

A single cohesin is believed to be sufficient to tether sister chromatids. Three examples of interallelic complementation are identified and characterized in which two different mutant cohesin complexes interact to promote cohesin functions. The results support a mechanism by which multiple cohesin complexes interact on DNA to mediate cohesion and condensation.
Abstract
The cohesin complex (Mcd1p, Smc1p, Smc3p, and Scc3p) has multiple roles in chromosome architecture, such as promoting sister chromatid cohesion, chromosome condensation, DNA repair, and transcriptional regulation. The prevailing embrace model for sister chromatid cohesion posits that a single cohesin complex entraps both sister chromatids. We report interallelic complementation between pairs of nonfunctional mcd1 alleles (mcd1-1 and mcd1-Q266) or smc3 alleles (smc3-42 and smc3-K113R). Cells bearing individual mcd1 or smc3 mutant alleles are inviable and defective for both sister chromatid cohesion and condensation. However, cells coexpressing two defective mcd1 or two defective smc3 alleles are viable and have cohesion and condensation. Because cohesin contains only a single copy of Smc3p or Mcd1p, these examples of interallelic complementation must result from interplay or communication between the two defective cohesin complexes, each harboring one of the mutant allele products. Neither mcd1-1p nor smc3-42p is bound to chromosomes when expressed individually at its restrictive temperature. However, their chromosome binding is restored when they are coexpressed with their chromosome-bound interallelic complementing partner. Our results support a mechanism by which multiple cohesin complexes interact on DNA to mediate cohesion and condensation.
INTRODUCTION
The protein complex cohesin has long been appreciated for its essential role in mediating chromosome architecture (reviewed in Onn et al., 2008). Cohesin is composed of four core subunits: structural maintenance of chromosomes (SMC) Smc1p, Smc3p, Mcd1p/Rad21p/Scc1p, and Scc3p/Irr1p/SAp (Losada et al., 1998). This large protein complex physically links sister chromatids to generate cohesion (Guacci et al., 1997; Michaelis et al., 1997). This cohesion enables each chromosome to achieve bipolar attachment to the spindle, which is critical for promoting high-fidelity segregation of sister chromatids at anaphase. Additional studies revealed that this same complex also plays a role in generating chromosome condensation, regulation of gene expression, and DNA repair (Guacci et al., 1997; Donze et al., 1999; Rollins et al., 1999; Ström et al., 2004; Unal et al., 2004; Yeh et al., 2008; Stephens et al., 2013). These different functions are believed be mediated by cohesin's ability to tether DNA, but using distinct target sites. Cohesion and DNA repair entail tethering two different DNA molecules (intermolecular), whereas condensation and gene regulation tether two different regions of a single DNA molecule (intramolecular) (Guacci et al., 1997; Hartman et al., 2000; Rollins et al., 2004; Ström et al., 2004). Despite cohesin's importance in chromosome architecture, the mechanism by which it generates intramolecular and intermolecular tethers remains an enigma.
The simplest model for chromosome tethering by cohesin is the “embrace” model, which is based on the ring-like or rod-like structure of cohesin that can be visualized in electron micrographs, as well as on a number of biochemical and genetic analyses (Melby et al., 1998; Gruber et al., 2003; Huis In ‘t Veld et al., 2014). In this model, a single cohesin ring entraps two sister chromatids, thereby tethering them together. For example, during DNA replication, the entrapment of sister chromatids by a cohesin “ring” generates sister chromatid cohesion (Uhlmann and Nasmyth, 1998; Gruber et al., 2003). Alternative models posit that interactions between multiple cohesin complexes (cohesin–cohesin interactions) are required for cohesion (Guacci et al., 1997; reviewed in Onn et al., 2008; Zhang et al., 2008b). For example, individual cohesin molecules bind a single sister chromatid, and, in turn, dimerization of two DNA-bound cohesins tethers two sister chromatids. It has been of great interest to distinguish between these models and elucidate cohesin's basic mode of action. One way to distinguish between these two models would be to visualize the structure of cohesin as it tethers DNA using high-resolution electron microscopy. Unfortunately, structural approaches like this have not yet been successful.
A cohesin-complex molecule contains a single copy of each of its four subunits (Losada et al., 1998; Tóth et al., 1999; Sumara et al., 2000; Ding et al., 2006; Holzmann et al., 2011). This simple stoichiometry should enable a straightforward biochemical assay to assess whether there are stable interactions between two or more cohesins. This assay entails coexpressing in cells differentially tagged copies of the same cohesin subunit, such as Smc1-3HA and Smc1-MYC, then assessing whether the differentially tagged subunits coimmunoprecipitate. Indeed, such experiments have been conducted in budding yeast and higher eukaryotes (Haering et al., 2002; Gruber et al., 2003; Hauf et al., 2005); one group detected an interaction, but the amounts were low and no biological relevance was shown (Zhang et al., 2008b). One caveat to these studies is that they analyzed solubilized cohesin from cell lysates, whereas putative cohesin–cohesin interactions could be restricted to DNA-bound cohesin. Although it is possible to solubilize chromatin by shearing, coimmunoprecipitation of differentially marked cohesins in the presence of DNA is difficult to interpret. Coimmunoprecipitation could result from a direct interaction between cohesin subunits or an indirect interaction from cohesin subunits bound independently to the same strand of DNA. The lack of robust biochemical or electron microscopic data for dimers or oligomers has led many to favor the idea that cohesin complexes function independently of one another.
A classic genetic signature of protein–protein interaction is often interallelic complementation, such as described for glutamate dehydrogenase, β-galactosidase, ATP-PRTase/His1, or assembly of the Rad52 heptameric ring (Perrin, 1963; Coddington and Fincham, 1965; Korch and Snow, 1973; Reha-Krantz, 1990; Boundy-Mills and Livingston, 1993). In these examples, the products of two mutant alleles in the same gene are defective for function when expressed individually in cells. However, when they are coexpressed in the same cell, they physically interact. The physical interaction restores function. Here we describe the existence of interallelic complementation between either two mutant mcd1 alleles or two mutant smc3 alleles. On their own, each mutant mcd1 or smc3 allele is inviable and defective for both sister chromatid cohesion and condensation. However, yeast cells coexpressing two defective mcd1 alleles or two defective smc3 alleles show robust restoration of viability, cohesion, and condensation. Furthermore, in both cases, the mutant protein of one allele changes the physical properties of the mutant protein from the second allele. These results are consistent with a mechanism by which two or more cohesin complexes directly interact to perform their functions.
RESULTS
Identification of interallelic complementation pairs in recessive cohesin alleles
The mcd1-Q266 allele of the cohesin's regulatory subunit Mcd1p contains an in-frame, 5–amino acid insertion immediately following residue Q266 (Eng et al., 2014). This allele does not support viability when present as the sole copy of this gene in yeast (Figure 1A; Eng et al., 2014). To study the effect of this lethal allele on cohesion and condensation, we previously introduced this allele into a strain harboring the MCD1-AID allele. Proteins fused to auxin-induced degron (AID) undergo ubiquitin-dependent degradation in the presence of the plant hormone auxin (Gray et al., 2001; Nishimura et al., 2009). When mcd1-Q266 MCD1-AID cells were treated with auxin, Mcd1-AIDp was degraded, revealing the phenotype of cells harboring only mcd1-Q266p. These cells were inviable as expected and defective in the maintenance of cohesion and the establishment of condensation (Eng et al., 2014).
FIGURE 1:

Analysis of single- and double-mutant alleles of mcd1 and smc3. (A) All four haploid yeast strains contain an MCD1 shuffle plasmid pVG201 {MCD1 URA3 CEN}. Two strains were deleted for the genomic MCD1 and contain a second centromere plasmid bearing MCD1 (MCD1; yTE474) or mcd1-Q266 (mcd1-Q266; yTE478). Two strains have the mcd1-1 allele at the endogenous locus: the parent strain alone (mcd1-1; yTE43) or containing a CEN TRP1 plasmid bearing mcd1-Q266 (mcd1-1 mcd1-Q266; yTE491). Liquid cultures of these four strains were grown to saturation overnight at 23°C and then plated in 10-fold serial dilutions on YPD or 5-FOA and incubated at 37°C for 3 d. Left, relevant MCD1 genotypes. MCD1 shuffle plasmid presence (+) or absence (–) is shown above YPD or 5-FOA plates, respectively. Failure to grow on 5-FOA indicates that the MCD1 shuffle plasmid (pVG201) must be retained. The mutant domains in mcd1-Q266p and mcd1-1p cohesin complexes are highlighted in red and marked with a snowflake. (B) All four haploid yeast strains contain SMC3 shuffle plasmid pEU42 {SMC3 URA3 CEN}. Two strains were deleted for the genomic SMC3 and contain a second SMC3 allele at LEU2 bearing either SMC3 (SMC3; yVG3486-00) or smc3-K113R (smc3-K113R; yVG3486-K113R). Two strains have the smc3-42 allele at the endogenous locus: the parent strain alone (smc3-42; yTE576) or containing smc3-K113R integrated at LEU2 (smc3-42 smc3-K113R; yTE578). Strains were grown and plated as described in A, except that plate incubation was at 34°C. Left, relevant SMC3 genotypes. SMC3 shuffle plasmid presence (+) or absence (–) is shown above YPD or 5-FOA plates, respectively. Failure to grow on 5-FOA indicates that pEU42 (wild-type SMC3) must be retained. The mutant domains in smc3-K113Rp and smc3-42p cohesin complexes are highlighted in red and marked with a snowflake. (C) Three haploid yeast strains bear the SMC3-AID608 at the genomic locus. The parent strain alone (SMC3-AID608; yVG3651-3D) or containing a second SMC3 allele integrated at the URA3 locus, either wild-type SMC3 (SMC3 SMC3-AID608; yMB81-1A) or smc3-K113R (smc3-K113R SMC3-AID608; yTE440), were grown at 23°C as described in A. Cells were plated in 10-fold serial dilutions on YPD alone (YPD) or YPD containing 500 μM auxin (YPD AUX) and then incubated for 2 d at 23°C. Left, relevant SMC3 genotypes. The AID degron near the Smc3 hinge and smc3-K113R cohesin complexes is highlighted in red and marked with a snowflake.
