

Abstract
A new series of novel opioid ligands have been designed and synthesized based on the 4-anilidopiperidine scaffold containing a 5-substituted tetrahydronaphthalen-2yl-methyl group with different N-phenyl-N-(piperidin-4-yl)propionamide derivatives to study the biological effects of these substituents on μ and δ opioid receptor interactions. Recently our group reported novel 4-anilidopiperidine analogues, in which several aromatic ring-contained amino acids were conjugated with N-phenyl-N-(piperidin-4-yl)propionamide and examined their biological activities at the μ and δ opioid receptors. In continuation of our efforts in these novel 4-anilidopiperidine analogues, we took a peptidomimetic approach in the present design, in which we substituted aromatic amino acids with tetrahydronaphthalen-2yl-methyl moiety with amino, amide and hydroxyl substitutions at the 5th position. In in vitro assays these ligands, showed very good binding affinity and highly selective towards the μ opioid receptor. Among these, the lead ligand 20 showed excellent binding affinity (2 nM) and 5000 fold selectivity towards the μ opioid receptor, as well as functional selectivity in GPI assay (55.20 +/− 4.30 nM) and weak or no agonist activities in MVD assays. Based on the in vitro bioassay results the lead compound 20 was chosen for in vivo assessment for efficacy in naïve rats after intrathecal administration. Compound 20 was not significantly effective in alleviating acute pain. This discrepancy between high in vitro binding affinity, moderate in vitro activity, and low in vivo activity may reflect differences in pharmacodynamics (i.e. engaging signaling pathways) or pharmacokinetics (i.e. metabolic stability). In sum, our data suggest that further optimization of this compound 20 is required to enhance in vivo activity.
Keywords: Opioids, Pain, Opioid receptors, 4-anilidopiperidines
Graphical Abstract
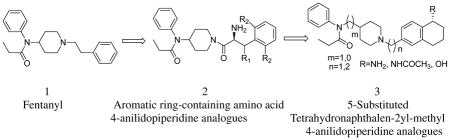
1. Introduction
Chronic pain is one of the major public health problems in the world, and more than one hundred million people in the United States alone are debilitated by chronic pain during their life time. Unfortunately, current treatments for pain are only partially effective, and many cause life style altering, debilitating, or dangerous side effects. Opioid receptors are an important class of GPCRs which modulate analgesic effects in humans. Among the three opioid receptors, MOR receptors are the most important receptor target for almost all commercially available opioid agonists.1 The other two opioid receptors (δ and κ) have also a role in analgesia but have other limitations. Opioids have been used throughout history for their antinociceptive activities. The presently available opioids have been effective in pain management, but they have restricted use because of several severe side effects including respiratory depression, constipation, analgesic tolerance, physical dependence, and addiction liabilities. Great efforts have been made to develop novel opioid analgesics2–13 and this has become a hot topic in medicinal chemistry. Therefore, there is a current need for novel opioid ligands that retain analgesic properties and devoid of side effect profile.
Since the discovery of fentanyl6 medicinal chemistry efforts have yielded thousands of 4-anilidopiperidine analogues. The 4-anilidopiperidines have a prominent place due to their high potency, low cardiovascular toxicity, fast onset, and short duration of action. The synthesis of new fentanyl analogues with high potency but reduced side effects is still one of the challenges of opioid research, and within this context, many 4-anilidopiperidines have been synthesized.8–14 Bagley et al14 summarized in great detail the evolution of the 4-anilidopiperidine class of opioid analgesics and structure-activity relationships of fentanyl analogues. Here we describe our approach and SAR of novel 4-anilidopiperidine analogues and their biological activities at μ and δ opioid receptors.
