

Abstract
The phosphite assimilating bacterium, P. glucosidilyticus DD6b, was isolated from the gut of the zooplankton Daphnia magna. Its 3,872,381 bp high-quality draft genome is arranged into 93 contigs containing 3311 predicted protein-coding and 41 RNA-encoding genes. This genome report presents the specific properties and common features of P. glucosidilyticus DD6b genome in comparison with the genomes of P. glucosidilyticus type strain DSM 23,534, and another five Pedobacter type strains with publicly available completely sequenced genomes. Here, we present the first journal report on P. glucosidilyticus genome sequence and provide information on a new specific physiological determinant of P. glucosidilyticus species.
Electronic supplementary material
The online version of this article (doi:10.1186/s40793-015-0086-x) contains supplementary material, which is available to authorized users.
Keywords: Pedobacter glucosidilyticus, Phosphite assimilation, Sphingobacteriia
Introduction
Pedobacter glucosidilyticus strain DD6b was isolated from the crustacean Daphnia magna gut microbial community. During a study on nutritional needs of D. magna, the gut microbiota was investigated over time, under starvation stress and after host death [1, 2].
Daphnia spp. are small members of the zooplankton and key organisms in freshwater food webs. Heterotrophic bacteria contribute significantly to the nutrition of Daphnia species [3, 4] and are often characterized by high P:C values [5], indicating that they are a rich source of phosphorus for zooplankton [6].
Furthermore, some bacteria can assimilate reduced inorganic and organic P compounds (phosphite [+III] and organophosphonates, [7–15]) under phosphate starvation. Organophosphonates have been found in a variety of biologically produced molecules including antibiotics, phosphonolipids, phosphonoproteins, phosphonoglycans [7]. The most common naturally occurring phosphonate 2-AEP or ciliatine serves as a precursor in the biosynthesis of phosphonolipids in marine single celled organisms, sea anemones or ciliated protozoan. Recently, a sphingophosphonolipid was found in Bacteriovorax stolpii, a facultative predator that parasitizes larger Gram-negative bacteria [16].
Tests with newly isolated P. glucosidilyticus DD6b revealed growth with phosphite as a single P-source. This ability of the strain DD6b, together with the lack of information in the literature concerning phosphite or organophosphonate assimilation abilities of the other members of genus Pedobacter was the reason to investigate the genome of P. glucosidilyticus DD6b.
Organism information
Classification and features
P. glucosidilyticus strain DD6b is an aerobic, Gram negative, non-spore-forming and rod-shaped gliding bacterium, isolated from homogenized guts of the crustacean Daphnia magna. Strain DD6b is mesophilic to psychrotolerant, chemoheterotrophic and assimilates phosphite as sole P-source under phosphate starvation.
The type strain of Pedobacter glucosidilyticus 1-2T (=CCTCC AB 206110T=KCTC 22438T=DSM 23,534) was isolated from a soil microbial community of a dry riverbed in the Xietongmen area (Tibet, China) in 2010 by Luo et al., [17].
The cells of P. glucosidilyticus strain DD6b are non-flagellated, non-spore-forming, flexible gliding rods with slightly rounded or tapered ends. They have protruded surfaces, and vary in size ranging from 1.0–1.2 μm in length and 0.2–0.3 μm in width (Fig. 1). Colonies (0.8–1 mm in diameter) appear after 6–7 days. They have orange-pink color on nutrient agar at 25 °C (Fig. 1, Right). Strain DD6b exhibits moderate growth, with a doubling time of 15–20 h, when grown on complex media such as nutrient broth. On chemically defined minimal medium MDS3 the strain had a doubling time of a) 7.5 h with phosphate and b) 20–23 h with phosphite as single P-source. Growth occurred at 15–28 °C, pH 7.0–7.2, and 0.2–0.5 % NaCl in the medium. Strain DD6b is motile via gliding.
Fig. 1.

Scanning electron micrograph of P. glucosidilyticus strain DD6b (Left) and colony morphology on solid medium (Right)
P. glucosidilyticus strain DD6b differs slightly from the type strain of P.glucosidilyticus DSM 23,534 showing a weak oxidation of amygdalin, arbutin, cellobiose, lactose, methyl-α-D-mannopyranoside, methyl-α-D-glucopyranoside and salicin [17]. In addition, strain DD6b cannot oxidize glycerol or melibiose coupled with production of acids whereas for the type strain P. glucosidilyticus DSM 23,534 weak growth on both substrates was reported [17].