An alternative and common strategy to study lethal cohesin subunit alleles like mcd1-Q266 has been to characterize them in strains bearing conditional temperature-sensitive alleles. Indeed for MCD1, we and others have used mcd1-1, a well-characterized temperature-sensitive allele of MCD1, which contains a serine-to-asparagine point mutation at residue 525 (termed mcd1-1; Guacci et al., 1997). Few if any cells in a mcd1-1 culture are viable at the nonpermissive temperature (Figure 1A; Guacci et al. 1997). To our surprise, the mcd1-1 mcd1-Q266 double mutant, containing mcd1-1 at the endogenous locus and mcd1-Q266 on a centromere plasmid, is viable at the nonpermissive temperature (Figure 1A). This full restoration of viability argues either for robust interallelic complementation between the mcd1 alleles or conversion of one of the two alleles to wild-type MCD1. We used PCR sequencing to confirm that the double mutant still contained both the mcd1-1 and mcd1-Q266 alleles in distinct copies. We also observed robust growth at the nonpermissive temperature in mcd1-1 mcd1-Q266 strains generated by integrating mcd1-Q266 at the URA3 locus of an mcd1-1 haploid and confirmed the presence of the insertion in MCD1 by PCR and diagnostic digest (Supplemental Figure S1, A and B). Importantly, when we grew double-mutant cells on 5-fluoroorotic acid (5-FOA) to select for cells that had lost mcd1-Q266 integrated at the URA3 locus, the 5-FOA–resistant cells became temperature sensitive (Supplemental Figure S1C). These results confirm that the double mutants still had the mcd1-1 allele. We also demonstrated allele specificity for complementation, as the lethal mcd1-V137K or mcd1-R135 alleles (Chan et al., 2013; Eng et al., 2014) failed to grow in the mcd1-1 background at the nonpermissive temperature (Supplemental Figure S2A). Thus this restoration of viability is observed only when cells contain both the mcd1-1 and mcd1-Q266 alleles in the trans configuration and is due to interallelic complementation.
We discovered two additional examples of interallelic complementation involving a different cohesin subunit, SMC3, through our study of the lethal smc3-K113R allele. This allele alters a key residue in Smc3p whose acetylation is required for the establishment of cohesion (Rolef Ben-Shahar et al., 2008; Unal et al., 2008; Zhang et al., 2008a). smc3-K113R is inviable as sole source from 23 through 37°C (Supplemental Figure S2B; Unal et al., 2008). Initial studies of the smc3-K113R allele involved placing it in a temperature-sensitive smc3-42 background and assessing its function at 37°C. We repeated this analysis at lower nonpermissive temperatures for smc3-42 than previously examined. Surprisingly, we found that the smc3-K113R smc3-42 double mutant shows robust viability at 34°C (Figure 1B and Supplemental Figure S2C), whereas the smc3-K113R alone or the smc3-42 allele alone cannot support viability as sole source at 34°C (Figure 1B). We sequenced the smc3-K113R smc3-42 double mutant and confirmed that both alleles were still present, ruling out gene conversion of either allele to wild-type SMC3. Counterselection against smc3-K113R by FOA selection confirmed linkage between smc3-K113R and suppression of the temperature sensitivity of smc3-42 cells (Supplemental Figure S1D). The complementation between smc3-K113R and smc3-42 is the second example of interallelic complementation, indicating that this phenomenon is a characteristic of the cohesin complex rather than a special feature of the MCD1 cohesin subunit.
The interallelic complementation in smc3-K113R smc3-42 strains prevented our analysis of smc3-K113Rp alone at lower temperatures. To circumvent this technical difficulty, we used a different conditional SMC3 allele (SMC3-AID608), which encodes an Smc3 protein with an internal auxin degron cassette immediately following residue N607 (Guacci et al., 2015). Strains expressing only the Smc3-AID608p are inviable in the presence of auxin at 23, 30, and 34°C (Figure 1C and Supplemental Figure S2; Guacci et al., 2015). Therefore, to assess smc3-K113R function at lower temperatures, we integrated it into the URA3 locus in an SMC3-AID608 strain. The smc3-K113R SMC3-AID608 strain is unable to suppress the auxin-induced lethality at 30 or 34°C (Supplemental Figure S3; unpublished data). However, in the presence of auxin at 23°C, the smc3-K113R SMC3-AID608 strain grew almost as well as the wild-type SMC3 strain (Figure 1C). We confirmed that both alleles were still present in the double mutant using PCR sequencing. As a control, we excised the smc3-K113R from the URA3 locus in the double-mutant strain by treatment with 5-FOA (Supplemental Figure S1E). These cells lacking smc3-K113R became sensitive to auxin, confirming that suppression of auxin sensitivity requires the smc3-K113R allele. Thus smc3-K113R complements both the auxin sensitivity of SMC3-AID608 at 23°C and the temperature sensitivity of smc3-42 at higher temperatures. The suppression of SMC3-AID608 and smc3-42 is allele specific, as the double acetyl-null allele smc3-K112R, K113R (K112/3-RR) fails to complement the smc3-42 temperature sensitivity or SMC3-AID608 on auxin plates (Supplemental Figures S2C and 3B, respectively).
These results demonstrate that three distinct allele pairs of cohesin-complex subunits exhibit interallelic complementation. Of note, the three pairs of complementing alleles occur in multiple regions of the cohesin complex (Figure 1, diagram). The amino acid changes encoded by mcd1-1, mcd1-Q266, smc3-42, and smc3-K113R alleles all cluster near cohesin's head domain, although mcd1-Q266 is located in the linker between the two heads (Figure 1, diagram). However, the smc3-K113R and SMC3-AID608 complementation involves one domain distal to the head, as the AID is in Smc3p's coiled coil proximal to the hinge domain (Figure 1, diagram). Thus interallelic complementation can occur between different domains of cohesin.
Interallelic complementation likely occurs by providing distinct activities of cohesin
On the basis of our observations of three diverse sets of interallelic complementation involving two cohesin subunits, we envision two mechanisms for complementation. One possibility is that the product of each mutant allele provides an activity that the other is missing. Alternatively, each mutant allele could have allowed assembly of a complex that was functional but with activity below a level required for viability. When both alleles are present, each contributes the same (reduced) activity, but the sum total of both would meet the threshold required for viability.
If any of the alleles capable of interallelic complementation simply provided cohesin activity below some critical level, then overexpression of the allele might restore cohesin function and viability. To test this possibility, we replaced the endogenous promoter of each mutant allele with the inducible GAL1 promoter. We then tested the ability of these mutant alleles when overexpressed to support viability as a sole source using the plasmid shuffle strategy. As expected, overexpression of wild-type MCD1 or SMC3 using the GAL1 promoter is viable (Figure 2, A and B, top row). In contrast, pGAL1-mediated overexpression of mcd1-1, smc3-K113R, or smc3-42 is unable to support viability in the absence of wild type at 34 or 37°C, respectively (Figure 2, A and B, second and third rows). We used Western blots to confirm that mcd1-1 protein levels were dramatically increased after galactose addition (Supplemental Figure S4A). There were no epitope tags on Smc3-K113Rp or smc3-42p, so we used quantitative reverse transcriptase PCR (qRT-PCR) to confirm that the amount of SMC3, smc3-K113R, and smc3-42 mRNA dramatically increased after galactose addition (Supplemental Figure S4B). Furthermore, overexpression of these lethal alleles is not sufficient to restore cohesion at the nonpermissive temperature (Supplemental Figure S4C). These results indicate that simply providing more of the product of any single allele (smc3-42, smc3-K113R, or mcd1-1) and its associated activity is insufficient for viability or cohesion. Furthermore, when the same mcd1 alleles are placed in the cis configuration, that is, within the same gene as a “chimeric” mcd1-1,Q266 allele, it fails to support viability, even when overexpressed (Supplemental Figure S5). Thus the restoration of viability mediated by interallelic complementation likely occurs by each mutant allele providing a distinct activity that is missing in its partner.
