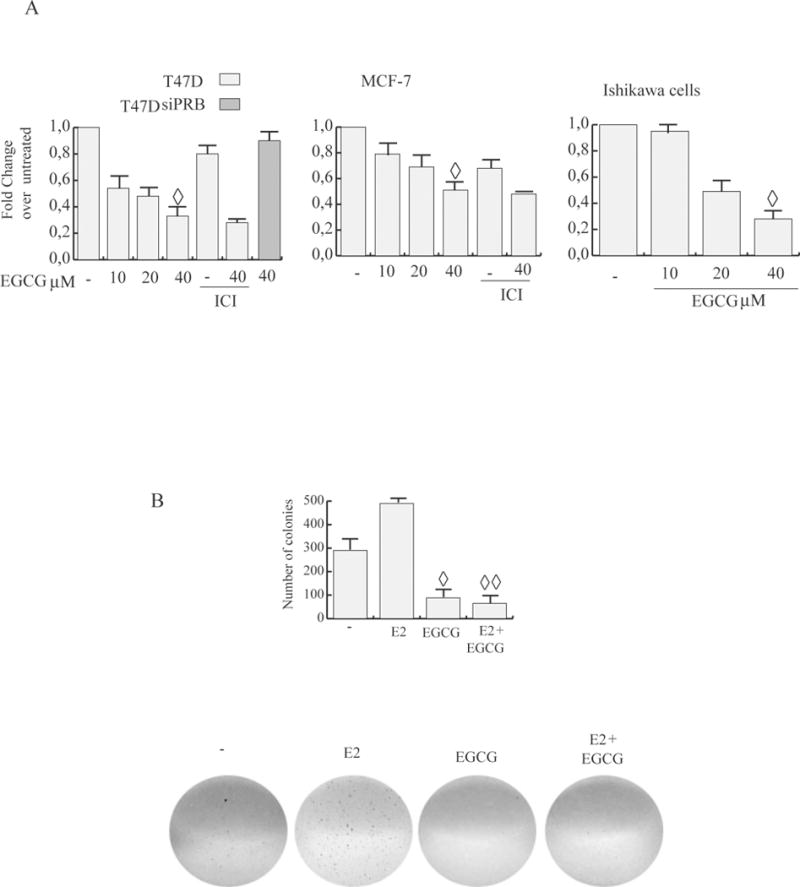
A, MTT assay. Cells, serum starved, were exposed to vehicle (−), or 1 μM ICI and/or different concentrations of EGCG in medium containing 1% dextran charcoal-stripped FBS for 4d (treatments were renewed every 2d). Results indicate mean of three independent experiments done in triplicate; bars SD;◊ P<0.05 compared with untreated cells. B, EGCG blocks E2 induced anchorage independent growth of T47D. Soft agar colony formation assay was performed in control conditions (−), or in the presence of 10 nM estrogen (E2) and/or 40 μM EGCG. Cells were allowed to grow for 14 d and the number of colonies ≥50 μm were quantified. bars SD;◊ P<0.05 compared with untreated cells; ◊◊ P<0.05 compared with E2 treated cells. Pictures at the bottom show typical well for each condition.
Of note is that the antiproliferative effects produced by EGCG occur earlier in T47D cells with respect of those observed in MCF-7 and ISK. Consistent with MTT assay results, 40μM EGCG treatment also markedly reduced basal and E2 induced colony formation of T47D cells growing in soft agar (Fig.1B).
EGCG down-regulates ERα expression and transcriptional activity
It is well known that increased expression of ERα is an early event in breast carcinogenesis, driving breast cancer cell proliferation. We thus hypothesized that EGCG’s effects in ERα-positive breast cancer cells could involve the regulation of ERα itself. As shown in Fig.2A, ERα protein expression levels were decreased after 24h of increasing EGCG concentrations, in MCF-7 and T47D. E2 treatment was used as control for down-regulation of ERα.
Figure 2. EGCG down-regulates ERα expression, genomic and non genomic signal.
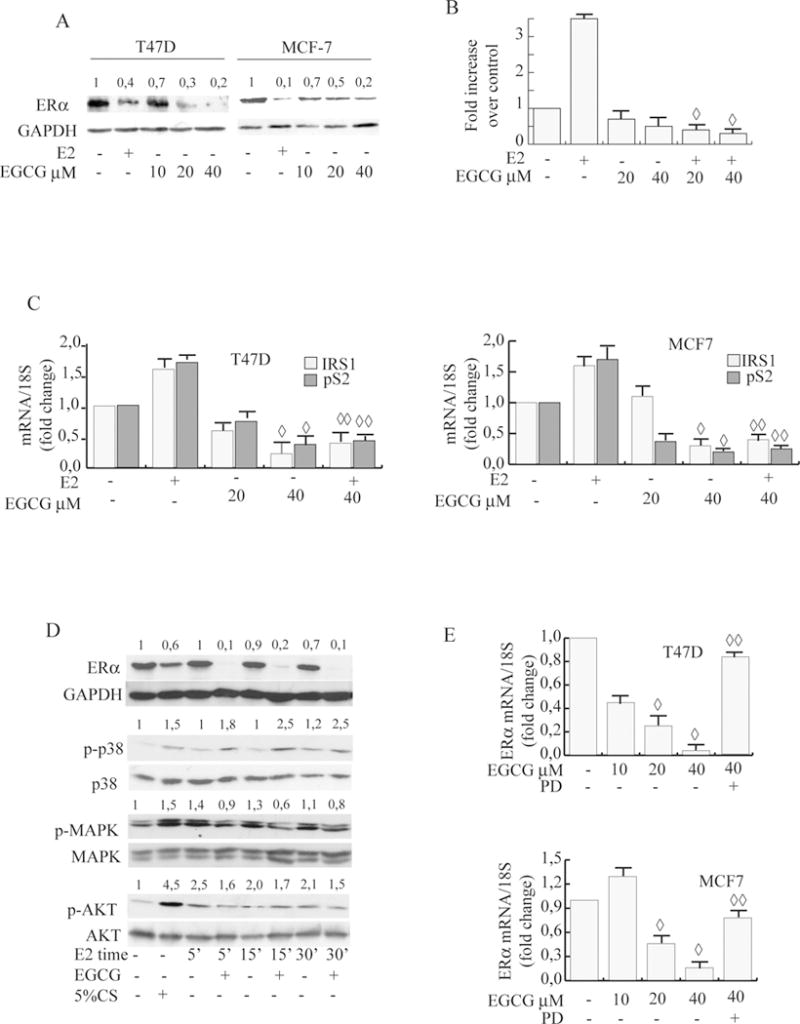
A, Immunoblot analysis. Cells were treated with different concentrations of EGCG for 24 h. The whole-cell lysates obtained were then collected and analyzed by WB using specific Abs. Numbers represent the average fold change in ERα and GAPDH levels. B, ERE luciferase reporter assay. XETL was transiently transfected into MCF-7 cells treated with vehicle (−) or 10 nM E2 and/or different concentrations of EGCG. Cells were then harvested, and luciferase activities were determined; Columns, mean of relative luciferase units (RLU); bars SD; ◊, P<0.05 compared with E2. C, Real time PCR assay. mRNA expression of pS2, IRS1 and CD1 in T47D and MCF-7 cells treated with vehicle (−) or 10 nM E2 and/or different concentrations of EGCG for 24 h; 18S was determined as a control. Graphs are the mean of three independent experiments run in triplicate; bars SD;◊ P<0.05 compared with untreated cells; ◊◊ P<0.05 compared with E2 treated cells. D, Immunoblot analysis. T47-D cells pre-treated with 40μM EGCG for 24 h were then treated with 10nM E2 for different times as indicated or with 5% CS for 24h treatment. The whole-cell lysates obtained were then collected and analyzed by WB using specific Abs. Numbers represent the average fold change in ERα and GAPDH, p-p38 and p38, p-MAPK and MAPK, p-AKT and AKT levels E, Real-time PCR assay. mRNA expression of ERα. Cells were treated with vehicle (−), different concentrations of EGCG for 24h as indicated in presence or absence of PD. Columns mean of three independent experiments; bars, SD. ◊, P<0.05 compared with untreated cells, ◊◊, P<0.05 compared with 40μM EGCG.