The ability of P. glucosidilyticus strain DD6b to grow on phosphite was proved by cultivating it successfully after 3 successive transfers on MDS3 medium, supplemented with 1 mM phosphite as sole P-source. The concentration of phosphite in the medium was monitored during the growth experiments as described previously [10].
Current taxonomic classification and general features of P. glucosidilyticus DD6b according to the minimum information about the genome sequence requirements are provided in Table 1. Additional information about the genome is available in the associated MIGS record table (Additional file 1: Table S1).
Table 1.
Classification and general features of P. glucosidilyticus strain DD6b [22]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [39] | |
| Phylum | TAS [40] | ||
| Class Sphingobacteriia | TAS [41] | ||
| Order Sphingobacteriales | TAS [42] | ||
| Family Sphingobacteriaceae | TAS [43] | ||
| Genus Pedobacter | TAS [43, 44] | ||
| Species Pedobacter glucosidilyticus | TAS [17] | ||
| strain: DD6b | |||
| Gram stain | negative | TAS [17] | |
| Cell shape | Rods | IDA,TAS [17] | |
| Motility | Gliding, non-flagelated | IDA | |
| Sporulation | Non-sporulating | TAS [17] | |
| Temperature range | 15–28 °C | IDA | |
| Optimum temperature | 25 °C | IDA | |
| pH range; Optimum | 6.5–7.5; 7.0 | IDA | |
| Carbon source | glucose | IDA | |
| MIGS-6 | Habitat | gut of D. magna | TAS [1] |
| MIGS-6.3 | Salinity | 0.2–0.5 % NaCl (w/v) | IDA |
| MIGS-22 | Oxygen requirement | Aerobic | IDA |
| MIGS-15 | Biotic relationship | commensal | TAS [1] |
| MIGS-14 | Pathogenicity | non-pathogen | NAS |
| MIGS-4 | Geographic location | Germany/Constance | TAS [1] |
| MIGS-5 | Sample collection | October 2008 | NAS |
| MIGS-4.1 | Latitude | 47.689081 | NAS |
| MIGS-4.2 | Longitude | 9.187099 | NAS |
| MIGS-4.4 | Altitude | 405 m a.s.l. | NAS |
aEvidence codes - IDA Inferred from Direct Assay, TAS Traceable Author Statement (i.e., a direct report exists in the literature), NAS Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [45]
The phylogenetic neighborhood of P. glucosidilyticus DD6b based on 16S rRNA sequence is shown in Fig. 2. A comparison of 16S rRNA of P. glucosidilyticus strain DD6b with the non-redundant nucleotide collection of NCBI using MegaBlast revealed 98 % sequence identity to 16S rRNA gene sequences of uncultured Pedobacter sp. clone BF 061 (1461/1484 bps; NCBI accession: KC994741) and Pedobacter glucosidilyticus strain HME8545 (1378/1399 bps; NCBI accession: KC157040), respectively.
Fig. 2.
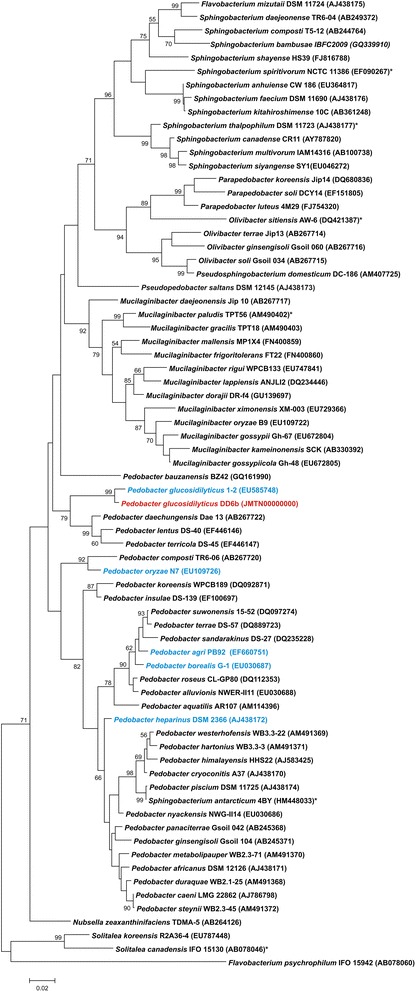
Phylogenetic tree based on 16S rRNA gene sequences of all types trains of Sphingobacteriaceae: The dendrogram was calculated with MEGA5 [18] using the Maximum Likelihood method based on the Jukes-Cantor model [19]. The analyzed sequences were aligned by CLUSTALW [21]. The clustering of the sequences was tested by the bootstrap approach with 1000 resamplings. The length of the tree branches was scaled according to the number of substitutions per site (see size bar). P. glucosidilyticus DD6b is marked in red, Pedobacter species with publicly available genome sequences are marked in blue. All other genome-sequenced species are marked with an asterisk
The phylogenetic tree was produced with MEGA5 [18] using the Maximum Likelihood method based on the Jukes Cantor model [19]. Sequences of all Sphingobacteriaceae type strains were downloaded from RDP [20], aligned by CLUSTALW [21] and tested by the bootstrap approach with 1000 resamplings. The length of the tree branches was scaled according the number of substitutions per site. Strain DD6b clustered clearly within the Pedobacter group and next to P. glucosidilyticus 1-2T.