FIGURE 2:
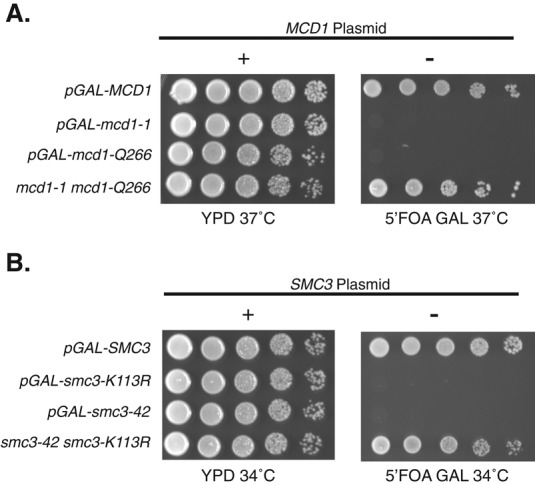
GAL1-induced overexpression of MCD1 and SMC3 alleles. (A) Four haploid yeast strains contain an MCD1 shuffle plasmid pVG201 {MCD1 URA3 CEN}. Three of these strains are deleted for endogenous MCD1 and contain a CEN TRP1 plasmid bearing different MCD1 alleles under pGAL control: pGAL-MCD1 (yTE480), pGAL-mcd1-1 (yTE502), and pGAL-mcd1-Q266 (yTE482). A positive control has mcd1-1 at the endogenous locus and mcd1-Q266 at LEU2 under their endogenous promoter (mcd1-1 mcd1-Q266; TE491). Strains were grown as described in Figure 1A and then plated in 10-fold serial dilutions on YPD or 5-FOA plus galactose (5-FOA GAL) and incubated 3 d at 37°C. MCD1 shuffle plasmid presence (+) or absence (–) is shown above YPD and 5-FOA GAL, respectively. Failure to grow on 5-FOA GAL indicates that pGAL overexpression alone is insufficient for viability. Left, relevant MCD1 genotypes. (B) Four haploid yeast strains containing an SMC3 shuffle plasmid pEU42 {SMC3 URA3 CEN}. Three of these strains are deleted for endogenous SMC3 and contain different SMC3 alleles under pGAL control integrated at LEU2: pGAL-SMC3 (yTE424), pGAL-smc3-K113R (yTE449), and pGAL-smc3-42 (yTE420). A positive control strain has both smc3-42 and smc3-K113R under the endogenous promoter integrated at the SMC3 and LEU2 loci, respectively (smc3-42 smc3-K113; yTE505). Strains were grown as described in Figure 1A and then plated in 10-fold serial dilutions on YPD or 5-FOA plus galactose (5-FOA GAL) and incubated 3 d at 34°C. SMC3 shuffle plasmid presence (+) or absence (–) is shown above YPD and 5-FOA GAL plates, respectively. Failure to grow on 5-FOA GAL indicates that pGAL overexpression alone is insufficient for viability. Left, relevant SMC3 genotype.
Interallelic complementation suppresses the condensation defects characteristic of the single mcd1 and smc3 lethal alleles
In budding yeast, cohesin is critical for viability, sister chromatid cohesion, and chromosome condensation. We asked whether the interallelic complementation we observed between pairs of alleles in MCD1 or SMC3 for viability reflected complementation of defects in cohesion, condensation, or both. We previously described several examples in budding yeast in which mutants in cohesin or cohesin regulators had little or no cohesion but were viable due to their ability to promote condensation (Guacci and Koshland, 2012). From these results, we concluded that condensation rather than cohesion is the major essential function of cohesin in budding yeast in an unperturbed cell cycle (Guacci and Koshland, 2012). This less-stringent requirement for cohesion compared with other eukaryotes is made possible in budding yeast because of its unusual initiation of spindle assembly during S phase (Guacci and Koshland, 2012; Guacci et al., 2015). This link between cohesin's condensation function and viability led to two predictions regarding interallelic complementation. First, the inviability of each of the single alleles in this study likely reflected a defect in condensation. Second, the viability promoted by interallelic complementation likely reflected their ability to complement each other's defect in condensation.
To test directly these two predictions, we compared condensation in single mutants or in double mutants that exhibited interallelic complementation. For this purpose, cells were synchronized in G1 phase, shifted to conditions nonpermissive for both single alleles, and then released from G1 under nonpermissive conditions and arrested in mid M phase using nocodazole (Materials and Methods). Chromosome spreads of these cells in mid M phase were prepared to examine the morphology of the rDNA (ribosomal DNA) locus, a well-established metric for condensation in budding yeast (Guacci et al., 1997; Lavoie et al., 2004; Eng et al., 2014). In this assay, proper chromosome condensation results in formation of rDNA loops (Figure 3A).
FIGURE 3:
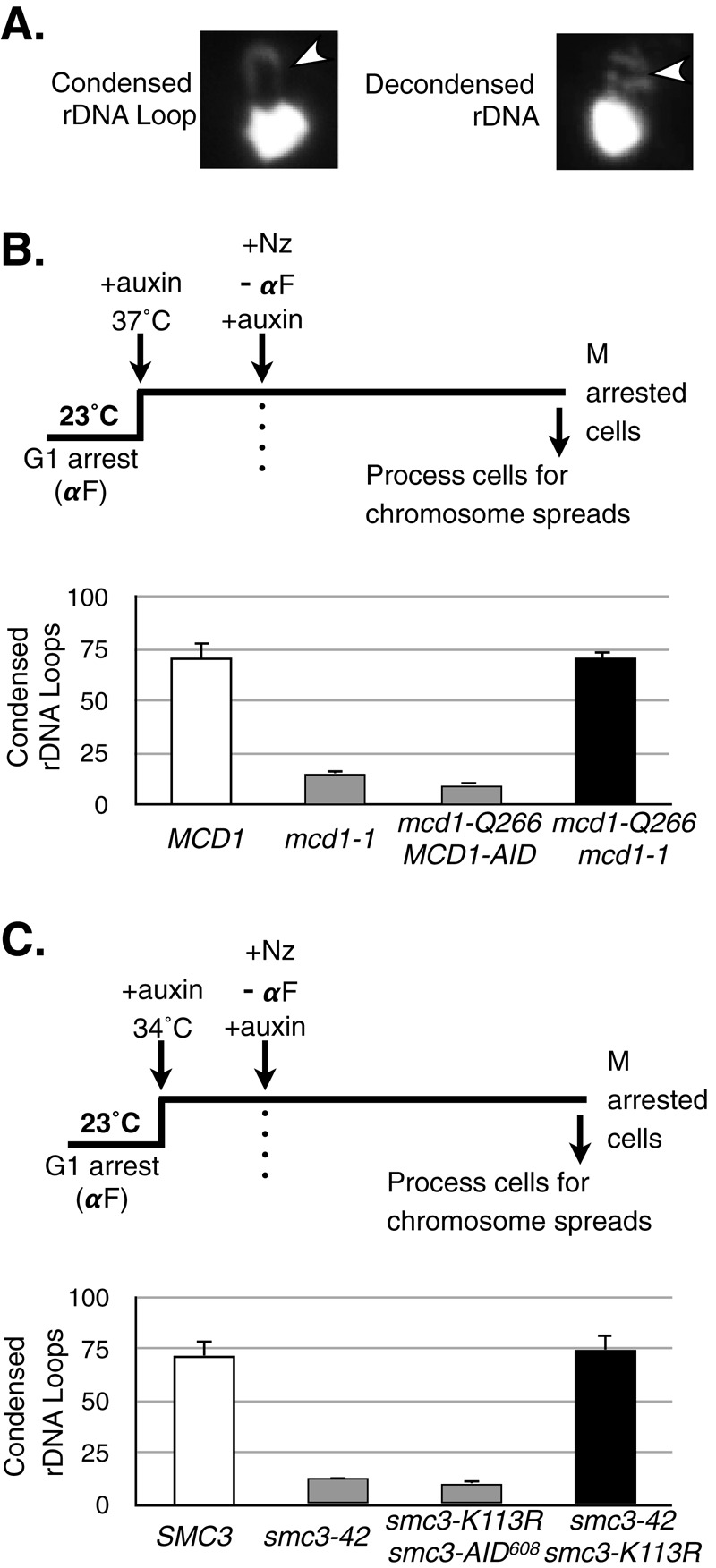
Analysis of chromosome condensation in MCD1 and SMC3 mutant allele strains. (A) Classification of rDNA morphology as assayed by chromosome spreads. Yeast spheroplasts were prepared, and chromosomes were spread on glass slides (see Materials and Methods). Chromosome masses were stained using DAPI and scored for morphology of the rDNA locus, which is adjacent to the bulk chromosome mass. A loop indicates a condensed rDNA locus (left), whereas a puff indicates a decondensed state (right), The rDNA is indicated by an arrowhead. (B) Haploid wild-type MCD1 (yVG3349-1B), mcd1-1 (yVG3312-7A), mcd1-Q266 MCD1-AID (yTE149), and mcd1-Q266-6MYC mcd1-1 (yTE42) strains were arrested in G1 phase at 23°C, auxin was added to degrade AID-tagged proteins, and shift to 37°C inactivated temperature-sensitive proteins. Cells were released from G1 at 37°C into medium containing auxin and nocodazole to arrest cells in mid M phase. Cells in mid M phase were processed for chromosome spreads and the rDNA morphology scored (Materials and Methods). The percentage of cells with condensed rDNA loops is plotted. At least 200 nuclei were scored for each genotype, and at least two biological replicates were completed for each genotype. (C) Haploid wild-type SMC3 (yTE45), smc3-42 (yVG3358-3B), smc3-K113R smc3-AID608 (yTE440), and smc3-42 smc3-K113R (yVG3473-1C) strains were synchronously arrested in mid M phase in auxin-containing medium as described in B, except that the temperature was up-shifted to 34°C. Cells were processed and scored for rDNA morphology and the data plotted as the percentage of cells with condensed rDNA loops as described in B. At least 200 DAPI-stained masses were scored for each genotype, and at least two biological replicates were completed for each genotype.
