

Abstract
The first solid phase extraction materials for selective lysophosphatidic acid (LPA) enrichment from human plasma are described. Molecularly imprinted polymers were designed, synthesized and evaluated as cartridge fillings. They enabled a relatively rapid and simple extraction protocol for LPA without any need for multiple liquid-liquid extraction steps. The five major subspecies of lysophosphatidic acid are readily separated from all other native plasma phospholipids, including those well-known to interfere with LPA quantitation, such as phosphatidylcholine and lysophosphatidylcholine. Outstanding LPA purity is obtained via these solid phase materials in a tandem extraction setup.
Introduction
Lysophosphatidic acid (LPA, Fig. 1) is a bioactive phospholipid involved in many cellular processes such as cell proliferation, migration and platelet aggregation.1 LPA influences many physiological processes, such as brain and vascular development, and is associated with numerous diseases including ovarian cancer, breast cancer, prostate cancer, colorectal cancer, hepatocellular carcinoma, multiple myeloma atherosclerotic disease, cardiovascular disease, pulmonary inflammatory disease and renal disease.2
Fig. 1.

Structures of major LPA subspecies. Non-native LPA 17:0 is used as an internal standard.
A wide variety of LPA sample preparation methods have been developed. Liquid-liquid extraction (LLE) is the most common methodology for LPA enrichment from biological samples. The LLE method developed by Bligh-Dyer3 has been modified and used in many LPA studies. These modified methods require one- or two-step extractions and are easy and fast.4-7 However, they can also be less than ideal because abundant potential interferences, such as lysophosphotidylcholine (LPC), can be co-extracted with LPA and affect LPA quantifications. For example, Zhao et al. found that LPC and lysophosphatidylserine (LPS) artificially generate LPA signals in electrospray ionization tandem mass spectrometry (ESI-MS/MS) at the ion source via loss of the head group.8 This fact may significantly affect quantification, since the biological concentration of total LPC is about 300 μM, which is two orders of magnitude higher than that of LPA (< 5 μM) in healthy individuals.9, 10 On the other hand, phospholipids such as phosphatidylcholine (PC) and LPC, may cause matrix effects that suppress or enhance ionization in the mass spectrometer detector shift retention times and elevate baselines during chromatographic separation.11-13 In addition to liquid chromatography-mass spectrometry (LC-MS), other analytical methods may be affected by the presence of interfering phospholipids in LPA samples because of their structural similarities. For example, phosphatidic acid (PA) has the same head group as LPA; lysophosphatidylethanolamine (LPE), LPS and LPC, all have the same or similar of alkyl chain length as LPA. Thus, if the detection is based on phosphate group binding or deaggregation of a binding group induced by the alkyl chain of LPA, the phospholipids that co-extract with LPA would likely interfere. Hence, removal of interferences prior to analysis is critical for an accurate quantification of LPA.
Our group has reported a method for LPA extraction and enrichment by using LLE followed by SPE.14 With that method, LPA could be extracted with high recovery and purity. To improve the efficiency of the sample preparation we eliminated the LLE step by using synthetic molecularly imprinted polymers (MIPs) as a stationary phase for SPE. Compared to LLE, SPE has several advantages including high recoveries without partition issues, less labor and time, and better potential for automation.