Genome sequencing information
Genome project history
This organism was selected for sequencing on the basis of its environmental relevance to issues in global P cycle and the source of isolation. The genome project is deposited in GenBank database (JMTN00000000) and an improved high-quality-draft genome sequence in IMG (2590828803). Genome sequencing and annotation were done at Göttingen Genomics Laboratory (Georg-August-University Göttingen), while cultivation and analysis were performed at the University of Konstanz. A summary of the project information and its association with MIGS version 2.0 compliance [22] is shown in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Improved-high-quality draft |
| MIGS-28 | Libraries used | Illumina paired-end library; Nextera XT |
| MIGS 29 | Sequencing platforms | Illumina XT |
| MIGS 31.2 | Fold coverage | 120 × Illumina |
| MIGS 30 | Assemblers | SPAdes |
| MIGS 32 | Gene calling method | Prodigal |
| Locus Tag | PBAC | |
| Genbank ID | JMTN00000000 | |
| GenBank Date of Release | December 8th, 2014 | |
| GOLD ID | Gp0043583 | |
| BIOPROJECT | PRJNA246159 | |
| MIGS 13 | Source Material Identifier | DD6b |
| Project relevance | Ecology |
Growth conditions and genomic DNA preparation
P. glucosidilyticus DD6b was grown in nutrient broth. A newly developed chemically defined medium MDS3 was used to study carbohydrate, phosphite and phosphate assimilation. Phosphite and phosphate assimilation tests were performed in MDS3 medium supplemented with 0.1 to 1 mM phosphite or phosphate as single P- source. Glucose 10 mM final concentration was used as C- source. The chemical composition and preparation of MDS3 medium are given in Additional file 2: Data S2. The DNA extraction procedure was performed in the following way: 4 ml of a well grown fresh culture reaching its maximum optical density (OD600nm = 0.291 ± 0.040) were spun down at 13 000 × g in a benchtop micro centrifuge for 5 min. The supernatant was discarded and the cell pellet was suspended in the cell lysis solution from the Purgene Core Kit B (Qiagen, Hilden, Germany). Further, the extraction was performed as per manufacturer’s instructions, following the protocol for Gram-negative bacteria. The genomic DNA yield was 47.7 ng/μl. The purity of the preparation was estimated with NanoDrop ND-1000 (Thermo Fisher Scientific, Germany), with an UV absorbance ratio at 260/280 nm of 2.33, and an UV absorbance ratio at 260/230 of 0.84.
Genome sequencing and assembly
The extracted genomic DNA was used for whole genome sequencing employing a Genome Analyzer II (Illumina, San Diego, CA, USA). Shotgun libraries were prepared according to the protocol of the manufacturer. Sequencing resulted in 12,380,618 paired-end Illumina reads of 112 bp. Reads were trimmed using Trimmomatic 0.32 [23] to get rid of sequences with quality scores lower than 20 (Illumina 1.9 encoding) and remaining adaptor sequences. SPAdes 2.5 software [24] was employed for the initial de novo assembly and 4,150,000 reads. The final assembly resulted in 93 contigs larger than 0.5 kb from which 84 were larger than 1 kb including 68 contigs larger than 3 kb. This assembly had an average coverage of 120, N50 value of 97,360 bp and N90 value of 24,905 bp, respectively.
Genome annotation
The software tool Prodigal [25] was used for automatic gene prediction. rRNA and tRNA genes were identified with RNAmmer and tRNAscan, respectively [26, 27]. Automatic annotation was carried out with the IMG-ER system [28, 29] and afterwards manually curated by employing BLASTP, Swiss-Prot, TrEMBL, and InterPro databases [30].