All mutant strains expressing only the single alleles showed a dramatic reduction in rDNA loops compared with wild type (Figure 3, B and C, gray bars), consistent with previous results (Guacci et al., 1997; Lavoie et al., 2004; Eng et al., 2014). Thus all single alleles caused a dramatic condensation defect, as expected, given their inviability (Figure 1, A and B, and Supplemental Figure S2). In contrast, both mutant strains expressing allele pairs exhibiting interallelic complementation for viability (mcd1-1/mcd1-Q266 or smc3-42/smc3-K113R strains) formed rDNA loops at levels indistinguishable from wild type (Figure 3, B and C, compare white and black bars). Thus, as expected, interallelic complementation suppresses both the inviability and condensation defects characteristic of the individual lethal mcd1 or smc3 alleles alone.
Interallelic complementation suppresses the cohesion maintenance and establishment defects inherent in the single mcd1 and smc3 lethal alleles, respectively
We next asked whether the interallelic complementation that restored both viability and condensation also restores cohesion. We used the regimen described for assaying condensation to obtain cells that progressed from G1 into arrest in mid M phase under conditions in which individual alleles are inactivated. We assayed cohesion in these cells arrested in mid M phase at a centromere-proximal locus (TRP1) and at a chromosomal arm locus (LYS4), using the LacO/LacI–green fluorescent protein (GFP) system (Straight et al., 1996). In this assay, chromosomes in mid M phase that have cohesion exhibit a single GFP spot, whereas if cohesion is defective, sister separation is detected by the appearance of two GFP spots. A severe cohesion defect is observed in cells expressing only the single-mutant alleles (Figure 4, A and B). In contrast, cells expressing each complementing pair of alleles (mcd1-1/mcd1-Q266 or smc3-42/smc3-K113R) showed near-wild-type levels of cohesion at both the centromere-proximal and chromosomal arm loci (Figure 4, A and B). Thus the interallelic complementation pairs suppress defects in cohesion, as well as defects in viability and condensation, that are inherent in each lethal mcd1 or smc3 allele alone.
FIGURE 4:
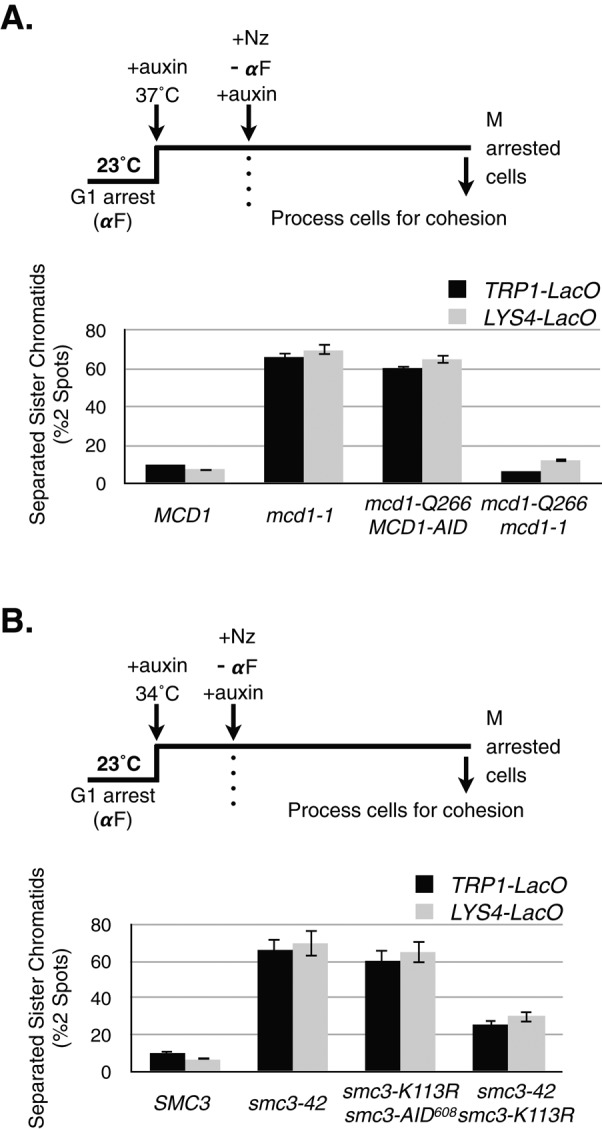
Analysis of sister chromatid cohesion at CEN-proximal (TRP1) and CEN-distal (LYS4) loci in MCD1 and SMC3 allele strains. (A) Haploid wild-type MCD1 (TRP1-LacO: yVG3460-2A and LYS4-LacO: yTE45), mcd1-1 (TRP1-LacO: yTE453 and LYS4-LacO: yVG3312-7A), mcd1-Q266 MCD1-AID (TRP1-LacO: yTE285 and LYS4-LacO: yTE149) and or mcd1-Q266 mcd1-1 (TRP1-LacO: yTE456 and LYS4-LacO: yTE42) were synchronously arrested in mid M phase at 37°C in medium containing auxin and nocodazole as described in Figure 3B. Cells were processed, fixed, and scored for cohesion at either the CEN-proximal TRP1 locus or the CEN-distal LYS4 locus (Material and Methods). The percentage of cells in which sisters had separated (two GFP spots) is plotted. At least 200 cells were scored for each genotype, and the experiment was repeated at least two times to generate data. (B) Haploid wild-type SMC3 (TRP1-LacO: yVG3460-2A and LYS4-LacO: yTE48), smc3-42 (TRP1-LacO: yTE494 and LYS4-LacO: yVG3358-3B), smc3-K113R smc3-AID608 (TRP1-LacO: yTE471 and LYS4-LacO: yTE440), and smc3-K113R smc3-42 (TRP1-LacO: yTE500 and LYS4-LacO: yVG3473-1C) were synchronously arrested in mid M phase at 34°C in medium containing auxin and nocodazole as described in Figure 3C. Cohesion was scored and plotted as described in A.
Previous studies showed that mutations in cohesin subunits or regulators can lead to defects in either the establishment or maintenance of cohesion (Skibbens et al., 1999; Stead et al., 2003; Noble et al., 2006; Eng et al., 2014). The mcd1-Q266 allele is defective only in the maintenance of cohesion (Eng et al., 2014), whereas the mcd1-1 allele is defective in both establishment and maintenance (Guacci et al., 1997; Noble et al., 2006). The robust cohesion observed in mcd1-1 mcd1-Q266 cells arrested at mid M phase (Figure 4A) indicates that mcd1-1p and mcd1-Q266p minimally must have complemented their shared defect in cohesion maintenance. It was previously shown that smc3-42 strains are defective for establishment and maintenance at 35.5 and 37°C (Michaelis et al., 1997; Unal et al., 2008; Guacci and Koshland, 2012). However, smc3-K113R smc3-42 interallelic complementation is robust at lower temperatures (Figure 1B and Supplemental Figure S2C). Therefore we analyzed cohesion loss at lower temperatures to assess whether, under complementation conditions, defects in cohesion establishment or maintenance are suppressed. The smc3-42 allele alone had a severe establishment defect, whereas the smc3-K113R smc3-42 strain established cohesion almost as well as the wild-type strain (Supplemental Figure S6A). The smc3-K113R smc3-42 strain showed similar restoration of cohesion at 30 and 34°C (Supplemental Figure S6B). Given that condensation is the essential cohesin function, it was formally possible that the smc3-K113R allele alone was competent for cohesion, whereas interallelic complementation with the smc3-42 mutant restored viability and condensation. To rule out this possibility, we needed to assess smc3-K113R under conditions lacking interallelic complementation. Therefore we examined cohesion in an smc3-K113R smc3-AID608 double mutant at 30 and 34°C, since there is no complementation of viability above 30°C (Supplemental Figure S3A; unpublished data). In the smc3-AID608 background, the smc3-K113R allele failed to establish cohesion at either 30 or 34°C (Supplemental Figure S6, C and 6). Therefore the robust cohesion in the smc3-42 smc3-K113R strain results from interallelic complementation restoring cohesion establishment. Thus interallelic complementation can lead to the restoration of cohesion establishment and maintenance, as well as to condensation and viability.
