

Abstract
Kindler syndrome (KS; OMIM 173650) is a rare autosomal recessive skin disorder, which results in symptoms including blistering, epidermal atrophy, increased risk of cancer, and poor wound healing. The majority of mutations of the disease-determining gene (FERMT1 gene) are single nucleotide substitutions, including missense mutations, nonsense mutations, etc. Large deletion mutations are seldom reported. To determine the mutation in the FERMT1 gene associated with a 7-year-old Chinese patient who presented clinical manifestation of KS, we performed direct sequencing of all the exons of FERMT1 gene. For the exons 2–6 without amplicons, we analyzed the copy numbers using quantitative real-time polymerase chain reaction (qRT-PCR) with specific primers. The deletion breakpoints were sublocalized and the range of deletion was confirmed by PCR and direct sequencing. In this study, we identified a new 17-kb deletion mutation spanning the introns 1–6 of FERMT1 gene in a Chinese patient with severe KS phenotypes. Her parents were carriers of the same mutation. Our study reported a newly identified large deletion mutation of FERMT1 gene involved in KS, which further enriched the mutation spectrum of the FERMT1 gene.
Keywords: Kindler syndrome, FERMT1 gene, Mutation
1. Introduction
Kindler syndrome (KS; OMIM 173650) is a rare autosomal recessive skin disorder characterized by blistering, extensive epidermal atrophy, photosensitivity, increased risk of cancer, and poor wound healing. The mucosal involvement included early and severe periodontitis, and/or esophageal, gastrointestinal, and genital involvement (Jobard et al., 2003; Siegel et al., 2003; Has and Bruckner-Tuderman, 2006; Kern et al., 2007). It was first described by Theresa Kindler (1954) in a 14-year-old girl who had suffered from acral blistering since childhood and subsequently developed poikiloderma and photosensitivity.
The KS disease-determining gene, termed FERMT1 gene (OMIM 607900), was mapped to chromosome 20p12.3 (Jobard et al., 2003; Siegel et al., 2003). This gene contains 15 exons and spans 48.5 kb of genomic DNA. The coding sequences extend from exon 2 to exon 15. The encoded protein (kindling-1, 677 amino acid (aa), molecular weight of 77.3 kDa) is implicated in linking the actin cytoskeleton to the extracellular matrix (Jobard et al., 2003; Siegel et al., 2003). Lack of kindling-1 in keratinocytes would result in decreased cell adhesion, reduced proliferation, increased apoptosis, and loss of cell polarity (Herz et al., 2006).
In our present study, genetic analysis, including the direct sequencing of all exons and quantitative real-time polymerase chain reaction (qRT-PCR) were performed in order to identify a new, larger deletion mutation spanning about 17 kb from the intron 1 to intron 6 of FERMT1 gene in a Chinese KS family.
2. Materials and methods
2.1. Patient and DNA samples
Genomic DNA was extracted from the peripheral blood leukocytes of the patient and her parents, using the phenol-chloroform method. Informed consent was obtained from the patient’s parents. The project was approved by the Ethics Committee of the Capital Institute of Pediatrics (Beijing, China).
2.2. Molecular genetic analysis
2.2.1 DNA sequence analysis
For the patient with clinically confirmed KS, we screened all exons (exons 1–15) and flanking sequences of the FERMT1 gene using Sanger sequencing. The primers for the PCR and the annealing temperatures were as described by Siegel et al. (2003). The PCR products were purified and direct sequencing of fragments was carried out with an ABI 3730 automatic sequencer (Applied Biosystems, USA).
2.2.2 Copy number analysis of FERMT1 gene
For the exons without amplification products by Sanger sequencing, we performed qRT-PCR in order to detect their copy numbers on an ABI Prism™ 7500 Sequence Detection System (Applied Biosystems, USA) using SYBR Green Master Mix (Applied Biosystems, Foster, USA). All data analysis was performed using the 2−ΔΔCT method in Microsoft Excel Version 2007. Primers were designed using the Primer Express 3.0 software (Applied Biosystems, USA) for the 9 exons of the FERMT1 gene and were evaluated using the National Center for Biotechnology Information’s BLAST program. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was selected as endogenous control. All samples were run in triplicate. We used the ΔΔC T method to analyze the copy numbers and compare them with those of 20 healthy samples. Experiments were replicated at least twice if a deletion/duplication was suspected to have occurred.