To investigate the functional effects of down-regulation of ERα by EGCG, we next analyzed ERα genomic activity after treatment. To this aim a luciferase reporter plasmid containing a consensus estrogen-responsive element (XETL) was transiently transfected into MCF-7 cells. EGCG treatment caused a significant decrease of basal, and with more intensity of E2 induced luciferase activity (7 fold) (Fig.2B). To confirm these results we also evaluated mRNA levels of known estrogen-regulated genes, such as IRS1, pS2 and cyclin D1 (CD1). As assessed by real-time PCR, EGCG at 40μM significantly decreased basal and E2 induced levels (Fig.2C) of IRSI, pS2 and CD1 in T47D and MCF-7 cells.
Stemming from our evidence that EGCG inhibited ERα levels, we hypothesized that it could also influence E2/ERα non genomic rapid effects within seconds to minutes [30]. Therefore to demonstrate the effects of EGCG on E2-induced non genomic activity, we pretreated T47D cells with EGCG for 24h causing ERα levels to decrease, and subsequently we treated the same cells with short exposures to E2. As shown in Fig.2D, EGCG treatment blocked E2 induced phosphorylation of MAPK and AKT. 5% CS treatments were used as positive controls for activation of downstream signaling.
The ERK/MAPK and PI3K/AKT signaling pathways that are rapidly activated by the ERα–E2 complex, also have critical roles in estrogen action as survival agents. In fact, these pathways enhance the expression of the anti-apoptotic protein Bcl-2 and block the activation of p38 kinase [31]. Western blot analysis in MCF-7 cells revealed that p38MAPK phosphorylation was significantly upregulated in EGCG-exposed cells, effectively opposing estrogen’s effect (Fig.2D). These results suggest that EGCG-induced ERα downegulation can affect non genomic events in the E2/ERα signaling cascade.
EGCG down-regulates ERα mRNA via a region between −2769 bp to −1000 bp of its promoter
To further investigate the molecular basis for regulation of ERα expression by EGCG, we examined its effects on ERα mRNA levels in T47D and MCF-7, treated for 24h. EGCG (20 μM and 40 μM) significantly down-regulated ERα as assessed by real-time-PCR (Fig.2E). Since EGCG down-regulated ERα mRNA, we next evaluated whether it could act at the level of gene transcription through regulatory regions within the ERα promoter. Therefore, we examined the ERα promoter region covered from −4100 bp to +212 bp first with a bioinformatics approach using the NCBI Genome data base (www.ncbi.nlm.nih.gov). The region examined in this study contains multiple regulatory elements, including binding sites for AP-1, NF-κB, Oct-1, Sp1, CCAAT binding proteins, CREB-2, USF1,1/2 PRE [20,25]. Five overlapping ERα promoter deletion constructs, −245 bp to +212 bp (A), −735 bp to +212 bp (B), −1000 bp to +212 bp (C), −2769 bp to +212 bp (D), and −4100 bp to +212 bp (E), all relative to the first transcriptional ATG start site (depicted in Fig.3A) and previously described [25], were transiently transfected into T47D and MCF-7 cells and the data are shown as relative promoter activity in luciferase units (Fig. 3B). We found that 40μM EGCG had no effect on the promoter activity of fragments A, B, or C. In contrast it reduced the activity of fragments D and E by 44% and 40%, respectively, indicating that the region between −2769 bp to −1000 bp may be responsible for down-regulation of ERα promoter activity after EGCG treatment.
Figure 3. EGCG down-regulates ERα promoter activity through an half PRE site.
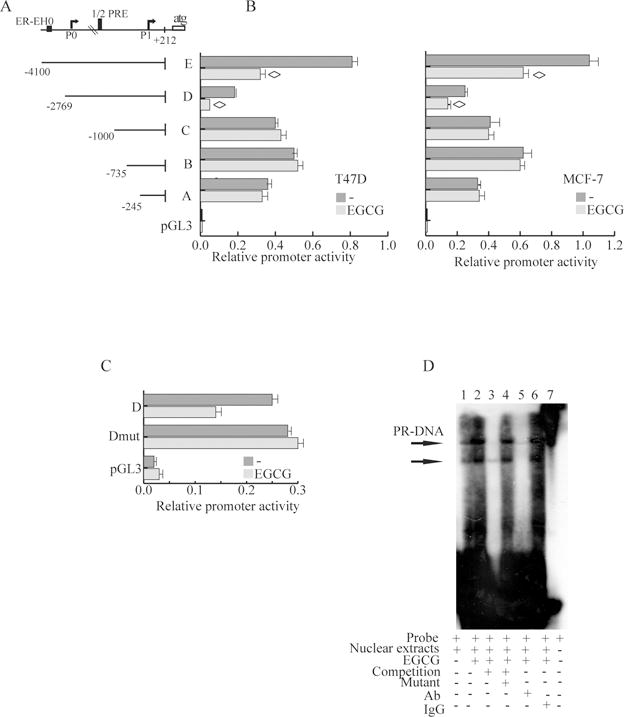
A, Schematic representation of deletion fragments of the ERα gene promoter. Fragments coordinates are expressed relative to the primary transcription start site. B, Promoter activity of the ERα 5′–flanking region. Constructs depicted in panel A were transiently transfected in T47D and MCF-7. cells, treated with vehicle (−) or 40μM EGCG. After 24h, cells were harvested and luciferase activities were determined; bars, SD; ◊, P<0.05 compared with untreated cells. C, Site-directed mutagenesis of the half-PRE site present in D fragment. Fragment D and fragment D mut promoter constructs were cotransfected into MCF-7 cells, and promoter activity was assessed in the absence or presence of 40μM EGCG.; bars, SD. D, EMSA assay. Nuclear extracts from T47-D cells treated with vehicle (−, lane 1) or 40μM EGCG (lanes 2–6) were incubated with a ERα promoter/half PRE annealed probe (lane 2) or, for competition analysis, 100-fold excess unlabeled annealed probe (lane 3), or mutated in half PRE site (lane 4). EGCG-treated nuclear extracts were also incubated with anti-PR-B (lane 5), IgG (lane 6), or probe alone (lane 7). Picture is representative of three independent experiments.