mcd1-1p is stabilized by the presence of its interallelic complementing partner, mcd1-Q266p
To address the molecular basis underlying these three examples of interallelic complementation, we asked whether the known molecular defects characteristic of each single allele are altered in cells expressing both alleles. A molecular defect of the mcd1-1p is its degradation at 37°C (Figure 5A). We asked whether mcd1-1p degradation is reduced in the presence of its complementing allelic partner, mcd1-Q266p. Cultures of mcd1-1 and mcd1-1 mcd1-Q266-6MYC strains were arrested in mid M phase at 23°C using nocodazole. Each culture was split, and half was incubated at 23°C and half at 37°C for an additional 1 h. Cells were analyzed by Western Blot to assess the mcd1-1p levels using a polyclonal antibody for Mcd1p. As expected for an mcd1-1 strain, mcd1-1p levels are reduced dramatically after incubation at 37°C compared with 23°C (Figure 5A). In the presence of mcd1-Q266-6MYCp, mcd1-1p levels remained high in cells incubated at 37°C, as seen by the lower–molecular weight band (Figure 5A). The higher–molecular weight band is mcd1-Q266-6MYCp. To rule out the possibility that the lower–molecular weight species detected by our Mcd1p antibody was a degradation product of mcd1-Q266-6MYC, we epitope tagged mcd1-1p with 3xFLAG (mcd1-1-3FLAGp; Materials and Methods). We could now unambiguously monitor mcd1-1-3FLAGp via antibodies to its FLAG tag. As expected for the single mcd1-1-3FLAG allele strain, anti-FLAG Western blots revealed that the mcd1-1-3FLAGp is degraded at 37°C (Figure 5B). In contrast, in the double-mutant mcd1-1-3FLAG mcd1-Q266-6MYC strain, the mcd1-1-3FLAGp is stabilized at 37°C in both S phase– and M phase–arrested cells (Figure 5B). To emphasize, merely increasing mcd1-1p levels by overexpression fails to suppress mcd1-1 strain inviability or cohesion at 37°C (Figure 2 and Supplemental Figure S4C). Therefore the increased mcd1-1p levels generated by the presence of mcd1-Q266p is not sufficient for viability, but instead the mcd1-Q266p cohesin complex itself is necessary for complementation.
FIGURE 5:

Analysis of Mcd1 protein levels in MCD1 allele strains. (A) Early log phase cultures of haploid strains mcd1-1 (yVG3312-7A) and mcd1-1 mcd1-Q266-6MYC (yTE42) were arrested in M phase with nocodazole for 2.5 h. After all cultures had arrested with 95% large- budded cells, cultures were incubated for an additional 1 h at either 23 or 37°C. Cells were then harvested, lysed, and prepared for protein analysis by SDS–PAGE and Western blotting. Tubulin was used as a loading control. (B) Early log phase cultures of haploid strains mcd1-1-3FLAG (yTE103) and mcd1-1-3FLAG mcd1-Q266-6MYC (yTE181) were arrested in S phase (hydroxyurea) or M phase (nocodazole) for 2.5 h. After arrest, cultures were split in half and incubated for an additional 1 h at either 23 or 37°C. Afterward, all cultures were harvested, lysed, and prepared for SDS–PAGE and Western blot analysis using mouse anti-FLAG or mouse anti-MYC antibodies to discriminate between both alleles of Mcd1p present. A nonspecific band with a faster mobility than Mcd1-3FLAGp was detected in cells lacking an epitope fusion protein (asterisk). Tubulin was used as a loading control.
The chromosome-binding defects of mcd1-1p and smc3-AIDp are suppressed by their interallelic complementing partners, mcd1-Q266p and smc3-K113Rp, respectively
mcd1-1p and smc3-42p fail to bind chromosomes at the nonpermissive temperature (Supplemental Figure S7; Haering et al., 2004; Unal et al., 2008), whereas their interallelic partners, mcd1-Q266p and smc3-K113Rp, are bound to chromosomes and cohesin-associated regions (CARs) similar to wild-type cohesin (Rolef Ben-Shahar et al., 2008; Eng et al., 2014). We epitope tagged mcd1-1p (mcd1-1-3FLAG) and smc3-42p (smc3-42-6HA) to assess whether their binding to chromosomes is restored by the presence of their respective interallelic complementing partners. We first characterized mcd1-1-3FLAG protein in strains bearing the mcd1-1-3FLAG allele alone or in the double mutant mcd1-1-3FLAG mcd1-Q266-6MYC. Cells were arrested in mid M phase at the nonpermissive temperature (37°C) and processed for global cohesin binding to chromosomes using chromosome spreads and for cohesin binding at CARs and centromere-proximal regions using chromatin immunoprecipitation (ChIP; Materials and Methods). As a positive control, we also examined wild-type Mcd1-3FLAGp. For chromosome spreads using anti-FLAG antibodies, as expected, wild-type Mcd1-3FLAGp showed robust chromosomal staining, whereas mcd1-1-3FLAGp did not bind chromosomes (Supplemental Figure S7A). However, when mcd1-Q266-6MYC was present in the same cell, the mcd1-1-3FLAGp bound chromosomes well (Supplemental Figure S7A). We also characterized the smc3-42-6HA protein in strains bearing the smc3-42-6HA allele alone or in the double mutant smc3-K113R smc3-42-6HA arrested in mid M phase at nonpermissive temperature. Chromosome spreads show that the smc3-42-6HA protein fails to bind chromosomes as the sole source but does bind chromosomes when the complementing smc3-K113R allele is also present (Supplemental Figure S7B). Thus the presence of the interallelic complementing partner complements the global chromosomal-binding defects of mcd1-1-3FLAGp and smc3-42-6HAp to a large extent.
ChIP analysis of mcd1-1p and smc3-42p at nonpermissive temperature allowed us to examine this altered binding at higher resolution at CARs on chromosomes I, III, XII, and XIV (Materials and Methods; Eng et al., 2014). As expected, wild-type Mcd1-3FLAGp and wild-type Smc3-6HAp show robust binding at CARs and near centromeres (Figure 6, A and B, top), whereas both mutant mcd1-1-3FLAGp and smc3-42-6HAp as sole source have little or no binding (Figure 6, A and B, middle). In contrast, when the complementing alleles are also present in cells, both mcd1-1-3FLAGp and smc3-42-6HAp (Figure 6, A and B, bottom) become enriched at all four regions. Note that this restoration of binding does not reach the levels seen with wild-type proteins. As a control, we show that mcd1-Q266-6MYCp is also bound at all four regions, indicating that it is not displaced by mcd1-1-3FLAGp binding (Supplemental Figure S8). Thus the presence of their interallelic complementing partner dramatically ameliorates the chromosome-binding defects of mcd1-1-3FLAGp and smc3-42-6HAp.
FIGURE 6:

Restoration of mcd1-1-3FLAG and smc3-42-6HA binding to chromosomes in interallelic complementation pairs. (A) Haploid MCD1-3FLAG MCD1-AID (yTE171), mcd1-1-3FLAG (yTE103), and double-mutant mcd1-1-3FLAG mcd1-Q266-6MYC (yTE181) strains were grown to early log phase and arrested in mid M phase at 37°C as described in Figure 3B, except that auxin was not added to yTE103 or yTE181. Cells were fixed and processed for chromatin immunoprecipitation using a mouse anti-FLAG antibody (Materials and Methods). After immunoprecipitation, CARs (left to right: CARL1, CARC1, and centromeres I and XIV) were examined by quantitative PCR (qPCR). The percentage of FLAG-tagged Mcd1p immunoprecipitated is plotted vs. total input for all strains (% FLAG IP). The MCD1 genotype for each strains of interest is indicated above each set of graphs. The average of two biological replicates and the SD between each are shown. (B) Haploid wild-type SMC3-6HA (yGC1-8A), smc3-42-6HA (yVG3523-1A), and double-mutant smc3-42-6HA smc3-K113R (yVG3527-1A) strains were grown to early log phase and then arrested in mid M phase at 34°C as described in Figure 3C, except without auxin addition. Cells were fixed and processed for chromatin immunoprecipitation using a mouse anti-HA antibody (Materials and Methods). The same CARs (left to right: CARL1, CARC1, and centromeres I and XIV) as in A were examined by qPCR. The percentage of HA-tagged Smc3p immunoprecipitated is plotted vs. total input for all strains (% HA IP). The SMC3 genotype for each strains of interest is indicated above each set of graphs. The average of two biological replicates and the SD between each are shown.
DISCUSSION
Here we provide the first demonstration that cohesin-complex subunits are capable of interallelic complementation. This complementation occurs between specific pairs of nonfunctional MCD1 alleles, as well as between nonfunctional SMC3 alleles. Yeast cells expressing only one of these mcd1 or smc3 alleles are inviable and defective for both sister chromatid cohesion and condensation. However, cells expressing pairs of these defective mcd1 or smc3 alleles become viable and generate cohesion and condensation. Overexpression of any of the individual mutants alone does not restore viability or cohesion, making it unlikely that restoration of viability was a consequence of increasing the activity of one of the two mutant alleles. Furthermore, only certain pairs of alleles are capable of interallelic complementation. Together these observations indicate that interallelic complementation occurs because the individual alleles cause defects in distinct molecular functions of the mutated subunit. It is possible that interallelic complementation had not been previously observed because most mutant alleles abrogate multiple properties of the subunit. The idea that an individual cohesin subunit could have several functional domains is not surprising, given that each subunit interacts with multiple other subunits, and that SMC ATPase regulation entails cross-talk between SMC heads (Hirano, 2001; Haering et al., 2002; Gruber et al., 2003).