Portoghese et al,15 incorporated enkephalin peptides into fentanyl, and synthesized 1-and 2- substituted fentanyl analogues. The resulting analogues showed very weak or no opioid activity. For the last decade our laboratory has been working on novel opioid ligands based on the 4-anilidopiperidine and enkephalin conjugate analogues.16–20 Recently our group reported a structural requirement of the 4-anilidopiperidine analogues16 in which the phenethyl group of fentanyl was replaced by several aromatic ring-containing amino acids. The biological effects of these analogues showed a broad range of affinity towards μ and δ opioid receptors. In continuation of our efforts in these novel 4-anilidopiperidine analogues in the present design (figure 1), we took a peptidomimetic approach; where in the phenethyl group is replaced by tetrahydronaphthalen-2yl methyl moiety having amino, acetamide and hydroxyl substitutions at the 5th position. Researchers at Neurosearch21 reported a design, synthesis and pharmacological evolution of 1-phenethylpiperidine derivatives, and their designs are based on N-phenyl-N-(piperidin-4-ylmethyl)propionamide moiety. Initially our designs are based on the 5-amino tetrahydronaphthalen-2yl-methyl group with N-phenyl-N-(piperidin-4-ylmethyl)propionamide derivatives. Further we expanded our design strategy to (figure 1) 5-substituted (amine, amide and hydroxyl) tetrahydronapthalene methyl moiety with different 4-anilidopiperidine cores such as N-phenyl-N-(piperidin-4-ylmethyl)propionamide and N-phenyl-N-(piperidin-4-yl)propionamide. Our interest here is to study the effect of the additional hydrophobic cyclohexyl group with hydroxyl and amine functional group and variation in the 4-anilidopiperidine core on opioid binding and selectivity.
Figure 1.

Design principle of 5-substiuted tetrahydronaphthalen-2yl-methyl 4-anilidopiperidine analogues
2. Chemistry
2.1 Amine and amide substituted 4-anilidopiperidine analogues (8-12)
Here we described general synthetic method for the preparation of the title compounds 8-12 (scheme 1). The 4-anilidopiperidine analogues 8-12 were synthesized according to Scheme 1. The intermediates 1, 3 and 4 were prepared following synthetic routes described in the literature, 21,22 and the intermediate 2 is commercially available. The treatment of intermediates (1 and 2) with iodo compound 3 in the presence of potassium carbonate in acetone gave the Boc-protected analogues 5 and 7 respectively. The desired compound 6 was prepared by a reductive amination of intermediate 2 with aldehyde 4. The Boc-protected analogues 5, 6 and 7 were converted to the corresponding amine trifluoroacetate derivatives 8, 9 and 10 by treating with 50% trifluoroacetic acid in dichloromethane. The amine trifluoroacetate derivatives 8 and 10 were transformed into the acetamide derivatives 11 and 12 by treating with acetyl chloride in the presence of triethyl amine in dichloromethane.
Scheme 1.

Preparation of amine and amide substituted 4-anilidopiperidine analogues.
Reagents and conditions: (a) K2CO3, Acetone, 4h; (b) Na(OAc)3BH, DCE, few drops of acetic acid, 12h; (c) 50% Trifluoroacetic acid in DCM, 0 °C to room temperature, 2h; (d) Acetyl Chloride, Et3N, 0 °C to room temperature, 3h.
2.2 Hydroxyl substituted 4-anilidopiperidine analogues (20-21)
The hydroxy substituted 4-anilidopiperidine analogues 20 and 21 were prepared by a route shown in Scheme 2. The S-alcohol 14 was prepared using the Corey-Bakshi-Shibata oxazaborolidine reduction23 previously reported in the literature. Treating S-alcohol 14 with 3, 4-Dihydro-2H-pyran in presence of PPTS (pyridinium p-toluenesulfonate) in dichloromethane gave the tetrahydropyran protected alcohol 15 in good yield. The protected alcohol 15 upon reduction with lithium aluminium hydride in tetrahydrofuran afforded the alcohol 16, and when followed by treatment with triphenylphosphine, imidazole and I2 gave the iodo compound 17. The treatment of iodo compound 17 with different piperidine analogues (1 and 2) in presence of potassium carbonate in acetone obtained the different 4-anildiopiperidine analogues 18 and 19. Finally the hydroxy substituted analogues 20 and 21 were achieved by deprotection of tetrahydropyranyl group with PPTS (pyridinium p-toluenesulfonate) in methanol at reflux temperature for 3 h.