Precisely within this region of ERα promoter we also previously reported the presence of a functional half-PRE site responsible for transcriptional repression of ERα mediated by the B isoform of the PR [20]. Green tea poliphenols have been reported to exert their biological effects through steroid receptors [15] therefore we hypothesized that EGCG down-regulatory effects on ERα gene transcription might involve this potential half-PRE site. To test this hypothesis we next used site-directed mutagenesis to alter this site. Mutation of 3bp of the half-PRE site of the ERα promoter D fragment resulted in the loss of EGCG-responsiveness, suggesting that this half-PRE site could be an effector of EGCG signaling (Fig.3C).
To investigate DNA-protein interactions induced by EGCG within this promoter region, we next performed EMSA, using oligonucleotide from the ERα promoter sequence bearing the half PRE site (Fig.3D). Cell extracts prepared in the absence of ligand (lane 1) generated double retarded bands corresponding to half-PRE complexes which were enhanced in EGCG treated extracts (lane 2), and whose appearance were effectively competed by a 100 fold molar excess of unlabelled probe (lane 3), thus demonstrating the specificity of the DNA-binding complexes. This inhibition was no longer observed using a mutated half-PRE oligonucleotide as competitor (lane 4). A rabbit antibody to PR also completely disrupted the DNA-protein complexed bands (lane 5) while normal rabbit IgG addition did not affect protein–DNA complex formation (lane 6).
EGCG inhibits Hsp90 client proteins and promotes PR-B translocation into the nucleus
The functions of PR are modulated by Hsp90 and its inhibition can be measured via the degradation of its many client proteins (such as c-Raf and Her2) [32]. As shown in Fig.4A we observed depletion of Hsp90 client proteins EGFR, Raf-1, and Her 2 after EGCG treatment.
Figure 4. EGCG inhibits Hsp90 client proteins and promotes PR translocation into the nucleus via p38MAPK activation.
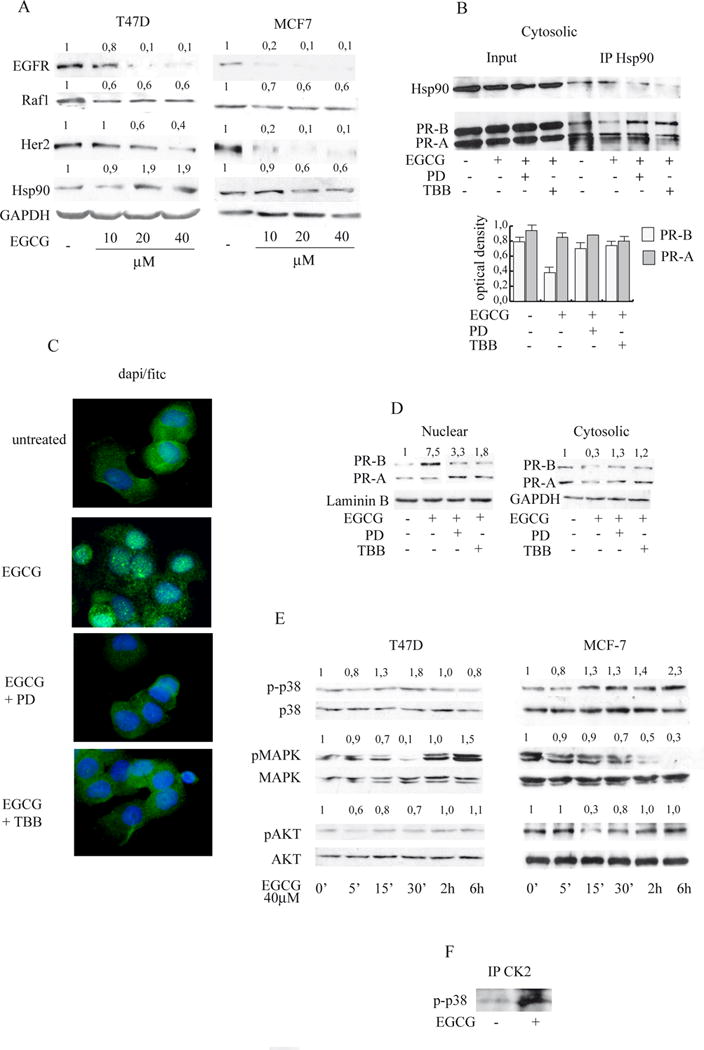
A, Immunoblot analysis. Cells were treated with different concentrations of EGCG for 24 h. The whole-cell lysates obtained were then collected and analyzed. Numbers represent the average fold change in EGFR or Raf1 or Her2 or Hsp90 and GAPDH levels B, Co-immunoprecipitation assay. T47-D cells were treated for 24h with vehicle (−) or 40μM EGCG in presence or absence of PD or TBB. Cytosolic cell lysates were immunoprecipitated using anti-Hsp90 and then blotted with anti Hsp90 or anti-PR antibody. Columns, mean of three independent experiments in which band intensities were evaluated in terms of optical density arbitrary units and expressed as IP/Input; bars, SD;◊ P<0.05 compared with untreated cells. C, Immunofluorescence with anti PR-B antibody. T47-D cells were treated for 12h with vehicle (−) or 40μM EGCG in presence or absence of PD or TBB, then were stained (Dapi/Fitc merge is presented). Immunofluorescent receptor is cytoplasmic in absence of treatment but shifted into the nucleus after EGCG administration. Pictures are representative of three independent experiments D, Immunoblot analysis. Cells were treated with 40μM of EGCG for 12h in presence or absence of PD or TBB, then nuclear and cytoplasmic protein extracts were analyzed. Numbers represent the average fold change in PR-B and Laminin B or GAPDH levels. E Immunoblot analysis. Cells were treated with vehicle (−) or 40μM EGCG at different times as indicated. The whole-cell lysates obtained were then collected and analyzed. Numbers represent the average fold change in p-p38 and p38, p-MAPK and MAPK, p-AKT and AKT levels. F Co-immunoprecipitation assay. Cells were treated for 24h with vehicle (−) or 40μM EGCG. Cell lysates were immunoprecipitated using anti-CK2 and then blotted with anti-p-p38.
Thus, we characterized the effects of EGCG on protein interaction between PR and Hsp90, performing co-immunoprecipitation assay from T47D cells cytosolic extracts. EGCG reduced the specific interactions between PR-B isoform and Hsp90 (Fig.4B), while it did not affect PR-A/Hsp90 complex. We also investigated the influence of EGCG treatment on PR-B cellular localization by immunofluorescence microscopy experiments using a specific antibody. As shown in Fig.4C, PR-B showed both a light nuclear and a stronger cytoplasmic distribution in untreated T47D cells. Interestingly, when cells were exposed to 40μM EGCG there was a remarkable redistribution of the PR-B from the cytosol to the nucleus. Results confirmed, by western blotting analysis of isolated cellular compartments (Fig 4D), that the PR-B nuclear isoform specifically was influenced by EGCG (7.5 fold nuclear vs. 0.3 fold cytoplasmic).