The molecular interpretation of our three examples of interallelic complementation is constrained significantly by the known stoichiometry of the cohesin complex. It is well established that each cohesin contains only one copy of each of the subunits (Losada et al., 1998; Tóth et al., 1999; Sumara et al., 2000; Ding et al., 2006; Holzmann et al., 2011). Given this stoichiometry, the interallelic complementation we observe must result from interplay or communication between the two defective cohesin complexes, each harboring one of the mutant allele products. This intercomplex complementation restores a core cohesin activity as it regenerates cohesin's functions in both cohesion and condensation.
One possible mechanism for intercomplex complementation is that one mutant complex indirectly activates the other mutant complex by removing some antagonist to cohesin function. This is hard to reconcile with the existing data, as such a putative factor would have to correct several different properties defective in many of the mutant cohesin alleles exhibiting interallelic complementation. For example, WPL1, the best-known cohesin inhibitor, cannot be this putative antagonist, as wpl1∆ is highly toxic to an mcd1-1 strain and has no obvious effect on an smc3-42 strain (Supplemental Figure S9).
Given that in both smc3-42 and mcd1-1 single mutants, the Mcd1p subunit is degraded (this study; Tóth et al., 1999), it could also be that the complementing allele titrates away an Mcd1-specific degradation factor. However, the remarkable suppression of the artificially induced degradation of Smc3-AID608p by smc3-K113R makes this hypothesis also unlikely.
We prefer a model in which the intercomplex complementation is a manifestation of a direct interaction between cohesins on chromosomes that is important for their normal function in cohesion and condensation. Once cohesin has assembled into its core complex, it is competent to bind chromosomes and subsequently undergo cohesin–cohesin interactions. We suggest that smc3-42p and mcd1-1p undergo transient nonproductive binding to chromosomes because cohesins with these mutant subunits are unstable. At nonpermissive temperatures, mcd1-1p is degraded and cohesin binding is lost. In mcd1-1 mcd1-Q266 cells, the chromosome-bound, mcd1-Q266p–containing cohesin complex interacts with the mcd1-1p containing cohesin complex on DNA (Figures 5 and 6). The same model can apply for the smc3-42p–containing cohesin complexes. Our results with the SMC3-AID608 complementation suggest that such an interaction must involve multiple subunits in intimate contact. The suppression of the temperature sensitivity of temperature-sensitive proteins through their assembly into larger complexes has many precedents, such as temperature-sensitive virion proteins, which become refractive to temperature inactivation after assembly into the virion (Gordon and King, 1994). By this rationale, cohesin–cohesin interactions could form higher-order structures to facilitate its function in both cohesion and condensation.
Cohesion can be established, but its maintenance can be compromised, without altering cohesin's stable binding to chromosomes (Eng et al., 2014). Other researchers have observed that cohesin at the silent mating locus is stably bound but does not tether sisters (Chang et al., 2005) and that inactivation of cohesin regulators such as Eco1p or Pds5p does not destabilize cohesin's chromosomal binding (Chan et al., 2012, 2013; Guacci and Koshland, 2012; Tong and Skibbens, 2014). This favors a two-step model for cohesin function, as cohesin first stably binds DNA and in turn undergoes a tethering step (Figure 7A). The three types of interallelic complementation pairs we described in this study suggest that an interaction between two or more cohesin molecules (dimerization or oligomerization) is part of this post–DNA binding/tethering activity. Two types of interactions fit these constraints. We call the first model the “intercohesin handcuff” (Figure 7, B and C). Two distinct cohesins could separately bind a sister, and the interaction of these two cohesins could tether the sisters together. In this case, the interaction is essential for cohesin's tethering activity. A second model that fits these constraints is the “cohesin stack” (Figure 7, D and E). Each cohesin molecule would bind both sisters but do so via two distinct DNA-binding sites rather than a single entrapment as postulated by the embrace model. This interaction is insufficient to stably tether sisters (Figure 7D). A second step involving oligomerization of similarly DNA bound cohesins would form an oligomer whose additive DNA binding would be robust enough to tether sisters (Figure 7E). Both of these models are amenable to long-range tethering described for condensation, as well as for gene regulation. Moreover, these new models provide a flexibility by which stably bound cohesin could be repurposed for other chromosome functions by altering oligomerization partners.
FIGURE 7:

Models for cohesin–cohesin interactions as mediators of sister chromatid cohesion. (A) Cohesin is loaded by the Scc2/Scc4 complex onto the unreplicated sister chromatid at the G1/S transition. A topological entrapment of DNA is shown, using the surface formed by the Smc1/Smc3 heads and Mcd1's linker domain. (B, C) Intercohesin handcuff model. (B) Concomitant with DNA replication, a second cohesin complex binds the newly replicated sister chromatid in close proximity to the G1/S-loaded cohesin. Both DNA-bound cohesins undergo a conformational change. (C). Cohesin handcuff state. The conformational change facilitates cohesin oligomerization to tether sister chromatids. Cohesin regulators are likely required to trigger the conformational change (B) and promote/stabilize the oligomeric form (C). (D, E) Cohesin stack model. (D) During DNA replication, the G1/S-loaded cohesin undergoes a conformational change to expose a second cohesin DNA binding motif that holds the sister chromatid. This dual DNA-binding cohesin conformation is not strong enough to tether sisters. (E) This cohesin complex facilitates an oligomerization with adjacent, similarly bound complexes, thus stacking cohesins into a robust tethering form. Cohesin regulators are likely required to promote the conformational change enabling a dual DNA-binding state (D) and to facilitate/stabilize stacking (E).
If cohesin dimerization or oligomerization is part of the mechanism of cohesin function, why have they not been broadly detected by biochemical methods? Previous biochemical studies examined soluble cohesin complexes and found no robust evidence for cohesin oligomers (Haering et al., 2002; Gruber et al., 2003; Hauf et al., 2005). This failure to observe physical interactions between soluble cohesins is consistent with the idea that such interactions inferred from this study must happen on chromosomes. Interestingly, when cohesin was subjected to a cross-linking reagent in vivo, a number of cross-links between different regions of Smc3p or Smc1p were observed (Huis In ‘t Veld et al., 2014). These cross-links could represent intramolecular interactions within a subunit. However, in light of our results regarding interallelic complementation, some could represent interactions between two Smc3ps or Smc1ps present in distinct but interacting cohesins. It would be interesting to compare the pattern of cross-links observed with purified soluble cohesin monomers and cohesin bound to chromosomes.
Is this intercomplex interaction a unique property of the cohesin complex, or does it represent a fundamental property conserved in all SMC-family complexes? We strongly suspect that interactions between condensins or other SMC complexes are likely to be important for their function as well. Indeed, a substantial fraction of purified condensin elutes from size exclusion columns at a molecular weight significantly greater than that of a monomeric condensin complex (St-Pierre et al., 2009). This multimeric fraction has robust DNA-supercoiling activity characteristic of condensin, which is more active than the monomeric fraction (Damien D’Amours, personal communication). Miniclusters of condensin complexes have also been observed in reconstituted Xenopus laevis chromosomes (König et al., 2007), and condensin may be configured in a “rosette” in the pericentromeric domain (Stephens et al., 2013). Intriguingly, it was recently reported that overexpression of either recessive inviable SMC4 allele smc4-7A or smc4-82 could restore viability in smc4-1 cells (Robellet et al., 2015). This result is consistent with interallelic complementation in the condensin complex.
Although the exact molecular basis for cohesin–cohesin interactions revealed in this study remains to be elucidated, their discovery reflects a major molecular component of cohesin function that has escaped previous studies. Their study is likely to provide important new insights into cohesin's molecular and biological functions.
MATERIALS AND METHODS
Yeast strains, media, and reagents
Yeast strains used in this study are in the A364a background, and their genotypes are listed in Supplemental Table S1. Synthetic dropout and yeast extract/peptone/dextrose (YPD) media were prepared as previously described (Guacci et al., 1997). Galactose (2% final [wt/vol]; Sigma-Aldrich, St. Louis, MO) was used as an alternative carbon source to induce genes under the GAL promoter as indicated. For experiments using the AID system, a 1 M stock of 3-indoleacetic acid (auxin; Sigma-Aldrich) dissolved in DMSO was added to plates or liquid cultures at a final concentration of 500 μM, cooling agar used in plates to ∼55°C before adding auxin to each batch. The pH of our YPD medium was approximately ∼7. To arrest cells in early S phase, we made a 2 M stock solution of hydroxyurea (Sigma-Aldrich), which we added to yeast cells in early log phase to a final concentration of 200 mM. Cell-cycle arrests were visually confirmed when cells were 95% large budded. 5-FOA was purchased from US Biological Life Sciences (Salem, MA) and used at a final concentration of 1 g/l in URA dropout plates supplemented with 50 mg/l uracil powder (Sigma-Aldrich).
Dilution plating assays
Cells were grown to overnight saturation in YPD medium at 23°C (or 30°C when listed), diluted to an OD660 of 1.0 using YPD, and then plated in 10-fold serial dilutions. Cells were incubated on different plates at the relevant temperatures indicated.
Shuffle strain construction
SMC3 shuffle strains.