MIPs are highly cross-linked polymers that can be used as artificial receptors for specific molecules.15-17 MIPs have been used in chromatography, electrophoresis, SPE, chemical sensing and catalysis, drug delivery, crystallization, artificial antibodies and cell culturing.18 Examples of current commercial MIPs include Affinilute™ (Biotage)19 and SupelMIP® (Sigma-Aldrich).20 To synthesize MIPs, the template and functional monomers are bound covalently or non-covalently before being polymerized in the presence of a high concentration of crosslinking monomer. After the removal of the template, a polymer with cavities and binding sites is formed and can be used to bind target molecules selectively. MIPs have many advantages as materials for the extraction of specific compounds. MIPs can be customized and modified simply by varying the selection and composition of the monomers. Functional monomers can be commercially available, or designed and synthesized for target compounds, which may enhance the selectivity of the MIPs. Furthermore, monomers that contain more than one functional group can bind with a higher affinity to the template molecule and with reduced nonspecific binding to other molecules.21-23 MIPs with urea moieties as receptors for the phosphate group have been designed and synthesized. The N-H groups in urea can bind to phosphate via hydrogen bonding and electrostatic interactions.24-26 MIPs for recognition of phospholipids have been prepared, but are specific for phosphatidylcholine.27 A commercially available material, HybridSPE™ (Sigma-Aldrich) has been reported to be effective for phospholipid removal, by using Zr as a Lewis acid to interact with the phosphate moiety of phospholipids.28 However, no prior SPE materials have been reported to enable selective LPA enrichment over all other classes of phospholipids.
Experimental Section
General Methods
NMR spectra were recorded on ARX-400 Advance spectrometer (Bruker Corporation). FTIR spectra were obtained on a Nicolet iS10 infrared spectrometer (Thermo Scientific, Madison, WI) in reflection geometry using a single bounce diamond attenuated total reflectance (ATR) accessory. LPA were separated on a Luna™ C-8 (50 × 2 mm, 3 μm) column connected to a guard cartridge with 2.0 to 3.0 mm internal diameters (Phenomenex, Torrance, CA) in an Accela™ UHPLC system (Thermo Scientific, Madison, WI). MS data were collected via an LTQ-Orbitrap™ XL Discovery instrument (Thermo Scientific, Madison, WI), equipped with an ESI ion max source. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification except when specifically mentioned. All phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL). Ethylene glycol dimethacrylate (EGDMA) was purified by vacuum distillation. Human plasma was collected by Lampire Biological Laboratories Inc., from female donors, processed to obtain platelet-free plasma, and frozen at −80 °C. Empty SPE tubes and frits were purchased from Sigma-Aldrich (St. Louis, MO). HPLC grade MeOH, CHCl3 and water were purchased from Honeywell International Inc.
Monomer Synthesis
Synthesis of monomer 1 was performed according to a literature procedure.29 2-Propenoic acid, 2-methyl-18-methyl-8-[2-[[[[2-[(2-methyl-1-oxo-2-propen-1-yl)oxy]ethyl]amino]carbonyl] amino] ethyl]-4,12,17-trioxo-16-oxa-3,5,8,11,13-pentaazanonadec-18-en-1-yl ester. Tris(2-aminoethyl) amine (2.2 g, 15.04 mmol) is dissolved in 110 mL CH2Cl2 and cooled to 0 °C before 2-isocyanatoethyl methacrylate (7.0 g, 45.12 mmol) is added dropwise. The mixture is stirred at rt for 4 h. The solvent is evaporated and dried under vacuum. The yield of the product was 9.2 g (100%). 1H NMR (400 MHz, CDCl3): δ 6.09 (s, 3H), 6.03 (bs, 3H), 5.80 (bs, 3H), 5.55 (s, 3H), 4.16 (bs, 6H), 3.43(bs, 6H), 3.12 (bs, 6H), 2.48 (bs, 6H), 1.90 (s, 9H). 13C NMR (400 MHz, CDCl3): δ 18.40, 38.72, 39.32, 55.36, 64.29, 126.06, 136.17, 159.45, 167.52.
Polymer Preparation
Octadecylphosphonic acid, OPA (1.00 g, 2.99 mmol), monomer 1 (1.83 g, 2.99 mmol) and methacrylic acid, MAA (0.257 g, 2.99 mmol) are dissolved in 37 mL CHCl3 and allowed to stand for 60 min for complete complex formation. EGDMA (11.9 g, 59.8 mmol) is added to the mixture in one portion. The mixture is purged by bubbling N2 for 5 min before and after azobisisobutyronitrile, AIBN (0.245 g, 1.49 mmol) is added. Polymerization is carried out at 55 °C for 16 h. Polymers are crushed into small pieces before being washed with MeOH and extracted in a Soxhlet apparatus with MeOH for 48 h before ground and sieved to 63-90 μm.