Therefore we next examined the effects of a short treatment of EGCG on the activation of kinase cascades. As shown in Fig4E, 40μM EGCG at different times modulated basal levels of p42/p44 MAPK and pAKT depending upon the cell type. At 15′, in both cell types, EGCG induced p38MAPK phosphorylation, which remained activated in MCF-7 cells even after 24h of treatment (data not shown). An important effector of p38MAPK is CK2 [33]. Immunoprecipitation assays revealed that EGCG treatment enhanced a physical interaction between CK2 and p-p38 (Fig.4F).
Stemming from these results, we next investigated the relevance of activated p38 in triggering the effects of EGCG treatment, by using a p38 inhibitor PD169316, known to block the p38 kinase activity [34]. Pretreatment with PD or with the CK2 inhibitor TBB, strongly restored the specific interaction between Hsp90 and PR-B (Fig.4B) indicating that activation of p38/CK2 signalling by ECGC modulates Hsp90/PR-B interaction.
Moreover PD and TBB clearly changed the cellular localization of PR-B produced by EGCG (Fig.4C,D) demonstrating that EGCG via p38/CK2 allows PR nuclear translocation; furthermore, PD attenuated ERα mRNA down-regulation by EGCG (Fig.2E).
Downregulation of ERα expression by EGCG is dependent on PR-B
We next investigated the role of the PR-B isoform in mediating EGCG effects on ERα. We co-transfected equal amounts of expression plasmids encoding PR-B or PR-A into the ER-PR-negative SKBR3 cell line, along with the ERα promoter reporter fragments. Only PR-B reduced ERα promoter activity (51%) with EGCG treatment (Fig.5A). Cotransfection of equal amounts of expression plasmids encoding either the PR-A isoform or the PR mutant with a disrupted DNA-binding domain (DBD) (Cys587 to Ala, called mDBD PR) did not affect ERα promoter activity either in presence or absence of EGCG, strongly suggesting that PR-B was responsible for EGCG down-regulatory effects on ERα, which occur at the transcriptional level.
Figure 5. PR-B mediates the EGCG effects on ERα promoter and recruits an NCoR corepressor complex.
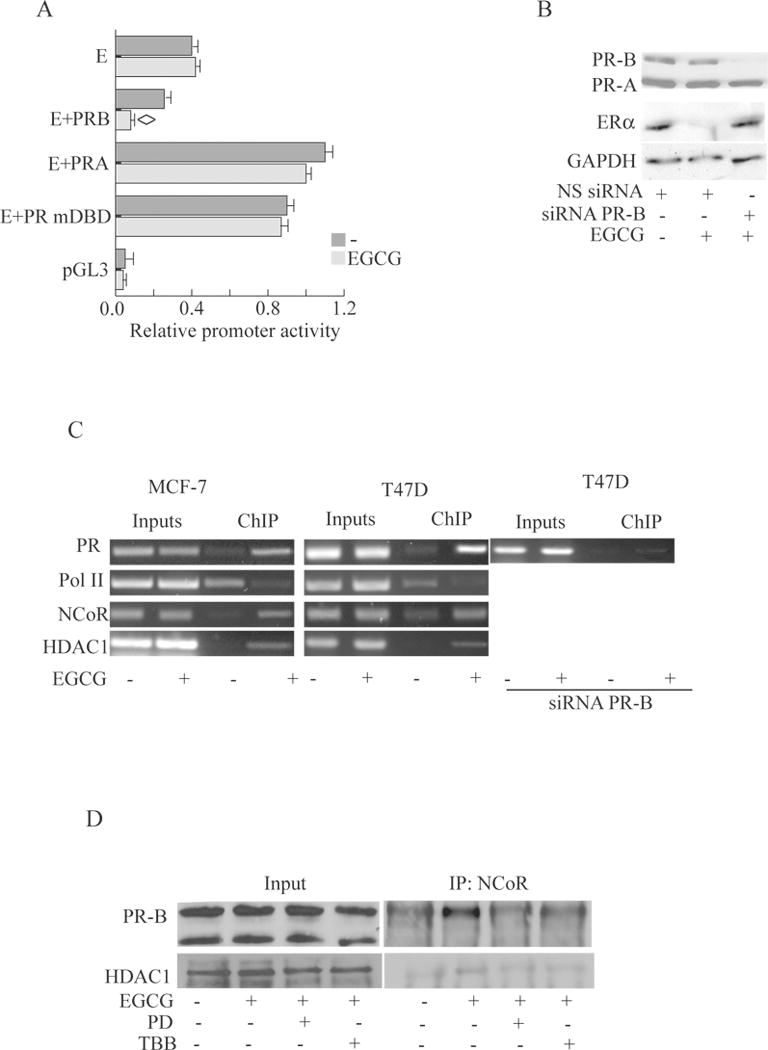
A, Promoter activity of the ERα 5′–flanking region. Fragment E was transiently transfected into Skbr3 cells in the presence or absence of full-length PR-B, or PR-A, or mDBD PR expression plasmid, then treated with 40μM EGCG or left untreated. After 24 h, cells were harvested, and luciferase activities were determined; bars, SD P<0.05 compared with untreated cells B, Immunoblot analysis.T47-D cells were transfected with NS siRNA or targeted against PR-B and treated with vehicle (−) or 40μM EGCG. C, ChIP assay. Cells were treated for 12h with vehicle (−) or 40 μM EGCG and then harvested. Antibodies used to immunoprecipitate the protein-DNA complex are indicated on the left. PCR primers encompass half PRE site within the ERα promoter. Results are representative of three independent experiments. D, Coimmunoprecipitation assay. T47-D cells were treated with vehicle (−) or 40μM EGCG in presence or absence of PD or TBB. Nuclear cell lysates were immunoprecipitated using anti-NCoR and then blotted with anti anti-PR or anti HDAC1 antibody.
We also performed PR-B siRNA knockdown experiments during EGCG treatment of T47D cells. Addition of a PR-B-targeting siRNA resulted in a remarkable decrease in PR-B protein levels (Fig.5B) which greatly blocked ERα downregulation by EGCG treatment. PR-B knockdown also blocked the growth inhibitory effect of EGCG in MTT assays (Fig 1A). Altogether these data strongly support the hypothesis that PR-B through the half PRE site within the ERα prmoter mediates EGCG effects on transcriptional regulation of ERα.