Genome properties
The statistics of the genome are given in Table 3. The high quality draft genome was assembled into 93 contigs with a total size of 3876 Mb and an overall GC content of 34.74 mol%. A total of 3352 genes were predicted of which 3311 were protein-encoding and 41 RNAs genes (3 rRNA and 38 tRNA). Of the protein-encoding genes 2610 (77.86 %) were assigned to a putative function and the remaining 701 (20.91 %) were annotated as hypothetical proteins. The distribution of the genes into COG functional categories [31] is shown in Table 4. One CRISPR array of 46 repeats with a direct repeat length of 46 nt adjacent to an incomplete cas cluster comprising cas1, cas2 and cas9 was identified in the genome of P. glucosidilyticus DD6b. However, potential prophage regions were not present in the genome sequence.
Table 3.
Genome statistics
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 3,872,381 | 100.00 |
| DNA coding (bp) | 3,510,386 | 90.65 |
| DNA G + C (bp) | 1,344,522 | 34.72 |
| DNA scaffolds | 93 | 100.00 |
| Total genes | 3352 | 100.00 |
| Protein coding genes | 3311 | 98.78 |
| RNA genes | 41 | 1.22 |
| Pseudo genes | 0 | 0 |
| Genes in internal clusters | 2465 | 73.54 |
| Genes with function prediction | 2610 | 77.86 |
| Genes assigned to COGs | 1910 | 73.54 |
| Genes with Pfam domains | 2646 | 56.98 |
| Genes with signal peptides | 482 | 14.38 |
| Genes with transmembrane helices | 749 | 22.34 |
| CRISPR repeats | 1 | 0 |
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | % age | Description |
|---|---|---|---|
| J | 149 | 7.17 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.00 | RNA processing and modification |
| K | 129 | 6.20 | Transcription |
| L | 102 | 4.91 | Replication, recombination and repair |
| B | 1 | 0.04 | Chromatin structure and dynamics |
| D | 20 | 0.96 | Cell cycle control, Cell division, chromosome partitioning |
| V | 35 | 1.68 | Defense mechanisms |
| T | 69 | 3.32 | Signal transduction mechanisms |
| M | 185 | 8.90 | Cell wall/membrane biogenesis |
| N | 4 | 0.19 | Cell motility |
| U | 27 | 1.30 | Intracellular trafficking and secretion |
| O | 76 | 3.66 | Posttranslational modification, protein turnover, chaperones |
| C | 123 | 5.92 | Energy production and conversion |
| G | 173 | 8.32 | Carbohydrate transport and metabolism |
| E | 154 | 7.41 | Amino acid transport and metabolism |
| F | 64 | 3.08 | Nucleotide transport and metabolism |
| H | 117 | 5.63 | Coenzyme transport and metabolism |
| I | 77 | 3.70 | Lipid transport and metabolism |
| P | 119 | 7.72 | Inorganic ion transport and metabolism |
| Q | 36 | 1.73 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 255 | 12.27 | General function prediction only |
| S | 164 | 7.89 | Function unknown |
| - | 1442 | 43.02 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Insights from the genome sequence
In the genome of P. glucosidilyticus DD6b two genes required for gliding motility in Flavobacteriumjonsoniae were identified: gldBDFGHJ and gldLMN [32, 33]. The presence of these genes indicates a gliding motility ability of P. glucosidilyticus DD6b, which was not reported for the P. glucosidilyticus type strain [17, 34–36].
Another specific property of P. glucosidilyticus DD6b in comparison with the P. glucosidilyticus type strain, is the presence of a complete DNRA pathway allowing the reduction of nitrate to ammonium, where the reduction of nitrate to nitrite proceeds through an assimilatory ferredoxin-nitrate reductase narB (PBAC_22000) and the reduction of nitrite to ammonia by a dissimilatory nitrite reductase nirBD (PBAC_21900; PBAC_21910). The positions of the gene clusters for DNRA pathway and phoPR are shown in Fig. 3 (circle 5, clusters 1 and 2, clockwise).
Fig. 3.