1H NMR Titrations
Stock solutions of monomer 1 (40 mM) and OPA (20 mM) were prepared in CDCl3. The initial concentration of OPA (template) was 5 mM and kept constant. The monomer concentrations were 0, 0.5, 1, 1.5, 2, 3, 4, 5, 7.5, 10, 15 mM, respectively. The proton chemical shifts in the urea and adjacent methylene groups were monitored. Curve fitting was performed with the Origin™ 8.0 program (OriginLab Corporation). Table S2 (ESI†) shows relevant titration data for the template-monomer complexation. Association constants are expressed as (M−1). The association constant (Ka) was calculated by non-linear regression using equation 1.
| (1) |
where δobs is the observed complexation induced chemical shift, S11 is the total chemical shift, Ka is the association constant and [L] is the ligand concentration.
LPA Enrichment Procedure
Six hundred μL of human plasma are mixed with 2 mL MeOH-CHCl3 2:1, vortexed at 2000 rpm for 30 s and incubated at 4 °C for 20 min. After warming to rt, the mixture is centrifuged at 2000 rpm for 10 min. The supernatant is decanted and loaded onto a cartridge packed with non-imprinted polymer (NIP). The cartridge is eluted with 2 mL 0.05% NH4OH in MeOH. The eluent is acidified to pH 3.0 with concentrated HCOOH and loaded into a cartridge packed with 30 mg MIP. The cartridge is washed with 2 mL CHCl3, followed by 2 mL MeOH. LPAs are eluted with 3 mL 0.05% NH4OH in MeOH. The solvent is evaporated under a N2 stream and the residue reconstituted in 0.2 mL MeOH-H2O 9:1.
LC-ESI/MS Procedure
Samples (10 μL) after SPE enrichment are injected into a Luna™ C-8 (50 × 2 mm, 3 μm) column at 40 °C. The mobile phase consisted in 9:1 MeOH:H2O (pH is adjusted to 2.5 by adding HCOOH) is delivered at a flow rate of 0.6 mL/min. Ions are created in negative ion mode by setting the sprayer voltage to 3.0 kV and the capillary temperature to 300 °C.
Polymer Swelling
Dry polymer is placed in a 10 mL graduated cylinder and weighed. The weight and volume of the dry polymer are used to calculate the density of the polymer. Excess CHCl3 is added and air bubbles removed by stirring. The polymer is allowed to swell for 24 h. The swelling factor is calculated as the ratio of the volume of the swollen polymer to the dry polymer.
Recovery Determination
Separation and quantification of recovered LPA and PA from enrichment procedures are achieved using LC-ESI/MS. Calibration curves and relevant statistical data is shown in Table S4, ESI†. Recoveries (%) of LPA and PA in plasma are calculated using equation 2.
| (2) |
where XR is the concentration of the analyte (LPA or PA) in the spiked sample, X is the concentration of the unspiked sample and XS is the known spike added concentration. For the polymer screenings, recovery was calculated using equation 2 with X = 0.
Results and Discussion
We synthesized MIPs that could serve as the stationary phase in an SPE cartridge. Functional monomers and crosslinking monomers were selected from a virtual library of twenty-two functional monomers containing functionalities for phosphate binding and seven cross-linking monomers (Figs. S2 and S3, ESI†). The Leapfrog™ algorithm (Certara, L. P.) was used to determine the binding of the functional monomer candidates and target analyte LPA 18:0. Functional monomers with the highest binding score and crosslinking monomers with the lowest binding score were selected as the best candidates for polymer preparation (Fig. 3). Multifunctional monomer 1 containing three urea groups, was selected for the purpose of this study. Fig. 2 shows an energy-minimized model of a 1:1 complex of a tris-urea scaffold and LPA using the SYBYL-X™ 2.0 Suite (Certara, L. P.). We proposed that because of the expected selectivity of this MIP, interfering phospholipids would be removed from LPA-containing biological samples.