The NCoR corepressor is recruited with PR-B to the ERα promoter upon EGCG treatment
To demonstrate the recruitment of PR to the ERα gene promoter, we also used ChIP assays. T47D and MCF-7 cells were exposed to either control vehicle or 40μM EGCG, after which chromatin was cross-linked with formaldehyde, and protein-DNA complexes were immunoprecipitated with antibodies directed against PR, RNA polymerase II, or the corepressors NCoR, DAX-1, and SMRT. Results obtained demonstrate an enhanced recruitment of PR to the ERα promoter with EGCG treatment (Fig.5C); as a control we did not see recruitment to an unrelated ERα promoter region located upstream of the half-PRE site (data not shown). Specific knockdown of PR-B also confirmed the recruitment of this isoform to the ERα promoter (Fig.5C).
Concomitant with the increased recruitment of PR-B to the ERα promoter after EGCG treatment, we saw the similar molecular events we described previously [20]: displacement of RNA polymerase II, indicating that the chromatin in this region is probably in a less permissive environment for gene transcription, recruitment of NCoR in T47D and MCF7 cells (Fig.5C). Since it is known that different corepressor complexes contain one or more HDACs, as candidate mediators of PR action [35] we looked for histone deacetylases, in particular the class I HDAC-1 and HDAC-3. Our results show that HDAC-1 was the most strongly recruited following EGCG treatment. These data indicate PR-B binds to the ERα promoter at half-PRE site and this results in enhancement of the tripartite complex containting NCoR and HDAC1 with EGCG treatment and a concomitant release of RNAPol II, thereby inhibiting ERα transcription. Thus the catechin may acquire pharmacological significance acting as an estrogen receptor down-regulator in breast cancer cells.
To further address the interaction between PR-B, HDAC1 and NCoR we performed coimmunoprecipitation assays from nuclear extracts of T47D cells. EGCG induced the complex formation between PR-B, NCoR and HDAC1 (Fig.5D) via p-38/CK2, since pretreatment with PD or TBB prevented the interaction upon EGCG.
Discussion
Epidemiological studies have shown that breast cancer incidence rates are lowest among Asians, suggesting that Asian diets may contain phytochemicals with anti-breast cancer activities. Accumulating clinical evidence indicates the protective effects of polyphenols derived from green tea in ER+/PR+/HER2− breast tumor patients [36], while recent in vitro studies demonstrate their specific cytotoxicity toward breast cancer cells regardless of their ER status [37]. This could be due to the use of high doses of phytochemicals which may produce misleading results with regard to DNA damage and in several other respects. Specifically it was reported that dietary chemopreventive compounds can increase oxidative stress and cause DNA damage at high concentrations [38], whereas they act as antioxidants to reduce DNA damage at dietary or pharmacologic concentrations [39]. Often, the doses utilized in these studies are not achievable in vivo, therefore, whether phytochemicals might exert translational promises and benefits in clinical settings and prevention of breast cancer remain unclear.
In this study we tested EGCG doses considerably lower than the dose of 100–120 μM used for most in vitro studies and thus are closer to levels with physiological relevance [40]. We demonstrate that at doses of 10,20,40 μM EGCG effectively inhibits the growth of ER+ PR+ cancer cells (Fig. 1), and it exerts its effects through multiple mechanisms of action including 1) modulation of Hsp90 chaperone activity (Fig. 4A,B) and 2) activation of p38MAPK/CK2 signalling (Fig. 4E,F), influencing the redistribution of the PR (Fig. 4C) from the cytosol to the nucleus 3) recruitment of PR-B onto the ERα promoter (Fig. 5C) leading to transcriptional inhibition of ERα (Fig.3B) thus reducing its expression (Fig. 2A) and E2/ERα genomic (Fig. 2B,C) and non genomic activity (Fig. 2D). We herein indicate that EGCG via modulation of ERα blocks cancer cell proliferation. These studies provide a better understanding of the specific factors that can mediate ERα down-regulation in breast cancer cells, which may be critical for future therapeutic strategies attempting to reduce its expression.
A body of evidences obtained by in vivo studies demonstrate the consequences of unregulated ERα levels at all stages of breast cancer development. For instance, transgenic mice with elevated ERα levels can develop ductal hyperplasias, lobular hyperplasias, and ductal carcinoma in situ [2]. Furthermore it is shown that ESR1 is amplified in subsets of breast cancers and in precancerous breast diseases [41]. Therefore it is reasonable to consider ERα down-regulators of significant clinical interest. In this concern we show that EGCG treatment decreased both ERα genomic and rapid non genomic activity as a consequence of ERα content depletion which occurs through PR-B nuclear translocation. These novel findings corroborate our previous studies [20] indicating that inhibition of ERα by PR-B is a critical regulatory pathway in ERα positive breast cancer cells, and highlight the protective role of PR-B as determinant of EGCG down-regulatory effects, since PR-B knockdown attenuated the inhibition of ERα levels after EGCG stimulus.
Steroid receptors such as PR in the absence of ligand, interact with Hsp90 [42]. Previous studies demonstrated that EGCG interferes with Hsp90 chaperone activity for PR, by binding to Hsp90 at or near a C-terminal ATP binding site [43]. We demonstrate that EGCG acts at both ERα mRNA and protein level via Hsp90/p-38MAPK signaling to PR-B in T47D and MCF-7 cells. We also define the molecular mechanisms through which EGCG interferes with ERα gene transcription. Our functional experiments using five deletion constructs of the ERα promoter showed that EGCG’s down-regulatory effects occur through a region between −2769 bp to −1000 bp of the promoter (Fig. 3A) which contains a known half-PRE site that is required for repression of the ERα promoter by PR-B [20]. Our data support previous clinical and in vitro studies [20,44] illustrating the protective role of PR-B expression in breast cancer. This concept is further reinforced by studies showing that HER-2/neu signaling, is also inversely associated with PR levels [45].
EGCG specifically reduced the expression of HSP70 and HSP90 [43] which overproduction, instead, results in the increased incidence of cell transformation [46]. Our results correlate with these data since EGCG decreased Hsp90 client proteins expression levels (Fig. 4A) with a concomitant reduction of cell proliferation and estradiol induced colony formation of MCF-7 and T47D (Fig. 1B). Hsp90 inhibition is known to impact cellular client protein homeostasis via different mechanisms [32,47]. For example, Hsp90 inhibition can arrest maturation of Hsp90-bound client proteins without disrupting the cochaperone complex, can block the release of the refolded protein, leading to ubiquitin-dependent degradation or can prevent the binding of the target client protein to Hsp90. Our results confirm that EGCG acts as an inhibitor of Hsp90 chaperone function in ER+ PR+ breast cancer cell types examined, since it produced a drastic reduction of Her 2, EGFR, raf1 levels in a dose dependent manner (Fig.4 A).
Hsp90 functions in part as a cytoplasmic retention factor for steroid receptors [42], and our results suggest a role of Hsp90 as mediator of EGCG action in the modulation of the subcellular localization of PR-B isoforms. The nucleo-cytoplasmic distribution of PR-B in T47D cells was affected by EGCG treatment (Fig. 4C). Unliganded receptors mainly localize to the cytoplasm. However, EGCG treatment leads to more PR-B nuclear localization, concomitant to a reduced cytoplasmic retention due inhibition of the PR-B/Hsp90 interaction (Fig. 4B). EGCG treatment via p38/CK2 activation indeed resulted in destabilization of Hsp90/PR-B complexes. We also demonstrated the specific involvement of the PR-B isoform in mediating EGCG action using the isoform-specific siRNA experiments (Fig. 5B,C).