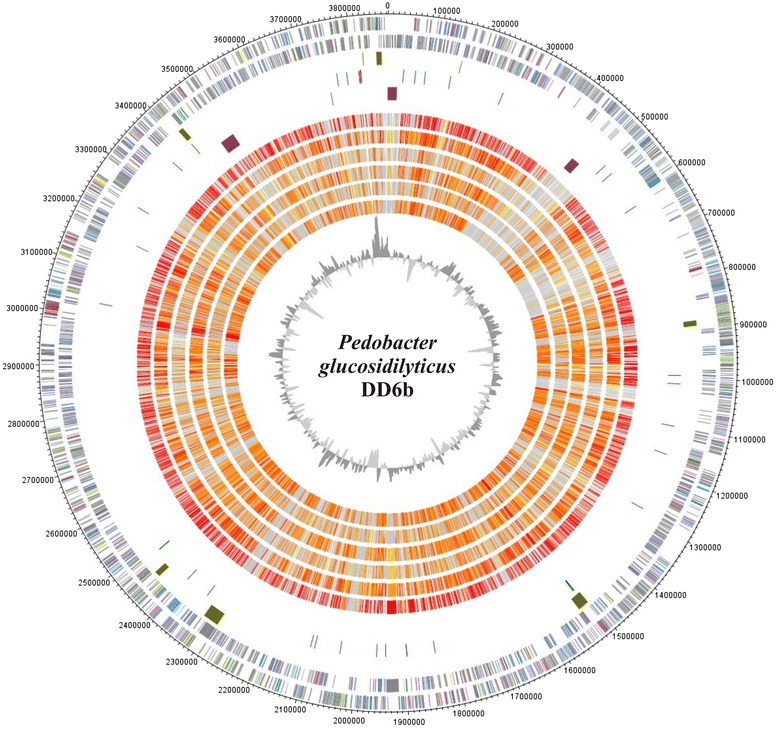
Genome comparison of P. glucosidilyticus DD6b with 6 completely genome-sequenced Pedobacter type strains: Genes encoded by the leading and the lagging strand (circle 1 and 2) of P. glucosidilyticus DD6b are marked in COG colors in the artificial chromosome map. Visualization was done with DNAPlotter [46]. Genomic islands (olive) identified with IslandViewer [47] are shown in circle 3, tRNAs (green) and rRNAs (pink) in circle 4. Special features of strain DD6b are marked in purple in circle 5 and described in the plain text. The presence of orthologs (circle 6 to 11) is indicated for the genomes of P. glucosidilyticus, DSM 23,534 (AULF00000000), P. borealis DSM 19,626 (JAUG00000000), P. heparinus HIM 762–3, DSM 2366 (CP001681.1), P. agri PB92 (AJLG00000000), P. oryzae DSM 19,973 (AUHA00000000) are illustrated in red to light yellow and singletons in grey (grey: >e−10-1; light yellow: <e−50- > e−10; gold: <e−50- > e−90; light orange: <e−90- > e−100; orange: <e−100- > e−120; red: <e−120-0). The innermost plot represents the GC-content. Paralogous genes were excluded from this analysis
Strain DD6b assimilates phosphite and most probably can assimilate phosphonoacetate under phosphate starvation to support its growth. Phosphite oxidation in strain DD6b proceeds via a sec-dependent periplasmic alkaline phosphatase (PBAC_31300), analogously to E.coli [15]. The genome sequence of strain DD6b suggests that this bacterium should be able to assimilate phosphonoacetate under phosphate starvation, as the gene encoding phosphonoacetate hydrolase phnA (PBAC_28850) catalyzing the hydrolysis of phosphonoacetate to acetate and phosphate is present. Strain DD6b does not harbor a complete 2-aminoethylphosphonate degradation pathway, despite the presence of a phosphonoacetate hydrolase encoding gene. Also, neither genes encoding carbon-phosphorus lyase complex, nor genes coding for phosphonopyruvate hydrolase or phosphonoacetaldehyde hydrolase were detected in the genome. The regulation of the phosphorus homeostasis in Gram negative bacteria is under the control of the two-component signal transduction pathway of the Pho regulon, encoded by phoPR. Also, 8 copies of the gene encoding alkaline phosphatase synthesis sensor protein PhoR were identified in the genome of P. glucosidilyticus DD6b one of which (PBAC_27380) was specific for this genome, without present orthologs in the rest of the Pedobacter type species whole genome sequences. The second component PhoP is present with two orthologous genes in the genome of DD6b.
In addition, a whole genome comparison of P. glucosidilyticus DD6b genome with the genomes of P. glucosidilyticus DSM 23,534 (AULF00000000), P. borealis DSM 19,626 (JAUG00000000), P. heparinus HIM762-3 DSM 2366 (CP001681), P. agri PB92 (AJLG00000000), P. oryzae DSM 19,973 ( AUHA00000000) was performed in order to define the size of the core and pan genomes of Pedobacter species. For this analysis Proteinortho software (blastp,[37]) with an identity cutoff of 50 %, and an E-value of 1e10 was used. The six compared species have a core genome of 1398 and a pan genome of 9962 orthologous groups. The highest number of orthologous groups (2387) was found for P. glucosidilyticus DD6b and P. glucosidilyticus DSM 23,534, whereas the lowest number (1675) was found for the pairwise comparison of P. glucosidilyticus DD6b with P. oryzae DSM 19,973. This genome comparison also revealed 225 specific orthologous groups only for both P. glucosidilyticus strains. Two of those specific orthologous CDS were a pectate lyase (PBAC_03170 and H510DRAFT_00682) and a periplasmic alkaline phosphatases, PhoD-like (PBAC_31300 and H510DRAFT_02447).