Scheme 2.

Preparation of hydroxyl substituted 4-anilidopiperidine analogues.
Reagents and conditions: (a) (R)-2-methyl-CBS-oxazaborolidine, BH3-SMe2,Toluene, THF, −15 °C, 5h; (b) DHP, PPTS, DCM, 4h; (c) 1N Lithium aluminium hydride in THF, 0 °C to room temperature, 12h; (d) Ph3P, Imidazole, I2, 1:1 Diethyl ether/DCM, 1h; (e) K2CO3, Acetone, 4h; (f) PPTS, MeOH, reflux temperature, 3h.
2.3 Phenyl substituted 4-anilidopiperidine analogues (38-40)
Scheme 3 outlines the procedures used to synthesize the phenyl substituted 4-anilidopiperidine analogues 38, 39 and 40. Reductive amination of different substituted phenylamine compounds 23, 24 and 25 with 1-benzylpiperidin-4-one using sodium triacetoxyborohydride in dichloroethane gave the desired compounds 26-28. The compounds 29, 30 and 31 were obtained from the respective amines by the treatment with propionyl chloride under basic conditions. Treatment of 29, 30 and 31 with 1-chloroethylchloroformate followed by refluxing the resulting carbamate in methanol afforded the N-debenzylated products 32, 33 and 34.
Scheme 3.

Preparation of phenyl substituted 4-anilidopiperidine analogues.
Reagents and conditions: (a) Na(OAc)3BH, Na2SO4, DCM, 12h; (b) Propionyl chloride, Et3N, DCM, 0 °C to room temperature, 4h; (c) 1-Chloroethyl chloroformate, 70 °C, DCE, 2h; (d) MeOH, reflux temperature; (e) K2CO3, acetone, 4h; (f) 50% TFA in DCM, 0 °C to room temperature.
The piperidine analogues 32, 33 and 34 were treated with iodo compound 3 in presence of potassium carbonate in acetone which yielded the Boc-protected analogues 35, 36 and 37. The subsequent removal of Boc-group provided the final 4-anilidopiperidine analogues 38, 39 and 40 in good yield. The final compounds were triturated with diethyl ether to get pure compounds.
3. Results and discussion
A series of novel 4-anilidopiperidine derivatives have been synthesized and tested for biological activities at the μ and δ opioid receptors, and the results are summarized below. The opioid binding affinities at the human δ opioid receptor (hDOR) and the rat μ opioid receptor (rMOR) were determined by competition analysis against [3H] DPDPE (δ) and [3H] DAMGO (μ) respectively using membrane preparations from transfected HN9.10 cells that constitutively express the respective receptors.24 Functional activities for δ and μ opioid receptors were evaluated in stimulated isolated mouse vas deferens (MVD, δ) and guinea pig isolated ileum (GPI, μ) bioassays, respectively, as previously published.25 The binding affinity results indicate that all these compounds are highly selective towards the μ opioid receptor. The compounds 8, 11 and 21 having amine, acetamide and hydroxyl substitution at the 5th position of 1,2,3,4 tetrahydronapthalene methyl group with N-phenyl-N-(piperidin-4-ylmethyl)propionamide core showed moderate binding affinity of 580 nM, 460 nM and 190 nM towards μ opioid receptor, respectively; poor binding affinity towards δ opioid receptor was observed. Replacement of amino group with hydroxy substitution increased the binding affinity towards the μ opioid receptor by three fold. In our further designs, compounds 10, 12, and 20, we replace the N-phenyl-N-(piperidin-4-ylmethyl) propionamide moiety with N-phenyl-N-(piperidin-4-yl) propionamide with amino, acetamide, and hydroxyl group substituted at the 5th position of the 1,2,3,4 tetrahydronapthalene moiety. Compounds 10 and 12 showed good binding affinity of 40 nM and 50 nM respectively, thus exhibiting 250 fold selective towards the μ opioid receptor. Compound 20 showed excellent binding affinity 2 nM with a 5000 fold selectivity towards the μ opioid receptor. The structural difference between compounds 8 and 10, 11 and 12 and 21 and 20 is the methylene group next to the piperidine ring. The compounds 10, 12 and 20 without the methylene group, show very good binding affinity compared to their methylene containing counter parts 8, 11 and 21.