2.2.3 Breakpoint analysis of FERMT1 gene
In order to sublocalize the deletion breakpoints, we designed 9 pairs of primers to amplify the non-overlapping fragments in introns 1 and 6. Subsequently, the junction fragment was amplified with the template DNA of the patient using a pair of primers, P10F in intron 1 (forward primer: 5'-AGGCAA GCAGTTAGGCCTAAT-3') and P10R in intron 6 (reverse primer: 5'-ATGACAGAGCCCATTTCCTG-3') to confirm the 5' and 3' breakpoints of the FERMT1 gene in the patient.
3. Results
3.1. Clinical features of the patient with Kindler syndrome
The patient, a 7-year-old female born to consanguineous parents, presented with recurrent blistering over the hands and feet (Fig. 1a). Her parents stated that blister generation might be associated with local friction. When given topical mupirocin ointment the sites of lesions were consequently healed without scarring, and it has been so since the early neonatal period. When she was eight months old, her teeth came through but dental caries were also observed. Subsequently, constipation, including dry stool and defecation difficulties, began at about the age of one. When exposed to sunlight she was found to be prone to photosensitivity. When she was seven years old, she was admitted to our department of dermatology. Upon physical examination, poikiloderma was noted on her face, neck, and upper chest, and erythematous lesions and skin atrophy were observed on the trunk and four extremities (Figs. 1b and 1c). In addition, pseudoainhum was obviously apparent. Examination of the oral cavity showed poor preservation of teeth and severe periodontitis, with easy bleeding (Figs. 1d–1f). She also stated that slight dysphagia existed. Electron microscopy showed a fracture of the lamina densa (Fig. 1g). Histopathological examinations of the skin biopsy with the abdominal involved area showed hyperkeratosis, atrophy of the epidermis, fracturing of the basal layer, and melanophages in the papillary dermis. A biopsy of the skin lesion showed the features of poikiloderma (Fig. 1h). The results of routine blood examinations, cranial computed tomography, and electroencephalogram were all normal.
Fig. 1.

Clinical features of the patient with Kindler syndrome
(a) Pedigree figure of the family (circle=female, square=male, black=carrier of the FERMT1 gene mutation; blank=normal); (b) Poikiloderma on her face; (c) Cigarette-paper-like atrophy on the skin of abdomen; (d) Erosion on the gingivitis; (e) Pseudoainhum and marked skin atrophy on hands; (f) Erosions and scars on the dorsal aspects of the feet; (g) Electron microscopic showed the fissure in lamina lucida (arrows); (h) Histopathological examinations of skin biopsy of the abdominal involved area showed hyperkeratosis and melanophages in the papillary dermis
3.2. Molecular genetic analysis
3.2.1 DNA sequence analysis
After the Sanger sequencing of the FERMT1 gene, no mutation was identified within 10 exons, including exons 1 and 7–15 in the patient. However, DNA sample from the patient failed to yield PCR amplification products for exon 2 to exon 6 (5 exons), whilst they could be amplified in the normal controls and her parents, which revealed that the amplification condition was optimal.
3.2.2 Gene copy number changes and breakpoint confirmation
Since no amplification products for exon 2 to exon 6 were observed with the patient, we were able to precisely identify the copy numbers of this gene (exons 1, 2–7, 14, and 15) using qRT-PCR on genomic DNA. This result further showed that, for exons 2–6, both of the patient’s parents carried one copy and the patient had no copy. However, for exons 1, 7, 14, and 15, the patient and her parents carried two copies (Fig. 2).
Fig. 2.