Also, P. glucosidilyticus DD6b has 447 singletons, the majority of which encode proteins of unknown function. Amongst the unique genes with assigned functions were found those encoding the synthesis of a green-light absorbing proteorhodopsin (PBAC_30230) playing a role in the generation of phototrophic energy (Fig. 3, position 3), proteins involved in the synthesis of EPS and capsule formation. Specific for the genome of P. glucosidilyticus DD6b are a rhamnogalacturonate lyase (PBAC_06010) and a second specific pectate lyase coding genes (PBAC_05900) participating in the degradation of rhamnogalacturonan I and pectin [38]. A comprehensive genome properties comparison of the 5 publicly available Pedobacter type strains genomes and the P. glucosidilyticus DD6b used in this study is presented in Additional file 3: Table S3.
Conclusions
In this work we report on the first whole genome sequence of P. glucosidilyticus, strain DD6b, its specific and common genome features as a member of the genus Pedobacter. The whole genome comparison of P. glucosidilyticus DD6b with 5 other publicly available whole genome Pedobacter type strains sequences (as on 5th of December 2014), revealed a core genome of 1398 orthologous genes or about 30 % of each genome. The number of common orthologous groups amongst all genomes varied in the range 2387 (71.2 %) for P. glucosidilyticus DD6b / P. glucosidilyticus DSM 23,534 genome couple and 1675 (49.97 %) for P. glucosidilyticus DD6b/ P. oryzae DSM 19,973 couple. This shows relatively wide genome plasticity within the genus Pedobacter.
Specific for P. glucosidilyticus DD6b genome is the presence of genes related to phytopathogenicity and pectine degradation, as well as for assimilative nitrate reduction.
Based on physiological experiments, we proved that P. glucosidilyticus DD6b assimilates phosphite as single phosphorus source, in agreement with the presence of a periplasmic alkaline phosphatase-encoding gene in the genome of the strain. Furthermore, the presence of an orthologous alkaline phosphatase gene in the genome of P. glucosidilyticus DSM 23,534 strongly suggests that the type strain might possess this ability too. Common and specific only for both P. glucosidilyticus genomes was the presence of a phosphonoacetate hydrolase (PhnA) encoding gene, suggesting phosphonoacetate utilization ability for both strains. However, their genomes do not encode a complete 2-aminoethylphosphonate degradation pathway. Finally, none of the 6 analyzed Pedobacter genomes encoded any of the rest known organophosphonate degradation pathways. Overall members of genus Pedobacter species are characterized by low diversity and distribution of inorganic and organophosphonate degradation pathways. However, in future studies the phosphite assimilation property of P. glucosidilyticus species can be regarded as specific physiological determinant within genus Pedobacter (Additional file 4).
Acknowledgements
We thank Heike M. Freese for supplying the strain, Bernhard Schink for support, Joachim Hentschel for the scanning electron micrograph of the strain, Bernd Gahr, Sylke Wiechmann, Frauke-Dorothee Meyer and Kathleen Gollnow for technical assistance. This work was supported by the University of Konstanz. D.D.S. was partially supported by a grant of the Deutsche Forschungsgemeinschaft, Bonn – Bad Godesberg, Germany (SI 1300/4-1).
Abbreviations
- 2-AEP
2-aminoethylphosphonate
Additional files
Associated MIGS record, P. glucosidilyticus DD6b (PDF 30 kb)
(DOCX 17 kb)
(PDF 64 kb)
The Annotation Summary; GenBank Accession Summary; Strain ID Summary; Plant Name Summary; Scientific Name Summary; Reference Search Summary (DOC 33 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DDS and AP designed research. DDS characterized the strain DD6b. AP and DDS carried out genome analyses. DDS, AP and RD wrote the manuscript. All authors read and approved the final manuscript.