Our results are consistent with previous studies [48] showing that disruption of the chaperone function of Hsp90 by treatment with different drugs increased polyubiquitylation and depletion of ERα levels and transcriptional activity resulting growth arrest and apoptosis [49, 50].
EGCG may indirectly affect steroid receptors nuclear translocation influencing their ability to interact with Hsp90 clients via MAP kinase superfamily. It has been reported that PR-A and PR-B stability and transcriptional activities is selectively regulated by p38 and p42/44 MAPK through specific phosphorylation sites [51] and MAPKs are known to play important roles in PR subcellular trafficking via Ser294 phosphorylation [52]. A significant ӿnding in our study is that sustained p38 and CK2 activation by EGCG influenced Hsp90/PR-B interactions, and played a critical role in the nuclear localization of PR-B as demonstrated with specific inhibitors of p38 and CK2 (Fig.4 C, D). Dissociation of PR with its chaperones and receptor dimerization, leads to its binding to PREs in the promoters of target genes and the recruitment of specific coregulators and general transcription factors, resulting in the modulation of transcription of those genes [53]. Recent studies in the human endometrium have demonstrated that PR-A and PR-B are distributed either evenly throughout the nucleus or into discrete nuclear foci, and that there is a link between the subnuclear distribution of PR and its transcriptional activity [54]. These data collectively indicate that these nuclear foci contain multiple activated PR-coregulator complexes which can influence the basic transcriptional machinery. Our ChIP assays (Fig. 5C) of the ERα promoter correlate well with these previous reports and also show a release of RNAPol II. This is concomitant to the enhancement of the tripartite corepressor complex PR-B/NCoR/HDAC1 (Fig. 5C) due to PR-B nuclear localization via p38/CK2 activation.
Our results support the molecular mechanism that EGCG’s inhibition of ERα gene transcription is mediated by a physical recruitment to the promoter of PR-B, which then exerts a repressive effect on chromatin by maintaining interactions between the corepressor NCoR and HDAC1 with the ERα gene (Fig. 5C). Thus, PR-B recruitment after EGCG treatment is to the PRE half-sites in the ERα promoter. We also show that EGCG induces PR-B binding directly to this PRE half-site. Results from transient cell transfection assays, with ERα reporter gene containing point mutations in the half PRE (Fig. 3C), further confirm the importance of PR DNA binding and the PRE half-site for a functional inhibitory response to EGCG. Furthermore siRNA knockdown of PR-B confirmed that recruitment of the PR-B isoform to the half-PRE site within the ERα gene promoter is a prerequisite in mediating EGCG down-regulatory effects on ERα expression in breast cancer cells.
We propose a novel model (Fig.6) for the inhibitory action of EGCG s on the proliferation of ER+ PR+ breast cancer cells, through regulation of ERα gene transcription. p38/CK2 activation by EGCG provides a mechanism for the downregulation of Hsp90/PR-B interactions, as well as PR-B nuclear localization which is necessary for its recruitment to the half-PRE site within the ERα promoter. The formation of a tripartite complex, containing PR-B, HDAC1, and NCoR which interacts with the half PRE, induces those alterations in chromatin structure such as deacetylation which then block promoter accessibility to the transcriptional machinery such as RNAPol II.
Figure 6. A proposed model for EGCG induced repression of ERα promoter in ER+ breast cancer cells.
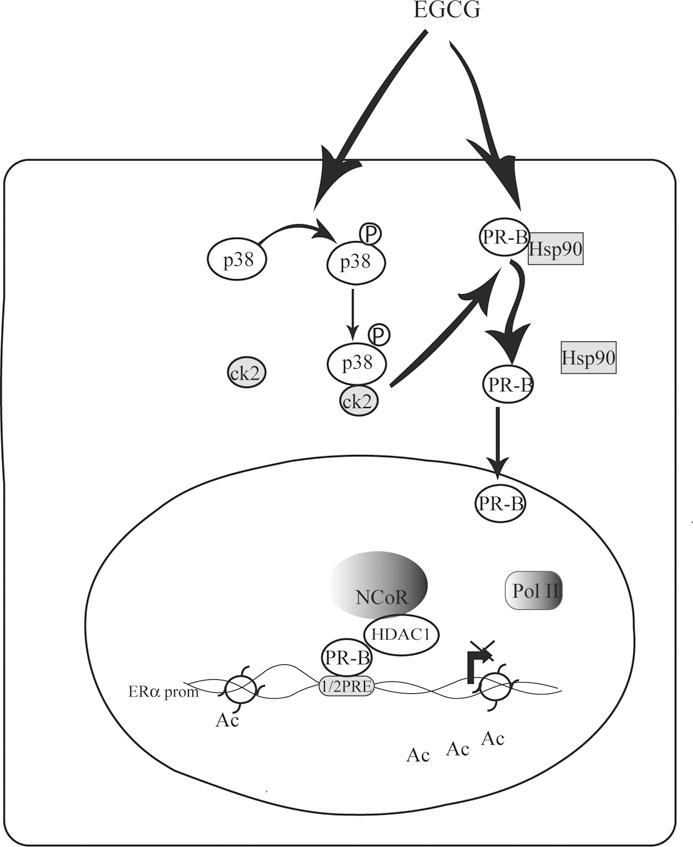
For details see the text. Ac, acetylation.
In conclusion we discovered that EGCG can function as ERα downregulator thus inhibiting ER+/PR+ breast cancer cell growth. Our results suggest that potentiating EGCG/PR-B signaling should be further exploited for clinical approach.
Acknowledgments
This work was supported by PRIN - MIUR and Ex 60 % 2011-and NIH/NCI R01 CA72038 to SAWF
Footnotes
Note
Authors declare that the experiments comply with the current laws of the country in which they were performed. The authors declare that they have no conflict of interest.