In this study we achieved the lead compound 20 with similar binding affinity (2 nM) as fentanyl (3.3 nM) at the μ opioid receptor. The binding affinities and functional activity of fentanyl molecule (Ki 5.9±1.4 and 568±159 nM)10 (3.45±0.45 and 9.45±4.05 nM)15 at μ and δ opioid receptors respectively. In our structure activity studies the lead compound 20 which has similar binding affinity of fentanyl at μ opioid receptor, and highly selective (5000 fold) in binding affinity as well as in functional selectivity towards the μ opioid receptor. Based on the design of 10, we further designed and synthesized analogues with fluoro, chloro and 3-methoxy substitutions on the N-phenyl aromatic group. The resulted ligands 38, 39 and 40 which showed moderate binding affinity towards the μ opioid receptor and very weak affinity towards the δ opioid receptor. From this SAR we concluded that substitution on the N-phenyl group is not well tolerated, and the fluoro substitution is better compared to other substitutions on the N-phenyl group.
Opioid agonist activities of all 4-anilidopiperidine analogues were evaluated in MVD and GPI assays. In case of amine and acetamide substituted 4-anilidopiperidine analogues 8-12 and 38-40 agonist activities in the MVD and the GPI assays were very low and did not correlate with the binding affinities at both μ and δ opioid receptors. Only the hydroxyl substituted 4-anilidopiperidine analogues 21 and 20 showed moderate to good agonist activities at GPI assays, and binding affinities very much correlated with the functional activity. The hydroxyl substituted analogue 21 showed moderate agonist activity(600+/−90 nM) in GPI assays, and the ligand 20 showed potent agonist activity (55+/−4 nM) in GPI assay. Similarly, both ligands 20 and 21 exhibit weak agonist activities in MVD assay that is correlated with their binding affinities at DOR.
Based on the in vitro bioassay results showing promising μ opioid activities, we chose to evaluate the lead compound 20 for in vivo antinociceptive activity. Using a rat model of acute nociception induced by application of radiant heat to the planar surface of the hind-paw of naïve animals, we determined the efficacy of 20 after spinal administration (0.1, 1, 3, 10, and 30 μg; n=5–11/treatment). Intrathecal administration of ligand 20 did not significantly raise paw withdrawal latencies (PWLs) at any dose evaluated compared to baseline values (p > 0.05, figure 2). Calculation of the area under the curve (AUC) confirmed a lack of efficacy of compound 20 against acute pain in non-injured rats. These in vivo observations after spinal administration of compound 20 are discordant with the high affinity and moderate potency in vitro. Detailed in vivo experimental procedures are given in the experimental section.
Figure 2.

(a) Compound 20 was evaluated in SD rats using a radiant heat assay (b) Compound 20 dose-dependency was assessed by constructing a dose response curve.