References
- 1.Freese HM, Schink B. Composition and stability of the microbial community inside the digestive tract of the aquatic crustacean Daphnia magna. Microb Ecol. 2011;62(4):882–894. doi: 10.1007/s00248-011-9886-8. [DOI] [PubMed] [Google Scholar]
- 2.Martin-Creuzburg D, Beck B, Freese HM. Food quality of heterotrophic bacteria for Daphnia magna: evidence for a limitation by sterols. FEMS Microbiol Ecol. 2011;76(3):592–601. doi: 10.1111/j.1574-6941.2011.01076.x. [DOI] [PubMed] [Google Scholar]
- 3.Taipale S, Kankaala P, Tiirola M, Jones RI. Whole-lake dissolved inorganic C-13 additions reveal seasonal shifts in zooplankton diet. Ecology. 2008;89(2):463–74. doi: 10.1890/07-0702.1. [DOI] [PubMed] [Google Scholar]
- 4.Taipale S, Kankaala P, Hamalainen H, Jones RI. Seasonal shifts in the diet of lake zooplankton revealed by phospholipid fatty acid analysis. Freshwater Biol. 2009;54(1):90–104. doi: 10.1111/j.1365-2427.2008.02094.x. [DOI] [Google Scholar]
- 5.Vadstein O.: Heterotrophic, planktonic bacteria and cycling of phosphorus - Phosphorus requirements, competitive ability, and food web interactions. In: Shink B, editorVol 16, Adv Microb Ecol. Springer US; Kluwer Academic/Plenum Publishers, New York 2000. p 115–167.
- 6.Hessen DO, Andersen T. Bacteria as a source of phosphorus for zooplankton. Hydrobiologia. 1990;206(3):217–223. doi: 10.1007/BF00014087. [DOI] [Google Scholar]
- 7.Kokonova SV, Nesmeyanova MA. Phosphonates and their degradation by microorganisms. Biochemistry (Moscow) 2002;67(2):184–195. doi: 10.1023/A:1014409929875. [DOI] [PubMed] [Google Scholar]
- 8.White AK, Metcalf WW. Microbial metabolism of reduced phosphorus compounds. Annu Rev Microbiol. 2007;61:379–400. doi: 10.1146/annurev.micro.61.080706.093357. [DOI] [PubMed] [Google Scholar]
- 9.Metcalf WW, Wanner BL. Involvement of the Escherichia coli phn (psiD) gene cluster in assimilation of phosphorus in the form of phosphonates, phosphite, Pi esters, and Pi. J Bacteriol. 1991;173(2):587–600. doi: 10.1128/jb.173.2.587-600.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simeonova DD, Wilson MM, Metcalf WW, Schink B. Identification and heterologous expression of genes involved in anaerobic dissimilatory phosphite oxidation by Desulfotignum phosphitoxidans. J. Bacteriol. 2010;192(19):5237–5244. doi: 10.1128/JB.00541-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poehlein A, Daniel R, Schink B, Simeonova DD. Life based on phosphite: a genome-guided analysis of Desulfotignum phosphitoxidans. BMC Genomics. 2013;14(1):753. doi: 10.1186/1471-2164-14-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villarreal-Chiu J.F., Quinn J.P., McGrath J.W.: The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Frontiers in Microbiology 2012, 3. http://www.frontiersin.org/Journal/Abstract.aspx?s=53&name=aquatic_microbiology&ART_DOI=10.3389/fmicb.2012.00019 [DOI] [PMC free article] [PubMed]
- 13.Borisova SA, Christman HD, Metcalf MEM, Zulkepli NA, Zhang JK, van der Donk WA, et al. Genetic and biochemical characterization of a pathway for the degradation of 2-aminoethylphosphonate in Sinorhizobium meliloti 1021. J Biol Chem. 2011;286(25):22283–90. http://www.jbc.org/content/286/25/22283. [DOI] [PMC free article] [PubMed]
- 14.Dumora C, Lacoste A-M, Cassaigne A. Phosphonoacetaldehyde hydrolase from Pseudomonas aeruginosa: Purification properties and comparison with Bacillus cereus enzyme. Biochim. Biophys. Acta (BBA) - Protein Structure and Molecular Enzymology. 1989;997(3):193–198. doi: 10.1016/0167-4838(89)90186-6. [DOI] [PubMed] [Google Scholar]
- 15.Yang K, Metcalf WW. A new activity for an old enzyme: Escherichia coli bacterial alkaline phosphatase is a phosphite-dependent hydrogenase. Proc Nat Acad Sci. 2004;101(21):7919–7924. doi: 10.1073/pnas.0400664101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe Y, Nakajima M, Hoshino T, Jayasimhulu K, Brooks E, Kaneshiro E. A novel sphingophosphonolipid head group 1-hydroxy-2-aminoethyl phosphonate in Bdellovibrio stolpii. Lipids. 2001;36(5):513–519. doi: 10.1007/s11745-001-0751-3. [DOI] [PubMed] [Google Scholar]
- 17.Luo X, Wang Z, Dai J, Zhang L, Li J, Tang Y, et al. Pedobacter glucosidilyticus sp. nov., isolated from dry riverbed soil. Int J Syst Evol Microbiol. 2010;60(1):229–33. http://ijs.sgmjournals.org/content/60/1/229.abstract. [DOI] [PubMed]
- 18.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro HN, editor. Mammalian Protein Metabolism. edition. New York: Academic; 1969. pp. 21–132. [Google Scholar]