Fig. 3.
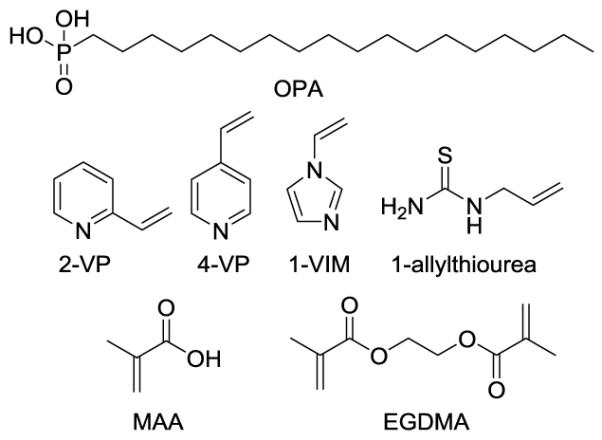
Structures of guest (OPA), functional monomers chosen (2-VP, 4-VP, 1-VIM, 1-allylthiourea and MAA) and crosslinking monomer (EGDMA).
Fig. 2.

Energy-minimized structure of the complex of a tris-urea scaffold and LPA 18:1. Atom colors: carbon=gray, hydrogen=white, oxygen=red, nitrogen=blue, phosphorus=cyan. Only polar hydrogens are shown for clarity.
Monomer 1 is synthesized by a procedure reported by Wu (Scheme 1).29 In order to study the binding properties of 1, OPA was used as the anionic guest.
Scheme 1.

Synthesis of monomer 1.
OPA was also selected as the template for imprinting instead of LPA to avoid any potential interference from residual LPA after template extraction. The binding affinity of the urea functional group in monomer 1 to the phosphate group in OPA (guest) was monitored using 1H NMR in CDCl3.
Based on the results from the Job plot (Table S1, Figs. S1 and S4, ESI†), the maximum variation in chemical shift was obtained when the mole ratio of monomer and guest was 1:1 (Fig. S1, ESI†). Downfield chemical shifts of the urea N-H and adjacent methylene protons were observed and monitored.30 A 1:1 binding model was used to determine the association constants (Ka). Results are shown in Table S2 (ESI†) and Fig. 4 with Ka calculated to be 83.2 M−1. In NMR titration experiments using MAA as the host and OPA as the guest, no chemical shift was observed for the proton in N-H group or the adjacent methylene groups.
Fig. 4.

1H NMR titration of monomer 1 using OPA as the guest.
In addition to 1, other functional monomers including 2-vinylpyridine (2-VP), 4-vinylpyridine (4-VP), 1-vinylimidazole (1-VIM) and 1-allylthiourea were also evaluated for OPA binding. However, OPA did not dissolve completely in CHCl3 under these conditions.
A series of MIP formulations (Table 1) were prepared by bulk polymerization. Polymers were crushed and subjected to Soxhlet extraction to remove the template from the imprinted polymer. After extraction, polymers were ground to a 63-90 μm particle size. SEM microphotographs of the ground material show these polymer particles are of irregular shape (Fig. S6, ESI†). LC-ESI/MS analysis of the extract shows that the OPA template was recovered in 95% yield in each formulation.
Table 1.
Formulations of MIP and non-imprinted polymer (NIP).a
| Polymer | Template | Functional monomers |
Recovery (%) | ||
|---|---|---|---|---|---|
| LPA | PA | ||||
| NIP-1 | — | 1 b | — | 39% | — |
| MIP-1 | OPAb | 1 b | — | 70% | 72% |
| NIP-2 | — | 1 b | MAAb | 50% | 10% |
| MIP-2 | OPAb | 1 b | MAAb | 92% | 32% |
| NIP-3 | — | 1 b | 4-VPb | 72% | 40% |
| MIP-3 | OPAb | 1 b | 4-VPb | 95% | 55% |
EGDMA (16 M), AIBN (0.4 M) and CHCl3 are used as crosslinking monomer, initiator and solvent, respectively, in all formulations.