Plasmid pEU42 (SMC3 URA3 CEN) was transformed into haploid strains. The endogenous SMC3 gene was deleted and replaced by the HPH cassette (encodes resistance to hygromycin B) using standard PCR-mediated homology-based recombination.
MCD1 shuffle strains.
Plasmid pVG210 (MCD1 URA3 CEN) was transformed into haploid strains. The endogenous MCD1 gene was deleted and replaced by the KanMx6 cassette.
Shuffle assays.
SMC3 or MCD1 under their endogenous promoters or under control of the GAL1 promoter was cloned into CEN TRP1 plasmids and transformed into SMC3 or MCD1 shuffle strains. Alternatively, SMC3 or MCD1 alleles were cloned into LEU2 integrating plasmids, linearized, and then transformed into shuffle stains to integrated constructs at the LEU2 genomic locus. Transformant clones were grown to saturation in YPD medium at 23°C to allow loss of plasmid pEU42 or pVG210 and then plated in 10-fold serial dilutions on medium containing 5-FOA. 5-FOA selectively kills URA3+ cells, thereby selecting for which of them have lost plasmid pEU42 or pVG210 to become URA3−. This plasmid loss allows assessment of the ability of test alleles on CEN TRP1 plasmids or integrated at LEU2 to support viability as the sole SMC3 or MCD1 in cells. As a control, cells were also plated on either YPD or URA dropout medium. When test alleles were placed under the GAL1 promoter, FOA GAL or YP GAL plates were used.
Cell-cycle staging (arrest) in G1 and release into arrest in mid M phase
Haploid yeast strains were grown to mid log phase at 23°C in YPD liquid, and then α factor (Sigma-Aldrich) was added to 10−8 M and cells incubated for 3 h to arrest cells in G1 phase. G1-phase arrest was confirmed by presence of unbudded “schmoo” morphology in 95% of the cells as scored by light microscopy and fluorescence-activated cell sorting (FACS). For stains bearing AID-tagged proteins, auxin was added (500 μM final) and cultures incubated 1 h more to induce degradation of AID-tagged proteins. For strains bearing temperature-sensitive alleles, cultures were transferred to the appropriate elevated temperature and then incubated 1 h more to inactivate the temperature-sensitive allele. Cells were released from G1 arrest by six washes in YPD containing 500 μM auxin and 0.1 mg/ml Pronase E (Sigma-Aldrich), resuspended in fresh YPD containing 500 μM auxin and nocodazole (Sigma-Aldrich) at 15 μg/ml final, and then incubated at 23°C for 3 h to allow cell-cycle progression until arrest in mid M phase. Arrest was confirmed by light microscopy for the classical large-budded cellular phenotype (∼95% of the population) and by FACS.
Monitoring cohesion using LacO-GFP
Cohesion was monitored using the LacO-LacI system, in which cells contained a GFP-LacI fusion protein and also tandem LacO repeats integrated at one chromosomal locus, which recruits the GFP-LacIp (Straight et al., 1996). CEN-distal cohesion was monitored by integrating LacO repeats at LYS4, located 470 kb from CEN4. CEN-proximal cohesion was monitored by integrating LacO at TRP1, located 10 kb from CEN4. Cells were fixed and processed to allow the number of GFP signals in each cell to be scored. Before replication, each chromosome exhibits one GFP spot. After replication, chromosomes that have sister chromatid cohesion still exhibit one GFP spot in cells, whereas chromosomes that have lost cohesion exhibit to two GFP spots due to sister separation. The percentage of cells with two GFP spots (separated sisters) was determined and plotted.
Chromosome spreads and microscopy
Chromosome spreads were performed as previously described (Wahba et al., 2013). Slides were incubated with a mouse anti-FLAG monoclonal (Sigma-Aldrich) at 1:4000, mouse anti-V5 (Life Technologies, Carlsbad, CA) at 1:2000, or polyclonal rabbit anti-Mcd1 at 1:4000 dilution. The primary antibody was diluted in blocking buffer (5% bovine serum albumin, 0.2% milk, 1× phosphate-buffered saline, 0.2% Triton X-100). The secondary Cy3-conjugated goat anti-mouse antibody (115-165-003) was obtained from Jackson ImmunoResearch (West Grove, PA) and diluted 1:2000 in blocking buffer. Indirect immunofluorescence was observed using an Olympus IX-70 microscope with a 100×/numerical aperture 1.4 objective and Orca II camera (Hamamatsu, Bridgewater, NJ).
Chromatin immunoprecipitation
Cells used for ChIP experiments were processed as described in Wahba et al. (2013). Briefly, after synchronous release from G1 into mitotic arrest, 5 × 108 large-budded cells were fixed for 2 h with 1% formaldehyde. After cell lysis, chromatin was sheared 20 times for 45 s each (settings at duty cycle, 20%; intensity, 10; cycles/burst, 200; 30s of rest between cycles) using a Covaris S2. Immunoprecipitation of epitope-tagged proteins was isolated using anti-FLAG (Sigma-Aldrich), anti-HA (12CA5; Roche Life Sciences, Indianapolis, IN), or anti-MYC (9E10; Roche Life Sciences) monoclonal antibodies. Mcd1p was alternatively immunoprecipitated using a polyclonal rabbit anti-Mcd1 antibody (Covance Biosciences, Princeton, NJ). A no-primary-antibody control was also run to ensure specificity. Appropriate dilutions of input and immunoprecipitated DNA samples were used for quantitative PCR analysis to ensure linearity of the PCR signal. PCR and data analysis was carried out as described previously (Wahba et al., 2013). All experiments were done at least twice, and a representative data set is shown. ChIP primers are available upon request.
Western blotting
Proteins from whole-cell extracts were prepared essentially as described in Foiani et al. (1994) with slight modifications. After initial lysis in 300 μl of 20% trichloroacetic acid (TCA; wt/vol), the solution was further diluted with another 300 μl of 5% TCA (wt/vol). The remaining insoluble material was resuspended once more in 500 μl of 5% TCA. These two lysates were combined together and then incubated on ice for 10 min before a 15-min centrifugation at 20,000 × g at 4°C. The resulting TCA pellet was resuspended in ∼100 μl of loading buffer and pH adjusted with ∼10 μl of 1 M Tris, pH 8.0. Resuspended pellets were boiled for 10 min at 70°C, and clarified whole-cell extracts were separated on 7% SDS–PAGE gels under standard laboratory conditions.
Microscopy
Images were acquired with an Axioplan2 microscope (100× objective, numerical aperture 1.40; Zeiss, Thornwood, NY) equipped with a Quantix charge-coupled device camera (Photometrics, Tucson, AZ).
Flow cytometry
DNA content of cells was analyzed using flow cytometry as described previously (Eng, 2014; Guacci et al., 2015).
Supplementary Material
Acknowledgments
We thank Anjali Krishnan, Hugo Tapia, Lorenzo Costantino, Gamze Camdere, and Elçin Ünal for helpful discussions, reagents, and constructive feedback on the manuscript. This work was supported by Grant R01 GM092813 from the National Institute of General Medical Sciences/National Institutes of Health to D.E.K. and a National Science Foundation Graduate Research Fellowship to T.E.
Abbreviations used:
- HA
hemagglutinin
- Nz
nocodazole
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-06-0331) on September 16, 2015.