- 20.Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 2005;33(suppl 1):D294–D296. doi: 10.1093/nar/gki038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 22.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A new genome assembly algorithm and Its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyatt D, Chen G-L, LoCascio P, Land M, Larimer F, Hauser L. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11(1):119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):0955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Pillay M, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42(D1):D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markowitz VM, Chen I-MA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40(D1):D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zdobnov EM, Apweiler R. InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 31.Tatusov R, Fedorova N, Jackson J, Jacobs A, Kiryutin B, Koonin E, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4(1):41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braun TF, McBride MJ. Flavobacterium johnsoniae GldJ is a lipoprotein that is required for gliding motility. J Bacteriol. 2005;187(8):2628–2637. doi: 10.1128/JB.187.8.2628-2637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braun TF, Khubbar MK, Saffarini DA, McBride MJ. Flavobacterium johnsoniae gliding motility genes identified by mariner mutagenesis. J Bacteriol. 2005;187(20):6943–6952. doi: 10.1128/JB.187.20.6943-6952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakane D, Sato K, Wada H, McBride MJ, Nakayama K. Helical flow of surface protein required for bacterial gliding motility. Proc Natl Acad Sci. 2013;110(27):11145–11150. doi: 10.1073/pnas.1219753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunnicutt DW, Kempf MJ, McBride MJ. Mutations in Flavobacterium johnsoniae gldF and gldG disrupt gliding motility and interfere with membrane localization of gldA. J Bacteriol. 2002;184(9):2370–2378. doi: 10.1128/JB.184.9.2370-2378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhodes RG, Nelson SS, Pochiraju S, McBride MJ. Flavobacterium johnsoniae sprB is part of an operon spanning the additional gliding motility genes sprC, sprD, and sprF. J Bacteriol. 2011;193(3):599–610. doi: 10.1128/JB.01203-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lechner M, FindeiSZ S, Steiner L, Marz M, Stadler P, Prohaska S. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011;12(1):124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laatu M, Condemine G. Rhamnogalacturonate lyase RhiE is secreted by the out system in Erwinia chrysanthemi. J Bacteriol. 2003;185(5):1642–1649. doi: 10.1128/JB.185.5.1642-1649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. 1990;87(12):4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krieg NR, Ludwig W, Euzeby J, Whitman WB. Phylum XIV. Bacteroidetes, phyl.nov. In: Krieg NR, Ludwig W, Whitman WB, Hedlund BP, Paster BJ, Staley JT, Ward N, Brown D, editors. Bergey’s Manual of Systematic Bacteriology. Volume 4. 2. New York: Springer; 2011. p. 25. [Google Scholar]
- 41.Kämpfer P. Class III. Sphingobacteriia class. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology. Volume 4. 2. New York: Springer New York; 2011. p. 330. [Google Scholar]
- 42.Kämpfer P. Order I. Sphingobacteriales ord. nov. In: Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology. Volume 4. 2. New York: Springer New York; 2011. p. 330. [Google Scholar]
- 43.Steyn PL, Segers P, Vancanneyt M, Sandra P, Kersters K, Joubert JJ. Classification of heparinolytic bacteria into a new genus, Pedobacter, comprising four species: Pedobacter heparinus comb. nov., Pedobacter piscium comb. nov., Pedobacter africanus sp. nov. and Pedobacter saltans sp. nov. Proposal of the family Sphingobacteriaceae fam. nov. Int J Syst Bacteriol. 1998;48(1):165–177. doi: 10.1099/00207713-48-1-165. [DOI] [PubMed] [Google Scholar]
- 44.Payza AN, Korn ED. The degradation of heparin by bacterial enzymes: I. Adaptation and lyophilized cells. J. Biol. Chem. 1956;223(2):853–858. [PubMed] [Google Scholar]
- 45.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25(1):119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langille MGI, Brinkman FSL. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25(5):664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]