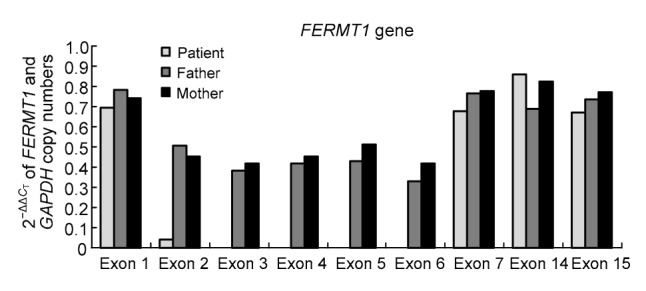
Gene copy number changes using
qRT-PCR qRT-PCR results showed zero copy at exon 2 to exon 6 for the patient
The results of DNA sequencing and copy numbers analysis revealed that the 5' and 3' breakpoints may be closed to exons 1 and 6, respectively. Several amplification results from intron 1 and intron 6 confirmed that 5' and 3' breakpoints were located in introns 1 and 6, respectively. With primers P10F and P10R, the truncated PCR product (763 bp) was obtained from the patient, which revealed that a homozygous large deletion mutation of 17 252 kb was observed in the patient. Meanwhile, her parents both carried the same heterozygous deletion mutation. However, no truncated fragments were observed within the normal controls (Fig. 3).
Fig. 3.

Breakpoint confirmation by genetic methods
(a) 763-bp product was obtained from the patient and her parents, while no amplicons were detected for the normal control since the 18 015-bp product from the normal allele might be too large to amplify in this experiment. M: DNA marker I; P: patient; PF: patient’s father; PM: patient’s mother; Con: control. (b) 763-bp fragment from the patient revealed the 5' and 3' breakpoints within introns 1 and 6 and the 14-bp short homology across the breakpoints is shown in the box. (c) FERMT1 genomic region spans exons 1–7, and the 17 252-bp deletion is delineated with a dashed line. P: primer
4. Discussion
In this paper, we have reported a Chinese patient with KS, who had been molecularly confirmed to carry a large homozygous deletion of the FERMT1 gene. To date, over 70 FERMT1 gene mutations have been reported (Lai-Cheong et al., 2007; Mansur et al., 2007; Has et al., 2008; Zhou et al., 2009; Mas-Vidal et al., 2010; Kartal et al., 2015; Youssefian et al., 2015). Many mutations are single nucleotide substitutions, including missense mutations, nonsense mutations, etc.
The predicted protein structure of kindlin-1 encoded by FERMT1 gene reveals several domains of interest, in particular the FERM domain which is interrupted by a pleckstrin homology (PH) domain (Jobard et al., 2003; Yates et al., 2012). The protein is expressed at high levels in epithelia, in particular in the epidermis and the gastro-intestinal tract (Sadler et al., 2006). Loss of kindlin-1 would interrupt the organization and anchorage of the actin cytoskeleton to integrin-associated platforms. To our knowledge, our patient carried the largest genomic deletion of the FERMT1 gene, spanning 17 kb from intron 1 to intron 6. We suspected that it was possible that this deletion might lead to a complete loss of exon 1 to exon 7 (1–319 aa). Moreover, it is noteworthy that the deletion range contained the translation initiation site (TIS) ATG located in exon 2. We used ORF (open reading frame) Finder (http://www.ncbi.nlm.nih.gov/gorf/ orfig.cgi) to predict the complementary DNA (cDNA) sequences lacking in the original TIS for potential protein encoding segments. The predicted TIS might be the new start codon, which was 690 bp in length and encoded a 230-aa protein. The predicted protein starting at ATG in exon 7 was 447 aa shorter than normal kindlin-1. It is therefore presumed that the truncated kindlin-1 fragment might lead to elimination of the entire N-terminal FERM domain and most of the PH domain of kindlin-1. It is also possible that no kindlin-1 is expressed at all. It is a pity that we cannot perform a mutant protein expression analysis since no further samples of the patient’s skin were available.