References
- 1.Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25:869–898. doi: 10.1210/er.2003-0010. [DOI] [PubMed] [Google Scholar]
- 2.Frech MS, Halama ED, Tilli MT, Singh B, Gunther EJ, Chodosh LA, Flaws JA, Furth PA. Deregulated estrogen receptor alpha expression in mammary epithelial cells of transgenic mice results in the development of ductal carcinoma in situ. Cancer Res. 2005;65:681–685. [PMC free article] [PubMed] [Google Scholar]
- 3.Poola I, Abraham J, Baldwin K, Saunders A, Bhatnagar R. Estrogen receptor h4 and h5 are ligand independent transcription factors and negative modulators of ERα: cloning from human ovary and functional characterization. Endocrine. 2005;27:227–38. doi: 10.1385/ENDO:27:3:227. [DOI] [PubMed] [Google Scholar]
- 4.van Agthoven T, Timmermans M, Dorssers LC, Henzen- Logmans SC. Expression of estrogen, progesterone and epidermal growth factor receptors in primary and metastatic breast cancer. Int J Cancer. 1995;63:790–793. doi: 10.1002/ijc.2910630607. [DOI] [PubMed] [Google Scholar]
- 5.Khan SA, Rogers MA, Obando JA, Tamsen A. Estrogen receptor expression of benign breast epithelium and its association with breast cancer. Cancer Res. 1994;54:993–997. [PubMed] [Google Scholar]
- 6.Hong WK, Sporn MB. Recent advances in chemoprevention of cancer. Science. 1997;278:1073–1077. doi: 10.1126/science.278.5340.1073. [DOI] [PubMed] [Google Scholar]
- 7.De Amicis F, Giordano F, Vivacqua A, Pellegrino M, Panno ML, Tramontano D, Fuqua SA, Andò S. Resveratrol, through NF-Y/p53/Sin3/HDAC1 complex phosphorylation, inhibits estrogen receptor {alpha} gene expression via p38MAPK/CK2 signaling in human breast cancer cells. FASEB J. 2011;25:3695–3707. doi: 10.1096/fj.10-178871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukhtar H, Wang ZY, Katiyar SK, Agarwal R. Tea components: antimutagenic and anticarcinogenic effects. Prev Med. 1992;21:351–360. doi: 10.1016/0091-7435(92)90042-g. [DOI] [PubMed] [Google Scholar]
- 9.Nakachi K, Suemasu K, Suga K, Takeo T, Imai K, Higashi Y. Influence of drinking green tea on breast cancer malignancy among Japanese patients. Jpn J Cancer Res. 1998;89:254–261. doi: 10.1111/j.1349-7006.1998.tb00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liao S, Umekita Y, Guo J, Kokontis JM, Hiipakka RA. Growth inhibition and regression of human prostate and breast tumors in athymic mice by tea epigallocatechin gallate. Cancer Lett. 1995;96:239–243. doi: 10.1016/0304-3835(95)03948-v. [DOI] [PubMed] [Google Scholar]
- 11.Belguise K, Guo S, Sonenshein GE. Activation of FOXO3a by the green tea polyphenol epigallocatechin-3-gallate induces estrogen receptor alpha expression reversing invasive phenotype of breast cancer cells. Cancer Res. 2007;67:5763–5770. doi: 10.1158/0008-5472.CAN-06-4327. [DOI] [PubMed] [Google Scholar]
- 12.Sah JF, Balasubramanian S, Eckert RL, Rorke EA. Epigallocatechin-3-gallate inhibits epidermal growth factor receptor signaling pathway. Evidence for direct inhibition of ERK1/2 and AKT kinases. J Biol Chem. 2004;279:12755–12762. doi: 10.1074/jbc.M312333200. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Hastak K, Ahmad N, Lewin JS, Mukhtar H. Inhibition of prostate carcinogenesis in TRAMP mice by oral infusion of green tea polyphenols. Proc Natl Acad Sci. 2001;98:10350–10355. doi: 10.1073/pnas.171326098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang YC, Lin-shiau SY, Chen CF, Lin JK. Suppression of extracellular signals and cell proliferation through EGF receptor binding by (−)-epigallocatechin gallate in human A431 epidermoid carcinoma cells. J Cell Biochem. 1997;67:55–65. doi: 10.1002/(sici)1097-4644(19971001)67:1<55::aid-jcb6>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 15.Siddiqui IA, Asim M, Hafeez BB, Adhami VM, Tarapore RS, Mukhtar H. Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. FASEB J. 2011;25:1198–1207. doi: 10.1096/fj.10-167924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qanungo S, Das M, Haldar S, Basu A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis. 2005;26:958–967. doi: 10.1093/carcin/bgi040. [DOI] [PubMed] [Google Scholar]
- 17.Tran PL, Kim SA, Choi HS, Yoon JH, Ahn SG. Epigallocatechin-3-gallate suppresses the expression of HSP70 and HSP90 and exhibits anti-tumor activity in vitro and in vivo. BMC Cancer. 2010;10:276. doi: 10.1186/1471-2407-10-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carson-Jurica MA, Schrader WT, O’Malley BW. Steroid receptor family: structure and functions. Endocr Rev. 1990;11:201–220. doi: 10.1210/edrv-11-2-201. [DOI] [PubMed] [Google Scholar]
- 19.Farabegoli F, Barbi C, Lambertini E, Piva R. (−)-Epigallocatechin-3-gallate downregulates estrogen receptor alpha function in MCF-7 breast carcinoma cells. Cancer Detect Prev. 2007;31:499–504. doi: 10.1016/j.cdp.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 20.De Amicis F, Zupo S, Panno ML, Malivindi R, Giordano F, Barone I, Mauro L, Fuqua SA, Andò S. Progesterone receptor B recruits a repressor complex to a half-PRE site of the estrogen receptor alpha gene promoter. Mol Endocrinol. 2009;23:454–465. doi: 10.1210/me.2008-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 22.Tora L, Mullick A, Metzger D, Ponglikitmongkol M, Park I, Chambon P. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J. 1989;8:1981–1986. doi: 10.1002/j.1460-2075.1989.tb03604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007;27:466–480. doi: 10.1128/MCB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, Chambon P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9:1603–1614. doi: 10.1002/j.1460-2075.1990.tb08280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.deGraffenried LA, Hilsenbeck SG, Fuqua SA. Sp1 is essential for estrogen receptor α gene transcription. J Steroid Biochem Mol Biol. 2002;82:7–18. doi: 10.1016/s0960-0760(02)00151-6. [DOI] [PubMed] [Google Scholar]
- 26.Vivacqua A, Lappano R, De Marco P, Sisci D, Aquila S, De Amicis F, Fuqua SA, Andò S, Maggiolini M. G protein-coupled receptor 30 expression is up-regulated by EGF and TGF alpha in estrogen receptor alpha-positive cancer cells. Mol Endocrinol. 2009;23:1815–1826. doi: 10.1210/me.2009-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Amicis F, Thirugnansampanthan J, Cui Y, Selever J, Beyer A, Parra I, Weigel NL, Herynk MH, Tsimelzon A, Lewis MT, Chamness GC, Hilsenbeck SG, Andò S, Fuqua SA. Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat. 2010;121:1–11. doi: 10.1007/s10549-009-0436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guido C, Panza S, Santoro M, Avena P, Panno ML, Perrotta I, Giordano F, Casaburi I, Catalano S, De Amicis F, Sotgia F, Lisanti MP, Andò S, Aquila S. Estrogen receptor beta (ERβ) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle. 2012;11:2911–21. doi: 10.4161/cc.21336. [DOI] [PubMed] [Google Scholar]