REFERENCES
- Boundy-Mills KL, Livingston DM. A Saccharomyces cerevisiae RAD52 allele expressing a C-terminal truncation protein: activities and intragenic complementation of missense mutations. Genetics. 1993;133:39–49. doi: 10.1093/genetics/133.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K-L, Gligoris T, Upcher W, Kato Y, Shirahige K, Nasmyth K, Beckouët F. Pds5 promotes and protects cohesin acetylation. Proc Natl Acad Sci USA. 2013;110:13020–13025. doi: 10.1073/pnas.1306900110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K-L, Roig MB, Hu B, Beckouët F, Metson J, Nasmyth K. Cohesin's DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell. 2012;150:961–974. doi: 10.1016/j.cell.2012.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-R, Wu C-S, Hom Y, Gartenberg MR. Targeting of cohesin by transcriptionally silent chromatin. Genes Dev. 2005;19:3031–3042. doi: 10.1101/gad.1356305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coddington A, Fincham JR. Proof of hybrid enzyme formation in a case of inter-allelic complementation in neurospora crassa. J Mol Biol. 1965;12:152–161. doi: 10.1016/s0022-2836(65)80289-3. [DOI] [PubMed] [Google Scholar]
- Ding D-Q, Sakurai N, Katou Y, Itoh T, Shirahige K, Haraguchi T, Hiraoka Y. Meiotic cohesins modulate chromosome compaction during meiotic prophase in fission yeast. J Cell Biol. 2006;174:499–508. doi: 10.1083/jcb.200605074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donze D, Adams CR, Rine J, Kamakaka RT. The boundaries of the silenced HMR domain in Saccharomyces cerevisiae. Genes Dev. 1999;13:698–708. doi: 10.1101/gad.13.6.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng T, Guacci V, Koshland D. ROCC, a conserved region in cohesin's Mcd1 subunit, is essential for the proper regulation of the maintenance of cohesion and establishment of condensation. Mol Biol Cell. 2014;25:2351–2364. doi: 10.1091/mbc.E14-04-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiani M, Marini F, Gamba D, Lucchini G, Plevani P. The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol Cell Biol. 1994;14:923–933. doi: 10.1128/mcb.14.2.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CL, King J. Genetic properties of temperature-sensitive folding mutants of the coat protein of phage P22. Genetics. 1994;136:427–438. doi: 10.1093/genetics/136.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray WM, Kepinski S, Rouse D, Leyser O, Estelle M. Auxin regulates SCF(TIR1)-dependent degradation of AUX/IAA proteins. Nature. 2001;414:271–276. doi: 10.1038/35104500. [DOI] [PubMed] [Google Scholar]
- Gruber S, Haering CH, Nasmyth K. Chromosomal cohesin forms a ring. Cell. 2003;112:765–777. doi: 10.1016/s0092-8674(03)00162-4. [DOI] [PubMed] [Google Scholar]
- Guacci V, Koshland D. Cohesin-independent segregation of sister chromatids in budding yeast. Mol Biol Cell. 2012;23:729–739. doi: 10.1091/mbc.E11-08-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci V, Stricklin J, Bloom MS, Guō X, Bhatter M, Koshland D. A novel mechanism for the establishment of sister chromatid cohesion by the ECO1 acetyltransferase. Mol Biol Cell. 2015;26:117–133. doi: 10.1091/mbc.E14-08-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haering CH, Löwe J, Hochwagen A, Nasmyth K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell. 2002;9:773–788. doi: 10.1016/s1097-2765(02)00515-4. [DOI] [PubMed] [Google Scholar]
- Haering CH, Schoffnegger D, Nishino T, Helmhart W, Nasmyth K, Löwe J. Structure and stability of cohesin's Smc1-kleisin interaction. Mol Cell. 2004;15:951–964. doi: 10.1016/j.molcel.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Hartman T, Stead K, Koshland D, Guacci V. Pds5p is an essential chromosomal protein required for both sister chromatid cohesion and condensation in Saccharomyces cerevisiae. J Cell Biol. 2000;151:613–626. doi: 10.1083/jcb.151.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, Peters J-M. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol. 2005;3:e69. doi: 10.1371/journal.pbio.0030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M. Bimodal activation of SMC ATPase by intra- and inter-molecular interactions. EMBO J. 2001;20:3238–3250. doi: 10.1093/emboj/20.12.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzmann J, Fuchs J, Pichler P, Peters J-M, Mechtler K. Lesson from the stoichiometry determination of the cohesin complex: a short protease mediated elution increases the recovery from cross-linked antibody-conjugated beads. J Proteome Res. 2011;10:780–789. doi: 10.1021/pr100927x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huis In ‘t Veld PJ, Herzog F, Ladurner R, Davidson IF, Piric S, Kreidl E, Bhaskara V, Aebersold R, Peters J-M. Characterization of a DNA exit gate in the human cohesin ring. Science. 2014;346:968–972. doi: 10.1126/science.1256904. [DOI] [PubMed] [Google Scholar]
- Korch CT, Snow R. Allelic complementation in the first gene for histidine biosynthesis in Saccharomyces cerevisiae. I. Characteristics of mutants and genetic mapping of alleles. Genetics. 1973;74:287–305. doi: 10.1093/genetics/74.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König P, Braunfeld MB, Sedat JW, Agard DA. The three-dimensional structure of in vitro reconstituted Xenopus laevis chromosomes by EM tomography. Chromosoma. 2007;116:349–372. doi: 10.1007/s00412-007-0101-0. [DOI] [PubMed] [Google Scholar]
- Lavoie BD, Hogan E, Koshland D. In vivo requirements for rDNA chromosome condensation reveal two cell-cycle-regulated pathways for mitotic chromosome folding. Genes Dev. 2004;18:76–87. doi: 10.1101/gad.1150404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A, Hirano M, Hirano T. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev. 1998;12:1986–1997. doi: 10.1101/gad.12.13.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melby TE, Ciampaglio CN, Briscoe G, Erickson HP. The symmetrical structure of structural maintenance of chromosomes (SMC) and MukB proteins: long, antiparallel coiled coils, folded at a flexible hinge. J Cell Biol. 1998;142:1595–1604. doi: 10.1083/jcb.142.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Noble D, Kenna MA, Dix M, Skibbens RV, Unal E, Guacci V. Intersection between the regulators of sister chromatid cohesion establishment and maintenance in budding yeast indicates a multi-step mechanism. Cell Cycle. 2006;5:2528–2536. doi: 10.4161/cc.5.21.3405. [DOI] [PubMed] [Google Scholar]
- Onn I, Heidinger-Pauli JM, Guacci V, Unal E, Koshland DE. Sister chromatid cohesion: a simple concept with a complex reality. Annu Rev Cell Dev Biol. 2008;24:105–129. doi: 10.1146/annurev.cellbio.24.110707.175350. [DOI] [PubMed] [Google Scholar]
- Perrin D. Immunological studies with genetically altered beta-galactosidases. Ann NY Acad Sci. 1963;103:1058–1066. doi: 10.1111/j.1749-6632.1963.tb53757.x. [DOI] [PubMed] [Google Scholar]
- Reha-Krantz LJ. Genetic evidence for two protein domains and a potential new activity in bacteriophage T4 DNA polymerase. Genetics. 1990;124:213–220. doi: 10.1093/genetics/124.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robellet X, Thattikota Y, Wang F, Wee T-L, Pascariu M, Shankar S, Bonneil É, Brown CM, D’Amours D. A high-sensitivity phospho-switch triggered by Cdk1 governs chromosome morphogenesis during cell division. Genes Dev. 2015;29:426–439. doi: 10.1101/gad.253294.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolef Ben-Shahar T, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science. 2008;321:563–566. doi: 10.1126/science.1157774. [DOI] [PubMed] [Google Scholar]
- Rollins RA, Korom M, Aulner N, Martens A, Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol Cell Biol. 2004;24:3100–3111. doi: 10.1128/MCB.24.8.3100-3111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins RA, Morcillo P, Dorsett D. Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics. 1999;152:577–593. doi: 10.1093/genetics/152.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbens RV, Corson LB, Koshland D, Hieter P. Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 1999;13:307–319. doi: 10.1101/gad.13.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead K, Aguilar C, Hartman T, Drexel M, Meluh P, Guacci V. Pds5p regulates the maintenance of sister chromatid cohesion and is sumoylated to promote the dissolution of cohesion. J Cell Biol. 2003;163:729–741. doi: 10.1083/jcb.200305080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AD, Quammen CW, Chang B, Haase J, Taylor RM, Bloom K. The spatial segregation of pericentric cohesin and condensin in the mitotic spindle. Mol Biol Cell. 2013;24:3909–3919. doi: 10.1091/mbc.E13-06-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Douziech M, Bazile F, Pascariu M, Bonneil É, Sauvé V, Ratsima H, D’Amours D. Polo kinase regulates mitotic chromosome condensation by hyperactivation of condensin DNA supercoiling activity. Mol Cell. 2009;34:416–426. doi: 10.1016/j.molcel.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Straight AF, Belmont AS, Robinett CC, Murray AW. GFP tagging of budding yeast chromosomes reveals that protein-protein interactions can mediate sister chromatid cohesion. Curr Biol. 1996;6:1599–1608. doi: 10.1016/s0960-9822(02)70783-5. [DOI] [PubMed] [Google Scholar]
- Ström L, Lindroos HB, Shirahige K, Sjögren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- Sumara I, Vorlaufer E, Gieffers C, Peters BH, Peters JM. Characterization of vertebrate cohesin complexes and their regulation in prophase. J Cell Biol. 2000;151:749–762. doi: 10.1083/jcb.151.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K, Skibbens RV. Cohesin without cohesion: a novel role for Pds5 in Saccharomyces cerevisiae. PLoS One. 2014;9:e100470. doi: 10.1371/journal.pone.0100470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth A, Ciosk R, Uhlmann F, Galova M, Schleiffer A, Nasmyth K. Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 1999;13:320–333. doi: 10.1101/gad.13.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F, Nasmyth K. Cohesion between sister chromatids must be established during DNA replication. Curr Biol. 1998;8:1095–1101. doi: 10.1016/s0960-9822(98)70463-4. [DOI] [PubMed] [Google Scholar]
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE. A molecular determinant for the establishment of sister chromatid cohesion. Science. 2008;321:566–569. doi: 10.1126/science.1157880. [DOI] [PubMed] [Google Scholar]
- Wahba L, Gore SK, Koshland D. The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. Elife. 2013;2:e00505. doi: 10.7554/eLife.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Haase J, Paliulis LV, Joglekar A, Bond L, Bouck D, Salmon ED, Bloom KS. Pericentric chromatin is organized into an intramolecular loop in mitosis. Curr Biol. 2008;18:81–90. doi: 10.1016/j.cub.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Shi X, Li Y, Kim BJ, Jia J, Huang Z, Yang T, Fu X, Jung SY, Wang Y, et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell. 2008a;31:143–151. doi: 10.1016/j.molcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Zhang N, Kuznetsov SG, Sharan SK, Li K, Rao PH, Pati D. A handcuff model for the cohesin complex. J Cell Biol. 2008b;183:1019–1031. doi: 10.1083/jcb.200801157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.