During the repair of double strand breaks (DSBs) in DNA, several different mechanisms are involved, including non-homologous end-joining (NHEJ), microhomology-mediated replication-dependent recombination (MMRDR), and homologous recombination (HR) (including non-allelic homologous recombination (NAHR) and single strand annealing (SSA)) (Chen et al., 2010; Schipler and Iliakis, 2013). Large genomic deletions of the FERMT1 gene can often occur through erroneous recombination of non-allelic homologous repetitive elements, such as Alu sequences (Has et al., 2006). By blasting these with the wide sequences, we found that the 5' and 3' breakpoints had 14 bp identical sequences. Meanwhile, analysis of the intron sequence repeat elements by the RepeatMasker program revealed that the 5' and 3' breakpoints were located in the AluSg and AluSp elements, respectively. Thus, we suspected that the deletion breakpoints embedded in Alu repeats might be mediated by Alu-mediated HR. Zhou et al. (2009) assumed that 3017-bp deletion in the FERMT1 gene may be caused by matrix attachment regions (MARs) (the short homologous sequence GA and the sequence TTTAAA), rather than by the Alu/Alu HR.
To date, the genomic Sanger sequencing has been the main detection method to screen the FERMT1 mutation for KS patients, since the majority of FERMT1 mutations are single nucleotide substitutions, including missense mutations, nonsense mutations, etc. However, in some cases, FERMT1 mutations could not be disclosed using the methods outlined above. In view of the presence of many repeat sequences in introns of the FERMT1 gene, we suspected that a certain proportion of cases might be due to large scale deletion of this gene. As more KS patients were confirmed with large deletion of the FERMT1 gene (Has et al., 2008; Zhou et al., 2009), many researches revealed that a rational mutation detection strategy of FERMT1 mutations would be necessary for the genetic diagnosis of KS patients (Takeichi et al., 2015). Thus, we proposed that a copy number analysis of FERMT1 gene might be an optimal procedure for the patients not fully confirmed by genomic Sanger sequencing. This could help us to preliminarily determine the deletion range. Subsequently, the 5' and 3' breakpoints would be sublocalized by PCR and direct sequencing. Primers designed near the two breakpoints would help us to confirm the deleted sequences.
Our study identified a new large deletion mutation of FERMT1 gene, which might expand the mutation database. Moreover, based on the experience of detecting the mutation, we would emphasize that a rational diagnostic procedure of FERMT1 gene mutations screening would be advisable.
Acknowledgements
We are grateful to the patient and her family for their participation in this study.
Footnotes
Compliance with ethics guidelines: Ying GAO, Jin-li BAI, Xiao-yan LIU, Yu-jin QU, Yan-yan CAO, Jian-cai WANG, Yu-wei JIN, Hong WANG, and Fang SONG declare that they have no conflict of interest.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from the patient for being included in the study. Additional informed consent was obtained from the patient for whom identifying information is included in this article.
References
- 1.Chen JM, Cooper DN, Ferec C, et al. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol. 2010;20(4):222–233. doi: 10.1016/j.semcancer.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Has C, Bruckner-Tuderman L. Molecular and diagnostic aspects of genetic skin fragility. J Dermatol Sci. 2006;44(3):129–144. doi: 10.1016/j.jdermsci.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Has C, Wessagowit V, Pascucci M, et al. Molecular basis of Kindler syndrome in Italy: novel and recurrent Alu/Alu recombination, splice site, nonsense, and frameshift mutations in the KIND1 gene. J Invest Dermatol. 2006;126(8):1776–1783. doi: 10.1038/sj.jid.5700339. [DOI] [PubMed] [Google Scholar]