Concentration: 0.81 M. For more details see the experimental section above.
LPA control solutions (5 μM) were prepared in MeOH and used to evaluate the performance of the MIPs for SPE. The first formulation studied (Table 1, MIP-1) included only 1 as the functional monomer. Screening results for MIP-1 showed that similar levels of PA and LPA coeluted with LPA from the MIP. In order to improve selectivity for LPA over PA, MAA and 4-VP were used in the formulation (MIP-2 and MIP-3) to add hydrogen bonding interactions to favor the binding of the relatively more polar LPA.
In the presence of MAA (MIP-2), the recovery of the relatively less polar PA was lowered by 40 %. The average recovery of LPAs was also increased to 92%. This is attributed to enhanced H-bonding to the LPA hydroxyl from the MAA carboxyl group. The formulation containing 4-VP as the second monomer (MIP-3) showed a similar LPA recovery to MIP-2, but with less selectivity. Thus, MIP-2 was chosen for further optimization. Optimization of the SPE protocol was carried out with control samples of LPA mixtures (Fig. S7, ESI†). In 1:1 CHCl3-MeOH, 75 % of the LPA loaded to MIP-1 remained bound. This value was increased to 95 % by adding HCOOH to protonate the LPA. To elute the bound LPA, either MeOH or CHCl3 as solvent recovered only 60-70% of the LPA from MIP-2. Addition of 0.05% NH4OH in MeOH increased the recovery to 90 %.
In order to investigate the retention and recovery of other phospholipids, a solution of LPA and PA, along with neutral phospholipids, was prepared. After loading with 1:1 CHCl3:MeOH and HCOOH, a CHCl3 elution followed by MeOH was found to remove neutral phospholipids, such as LPC and PC, with > 99 % recovery.
During the course of these studies, control non-imprinted polymers NIPs 1-3 were investigated to evaluate the effectiveness of the imprinting method (Table 1). Interestingly, NIP-2 showed stronger binding to PA than MIP-2. We thus reasoned that optimal LPA recovery and selectivity would be obtained via an SPE method involving sample treatment with NIP-2 (to remove PAs) and MIP-2 (to remove other phospholipids) in tandem.
Two LPA mixtures (0.5 μM and 2.5 μM) were evaluated (three runs each) using the sequential extraction method involving NIP-2 and MIP-2. Good recoveries of LPA were exhibited as determined by LC-ESI/MS using LPA control samples (Table 2) and hexadecylphosphonic acid (5 μM) as the internal standard.
Table 2.
Recoveries of individual LPA species after SPE. (n = 3)
| LPA species | Recovery (%) | σ |
|---|---|---|
| 14:0 | 87.1 | 1.5 |
| 20:4 | 86.2 | 1.6 |
| 16:0 | 89.5 | 1.4 |
| 18:1 | 87.6 | 1.5 |
| 17:0 | 85.1 | 1.6 |
| 18:0 | 87.3 | 1.3 |
Commercial human plasma samples, non-spiked and spiked with 1 μM of each LPA subspecies, were used to evaluate the SPE enrichment method and determine potential matrix interferences (Table 3). Recoveries of the LPA subspecies were excellent, ranging from 94.6 % to 105.2 %. Representative LC-ESI/MS traces of a non-spiked plasma sample and a LPA control sample containing all the subspecies at the same ratio (10 μM) are shown in Fig. 5. As expected, ratios of LPAs in the plasma sample are different and dependent on the plasma source.
Table 3.