- 29.Hu XF, Veroni M, De Luise M, Wakeling A, Sutherland R, Watts CK, Zalcberg JR. Circumvention of tamoxifen resistance by the pure anti-estrogen ICI 182,780. Int J Cancer. 1993;55:873–876. doi: 10.1002/ijc.2910550529. [DOI] [PubMed] [Google Scholar]
- 30.Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, et al. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001;20:6050–9. doi: 10.1093/emboj/20.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Acconcia F, Totta P, Ogawa S, Cardillo I, Inoue S, Leone S, et al. Survival versus apoptotic 17b-estradiol effect: role of ERα and ERβ activated non-genomic signalling. J Cell Physiol. 2005;203:193–20. doi: 10.1002/jcp.20219. [DOI] [PubMed] [Google Scholar]
- 32.Tillotson B, Slocum K, Coco J, Whitebread N, Thomas B, West KA, MacDougall J, Ge J, Ali JA, Palombella VJ, Normant E, Adams J, Fritz CC. Hsp90 (heat shock protein 90) inhibitor occupancy is a direct determinant of client protein degradation and tumor growth arrest in vivo. J Biol Chem. 2010;285:39835–39843. doi: 10.1074/jbc.M110.141580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hildesheim J, Salvador JM, Hollander MC, Fornace AJ. Jr Casein kinase 2- and protein kinase A-regulated adenomatous polyposis coli and beta-catenin cellular localization is dependent on p38 MAPK. J Biol Chem. 2005;280:17221–17226. doi: 10.1074/jbc.M410440200. [DOI] [PubMed] [Google Scholar]
- 34.Duval D, Malaisé M, Reinhardt B, Kedinger C, Boeuf H. A p38 inhibitor allows to dissociate differentiation and apoptotic processes triggered upon LIF withdrawal in mouse embryonic stem cells. Cell Death Differ. 2004;11:331–341. doi: 10.1038/sj.cdd.4401337. [DOI] [PubMed] [Google Scholar]
- 35.Jepsen K, Rosenfeld MG. Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci. 2006;115:689–698. doi: 10.1242/jcs.115.4.689. [DOI] [PubMed] [Google Scholar]
- 36.Ogunleye AA, Xue F, Michels KB. Green tea consumption and breast cancer risk or recurrence: a meta-analysis. Breast Cancer Res Treat. 2010;119:477–484. doi: 10.1007/s10549-009-0415-0. [DOI] [PubMed] [Google Scholar]
- 37.Thangapazham RL, Passi N, Maheshwari RK. Green tea polyphenol and epigallocatechin gallate induce apoptosis and inhibit invasion in human breast cancer cells. Cancer Biol Ther. 2007;6:1938–1943. doi: 10.4161/cbt.6.12.4974. [DOI] [PubMed] [Google Scholar]
- 38.De Amicis F, Santoro M, Guido C, Russo A, Aquila S. Epigallocatechin gallate affects survival and metabolism of human sperm. Mol Nutr Food Res. 2012 Sep 13; doi: 10.1002/mnfr.201200190. [DOI] [PubMed] [Google Scholar]
- 39.Moiseeva EP, Almeida GM, Jones GD, Manson MM. Extended treatment with physiologic concentrations of dietary phytochemicals results in altered gene expression, reduced growth, and apoptosis of cancer cells. Mol Cancer Ther. 2007;6:3071–3079. doi: 10.1158/1535-7163.MCT-07-0117. [DOI] [PubMed] [Google Scholar]
- 40.Hsieh TC, Wu JM. Suppression of cell proliferation and gene expression by combinatorial synergy of EGCG, resveratrol and gamma-tocotrienol in estrogen receptor-positive MCF-7 breast cancer cells. Int J Oncol. 2008;33:851–859. [PubMed] [Google Scholar]
- 41.Holst F, Stahl PR, Ruiz C, Hellwinkel O, Jehan Z, Wendland M, Lebeau A, Terracciano L, Al-Kuraya K, Janicke F, Sauter G, Simon R. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat Genet. 2007;39:655–660. doi: 10.1038/ng2006. [DOI] [PubMed] [Google Scholar]
- 42.Schowalter DB, Sullivan WP, Maihle NJ, Dobson AD, Conneely OM, O’Malley BW, Toft DO. Characterization of progesterone receptor binding to the 90- and 70-kDa heat shock proteins. J Biol Chem. 1991;266:21165–21173. [PubMed] [Google Scholar]
- 43.Yin Z, Henry EC, Gasiewicz TA. Epigallocatechin-3-gallate is a novel Hsp90 inhibitor. Biochemistry. 2009;48:336–345. doi: 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hopp TA, Weiss HL, Hilsenbeck SG, Cui Y, Allred DC, Horwitz KB, Fuqua SA. Breast cancer patients with progesterone receptor PR-A-rich tumors have poorer disease-free survival rates. Clin Cancer Res. 2004;10:2751–2760. doi: 10.1158/1078-0432.ccr-03-0141. [DOI] [PubMed] [Google Scholar]
- 45.Huang HJ, Neven P, Drijkoningen M, Paridaens R, Wildiers H, Van Limbergen E, Berteloot P, Amant F, Christiaens MR, Vergote I. Association between HER-2/neu and the progesterone receptor in oestrogen-dependent breast cancer is age-related. Breast Cancer Res Treat. 2005;91:81–87. doi: 10.1007/s10549-004-8235-8. [DOI] [PubMed] [Google Scholar]
- 46.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90(Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- 48.Fiskus W, Ren Y, Mohapatra A, Bali P, Mandawat A, Rao R, Herger B, Yang Y, Atadja P, Wu J, Bhalla K. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res. 2007;13:4882–90. doi: 10.1158/1078-0432.CCR-06-3093. [DOI] [PubMed] [Google Scholar]
- 49.Bagatell R, Khan O, Paine-Murrieta G, et al. Destabilization of steroid receptors by heat shock protein 90-binding drugs: a ligand-independent approach to hormonal therapy of breast cancer. Clin Cancer Res. 2001;7:2076–84. [PubMed] [Google Scholar]
- 50.Lee M, Kim E, Kwon HJ, et al. Radicicol represses the transcriptional function of the estrogen receptor by suppressing the stabilization of the receptor by heat shock protein 90. Mol Cell Endocrinol. 2002;188:47–54. doi: 10.1016/s0303-7207(01)00753-5. [DOI] [PubMed] [Google Scholar]
- 51.Khan JA, Amazit L, Bellance C, Guiochon-Mantel A, Lombès M, Loosfelt H. p38 and p42/44 MAPKs differentially regulate progesterone receptor A and B isoform stabilization. Mol Endocrinol. 2011;25:1710–1724. doi: 10.1210/me.2011-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu M, Olsen A, Faivre E, Horwitz KB, Lange CA. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol. 2003;17:628–642. doi: 10.1210/me.2002-0378. [DOI] [PubMed] [Google Scholar]
- 53.Arnett-Mansfield RL, Graham JD, Hanson AR, Mote PA, Gompel A, Scurr LL, Gava N, de Fazio A, Clarke CL. Focal subnuclear distribution of progesterone receptor is ligand dependent and associated with transcriptional activity. Mol Endocrinol. 2007;21:14–29. doi: 10.1210/me.2006-0041. [DOI] [PubMed] [Google Scholar]