- 4.Has C, Yordanova I, Balabanova M, et al. A novel large FERMT1 (KIND1) gene deletion in Kindler syndrome. J Dermatol Sci Dec. 2008;52(3):209–212. doi: 10.1016/j.jdermsci.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Herz C, Aumailley M, Schulte C, et al. Kindlin-1 is a phosphoprotein involved in regulation of polarity, proliferation, and motility of epidermal keratinocytes. J Biol Chem. 2006;281(47):36082–36090. doi: 10.1074/jbc.M606259200. [DOI] [PubMed] [Google Scholar]
- 6.Jobard F, Bouadjar B, Caux F, et al. Identification of mutations in a new gene encoding a FERM family protein with a pleckstrin homology domain in Kindler syndrome. Hum Mol Genet. 2003;12(8):925–935. doi: 10.1093/hmg/ddg097. [DOI] [PubMed] [Google Scholar]
- 7.Kartal D, Borlu M, Has C, et al. A novel mutation in the FERMT1 gene in Turkish siblings with Kindler syndrome. J Eur Acad Dermatol Venereol, in press. 2015 doi: 10.1111/jdv.13163. [DOI] [PubMed] [Google Scholar]
- 8.Kern J, Herz C, Haan E, et al. Chronic colitis due to an epithelial barrier defect: the role of kindlin-1 isoforms. J Pathol. 2007;213(4):462–470. doi: 10.1002/path.2253. [DOI] [PubMed] [Google Scholar]
- 9.Kindler T. Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy. Br J Dermatol. 1954;66(3):104–111. doi: 10.1111/j.1365-2133.1954.tb12598.x. [DOI] [PubMed] [Google Scholar]
- 10.Lai-Cheong JE, Liu L, Sethuraman G, et al. Five new homozygous mutations in the KIND1 gene in Kindler syndrome. J Invest Dermatol. 2007;127(9):2268–2270. doi: 10.1038/sj.jid.5700830. [DOI] [PubMed] [Google Scholar]
- 11.Mansur AT, Elcioglu NH, Aydingoz IE, et al. Novel and recurrent KIND1 mutations in two patients with Kindler syndrome and severe mucosal involvement. Acta Derm Venereol. 2007;87(6):563–565. doi: 10.2340/00015555-0314. [DOI] [PubMed] [Google Scholar]
- 12.Mas-Vidal A, Miñones-Suárez L, Toral JF, et al. A novel mutation in the FERMT1 gene in a Spanish family with Kindler’s syndrome. J Eur Acad Dermatol Venereol. 2010;24(8):978–979. doi: 10.1111/j.1468-3083.2009.03554.x. [DOI] [PubMed] [Google Scholar]
- 13.Sadler E, Klausegger A, Muss W, et al. Novel KIND1 gene mutation in Kindler syndrome with severe gastrointestinal tract involvement. Arch Dermatol. 2006;142(12):1619–1624. doi: 10.1001/archderm.142.12.1619. [DOI] [PubMed] [Google Scholar]
- 14.Schipler A, Iliakis G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013;41(16):7589–7605. doi: 10.1093/nar/gkt556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel DH, Ashton GH, Penagos HG, et al. Loss of kindlin-1, a human homolog of the Caenorhabditis elegans actin-extracellular-matrix linker protein UNC-112, causes Kindler syndrome. Am J Hum Genet. 2003;73(1):174–187. doi: 10.1086/376609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeichi T, Liu L, Fong K, et al. Whole-exome sequencing improves mutation detection in a diagnostic epidermolysis bullosa laboratory. Br J Dermatol. 2015;172(1):94–100. doi: 10.1111/bjd.13190. [DOI] [PubMed] [Google Scholar]
- 17.Yates LA, Lumb CN, Brahme NN, et al. Structural and functional characterization of the kindlin-1 pleckstrin homology domain. J Biol Chem. 2012;287(52):43246–43261. doi: 10.1074/jbc.M112.422089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youssefian L, Vahidnezhad H, Barzegar M, et al. The Kindler syndrome: a spectrum of FERMT1 mutations in Iranian families. J Invest Dermatol. 2015;135(5):1447–1450. doi: 10.1038/jid.2015.9. [DOI] [PubMed] [Google Scholar]
- 19.Zho C, Song S, Zhang J, et al. A novel 3017-bp deletion mutation in the FERMT1 (KIND1) gene in a Chinese family with Kindler syndrome. Br J Dermatol. 2009;160(5):1119–1122. doi: 10.1111/j.1365-2133.2009.09052.x. [DOI] [PubMed] [Google Scholar]