LPA analysis in human plasma using LC-ESI/MS. (n=3)
| LPA species |
Non-spiked | Spikeda | |||
|---|---|---|---|---|---|
|
|
|||||
| average (μM) |
σ | average (μM) |
σ | recovery (%) |
|
| 14:0 | 0.25 | 0.02 | 1.19 | 0.03 | 94.6 |
| 20:4 | 0.23 | 0.02 | 1.25 | 0.06 | 101.8 |
| 16:0 | 0.37 | 0.02 | 1.42 | 0.05 | 105.2 |
| 18:1 | 0.26 | 0.03 | 1.29 | 0.04 | 103.3 |
| 18:0 | 0.34 | 0.01 | 1.36 | 0.05 | 101.7 |
| Total | 1.45 | 0.05 | 6.51 | 0.10 | 101.3 |
Samples are spiked with 1 μM of the respective LPA subspecies.
Fig. 5.

LC-ESI/MS traces. Left: 10 μM standard mixtures of LPAs. Right: plasma sample. Column: Luna™ C-8 (50 × 2 mm, 3 μm) at 40 °C. Injection volume: 10 μL. Mobile phase: 9:1 MeOH–HCOOH (pH 2.5). Flow rate: 0.6 mL/min. Ions are detected in negative ion mode. Sprayer voltage: 3.0 kV and capillary temperature: 300 °C.
To identify non-LPA phospholipids in plasma, an LC-ESI/MS full scan in negative and positive modes was used. A mixture of twenty-two phospholipids was employed as the control sample and each was confirmed to be detectable in either negative or positive mode. The plasma sample was tested after using the new tandem extraction exactly as described above. Signals were compared to the LIPID MAPS Structure Database (LMSD).31 No interfering phospholipids, except for trace LPC, could be detected. Using LPC 15:0 as the internal standard, concentrations of LPC 16:0, LPC 18:1 and LPC 18:0 were estimated to be 0.05 μM, 0.01 μM and 0.02 μM, respectively. The levels represent less than 0.1 % of the total LPC content in human plasma.9 This result shows that the solid phase extraction results in clean samples with minimum interferences from other relevant phospholipids.
Conclusions
In conclusion, the polymers prepared using 1 and MAA as functional monomers showed significant advantages in selectivity for LPA extraction and enrichment. Their use as SPE cartridge stationary phases allows the relatively fast and simple extraction of LPA from human plasma at high recoveries and purities. Compared to the extraction method we developed earlier,14 the current method is faster and less labor intensive, as most of the vortexing and centrifugation steps required in LLE are eliminated. The solid phase method should also be easier to automate and use in clinical settings.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01CA136491).
Footnotes
Electronic Supplementary Information (ESI) available: Additional tables and characterization data. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Stähle M, Veit C, Bachfischer U, Schierling K, Skripczynski B, Hall A, Gierschik P, Giehl K. J. Cell Sci. 2003;116:3835–3846. doi: 10.1242/jcs.00679. [DOI] [PubMed] [Google Scholar]
- 2.Jesionowska A, Cecerska E, Dolegowska B. Anal. Biochem. 2014;453:38–43. doi: 10.1016/j.ab.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 3.Bligh EG, Dyer WJ. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 4.Baker DL, Desiderio DM, Miller DD, Tolley B, Tigyi GJ. Anal. Biochem. 2001;292:287–295. doi: 10.1006/abio.2001.5063. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y-L, Xu Y. J. Chromatogr. B Biomed. Appl. 2001;753:355–363. doi: 10.1016/s0378-4347(00)00582-x. [DOI] [PubMed] [Google Scholar]
- 6.Yoon H-R, Kim H, Cho S-H. J. Chromatogr. B. 2003;788:85–92. doi: 10.1016/s1570-0232(02)01031-0. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Z, Xu Y. J. Lipid Res. 2010;51:652–659. doi: 10.1194/jlr.D001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao Z, Xu Y. J. Chromatogr. B. 2009;877:3739–3742. doi: 10.1016/j.jchromb.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takatera A, Takeuchi A, Saiki K, Morisawa T, Yokoyama N, Matsuo M. J. Chromatogr. B. 2006;838:31–36. doi: 10.1016/j.jchromb.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Bese T, Barbaros M, Baykara E, Guralp O, Cengiz S, Demirkiran F, Sanioglu C, Arvas M. J. Gynecol. Oncol. 2010;21:248–254. doi: 10.3802/jgo.2010.21.4.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin C, Zhang ZP, Karnes HT. J. Pharm. Biomed. Anal. 2004;35:1149–1167. doi: 10.1016/j.jpba.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Fu I, Woolf EJ, Matuszewski BK. J. Pharm. Biomed. Anal. 1998;18:347–357. doi: 10.1016/s0731-7085(98)00048-x. [DOI] [PubMed] [Google Scholar]
- 13.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Anal. Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Sibrian-Vazquez M, Escobedo JO, Lowry M, Wang L, Chu Y-H, Moore RG, Strongin RM. Analyst. 2013;138:6852–6859. doi: 10.1039/c3an01168b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wulff G. Angew. Chem., Int. Ed. 1995;34:1812–1832. [Google Scholar]
- 16.Wulff G, Gross T, Schönfeld R. Angew. Chem., Int. Ed. 1997;36:1962–1964. [Google Scholar]
- 17.Zimmerman SC, Lemcoff NG. Chem. Commun. 2004:5–14. doi: 10.1039/b304720b. [DOI] [PubMed] [Google Scholar]
- 18.Schirhagl R. Anal. Chem. 2013;86:250–261. doi: 10.1021/ac401251j. [DOI] [PubMed] [Google Scholar]
- 19. [Accessed May 30, 2015];MIPs - Molecularly Imprinted Polymers. http://www.biotage.com/product-page/mips---molecularly-imprinted-polymers.
- 20. [Accessed June 4, 2015];SupelMIP Molecularly Imprinted Polymer SPE Cartridges. http://www.sigmaaldrich.com/analytical-chromatography/sample-preparation/spe/supelmip.html.
- 21.Schirhagl R, Podlipna D, Lieberzeit PA, Dickert FL. Chem. Commun. 2010;46:3128–3130. doi: 10.1039/c000936a. [DOI] [PubMed] [Google Scholar]
- 22.Lee J-D, Greene NT, Rushton GT, Shimizu KD, Hong J-I. Org. Lett. 2005;7:963–966. doi: 10.1021/ol047618o. [DOI] [PubMed] [Google Scholar]
- 23.Tanabe K, Takeuchi T, Matsui J, Ikebukuro K, Yano K, Karube I. J. Chem. Soc., Chem. Commun. 1995:2303–2304. [Google Scholar]
- 24.Gale PA. Coord. Chem. Rev. 2003;240:191–221. [Google Scholar]
- 25.Brooks SJ, Edwards PR, Gale PA, Light ME. New J. Chem. 2006;30:65–70. [Google Scholar]
- 26.Nishizawa S, Kato Y, Teramae N. J. Am. Chem. Soc. 1999;121:9463–9464. [Google Scholar]
- 27.Pegoraro C, Silvestri D, Ciardelli G, Cristallini C, Barbani N. Biosens. Bioelectron. 2008;24:748–755. doi: 10.1016/j.bios.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 28.Ahmad S, Kalra H, Gupta A, Raut B, Hussain A, Rahman MA. J. Pharm. Bioallied Sci. 2012;4:267–275. doi: 10.4103/0975-7406.103234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu X, Goswami K, Shimizu KD. J. Mol. Recognit. 2008;21:410–418. doi: 10.1002/jmr.912. [DOI] [PubMed] [Google Scholar]
- 30.Schmidtchen FP, Berger M. Chem. Rev. 1997;97:1609–1646. doi: 10.1021/cr9603845. [DOI] [PubMed] [Google Scholar]
- 31.Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, Merrill AH, Jr., Murphy RC, Raetz CR, Russell DW, Subramaniam S. Nucleic Acids Res. 2007;35:D527–532. doi: 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.