4. Conclusions
A series of novel opioid ligands designed and synthesized based on 4-anilidopiperidine analogues containing 5-substituted tetrahydronaphthalen-2yl-methyl group with different 4-anilidopiperidine core moieties. These 4-anilidopiperidine analogues showed good binding affinity and highly selective towards μ opioid receptor. 5-substituted tetrahydronaphthalen-2yl- methyl group with N-phenyl-N-(piperidin-4-ylmethyl)propionamide analogues 8, 9, 11 and 21 showed moderate binding affinity and very weak agonist activities at μ and δ receptors, and with similar moiety the amine, acetamide and hydroxyl substitution at the 5th position with N-phenyl-N-(piperidin-4-yl)propionamide analogues 10, 12 and 20 showed good binding affinity and high selectivity towards the μ opioid receptor. In the case of the amine and the acetamide compounds 10 and 12, agonist activities are very low in the MVD and the GPI assays which did not correlate with the binding affinities at both μ and δ opioid receptors. On the contrary, the hydroxyl substituted 4-anilidopiperidine analogues 20 and 21 are highly selective towards the MOR and their agonist activities correlate with their binding affinities. The lead compound 20 is highly selective (5000 fold) towards the MOR receptor in binding affinity as well as selective in functional activity at GPI (μ) assays. The ligands 38, 39 and 40 substitutions on the N- phenyl group with fluoro, chloro and methoxy are not tolerated and these ligands showed very weak binding affinity. Based on the in vitro bioassay results the lead compound 20 was chosen for in vivo assessment for efficacy in naïve rats after intrathecal administration. In this assay of acute antinociception, the direct activation of central opioid receptors at the spinal level those are often required to block nociceptive transmission. Compound 20 was not significantly effective in alleviating acute pain. This discrepancy between high in vitro binding affinity, moderate in vitro activity, and low in vivo activity may reflect differences in pharmacodynamics (i.e. engaging signaling pathways) or pharmacokinetics (i.e. metabolic stability). In sum, our data suggest that further optimization of this compound 20 is required to enhance in vivo activity.
Experimental section
General considerations
All reactions were performed under N2 unless otherwise noted. All 1H NMR and 13C NMR were recorded at 500 and 125 MHz, respectively, on a Bruker DRX 500. Chemical shifts were referred to TMS as internal standard in the case of CDCl3 solution and to the residual proton signal of DMSO at 2.5 ppm in the case of DMSO-d6 solution. The following abbreviations were used in reporting spectra: s=singlet, d=doublet, t=triplet, m=multiplet. Mass spectra were recorded on a Thermo Fisher LCQ ion trap mass spectrometer. High-resolution mass spectra (HRMS) were obtained using a Bruker Apex ion cyclotran resonance mass spectrometer in ESI positive mode. Unless otherwise mentioned, all of the solvents used were of laboratory reagent grade. Usually, the flash chromatography was performed using 100–200 mesh silica gel. All of the organic extracts were dried over sodium sulfate after work up. For analytical reversed-phase HPLC a Vydac 218-TP54 column (5 _ 250 mm) was used with a l gradient of 10–90% acetonitrile in 0.1% TFA/H2O over 40 min at a flow rate of 1 mL/min.
In vivo Assay
Adult male Sprague-Dawley rats (225–300 g; Harlan, Indianapolis, IN) were kept in a temperature-controlled environment with lights on 07:00–19:00 with food and water available ad libitum. All animal procedures were performed in accordance with the policies and recommendations of the International Association for the Study of Pain, The National Institutes of Health, and with approval from the Animal Care and Use Committee of the University of Arizona for the handling and use of laboratory animals.
Surgical methods
Rats were anesthetized (ketamine/xylazine anesthesia, 80/12 mg/kg i.p.; Sigma-Aldrich) and placed in a stereotaxic head holder. The cisterna magna was exposed and incised, and an 8-cm catheter (PE-10; Stoelting) was implanted as previously reported,26 terminating in the lumbar region of the spinal cord. Catheters were sutured (3-0 silk suture) into the deep muscle and externalized at the back of the neck. Seven days after implantation of an indwelling cannula, vehicle (10% DMSO:10% Tween 80:80% saline) and ligand 20 (1, 3, 10, and 30 μg; n=6–9/treatment) were injected in a 5μL volume followed by a 9μL saline flush. Catheter placement was verified at completion of experiments.
Behavioral test used
Paw-flick latency27 was determined as follows. Rats were allowed to acclimate to the testing room for 30 minutes prior to testing. Basal paw withdrawal latencies (PWLs) to an infrared radiant heat source were measured and ranged between 16.0 and 20.0 seconds. A cutoff time of 33.0 seconds was used to prevent tissue damage. After a single, intrathecal injection of ligand 20 (i.t.) or vehicle, PWLs were re-assessed 7 times over a duration of to 4 hours.
(R)-tert-butyl (6-((4-((N-phenylpropionamido)methyl)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (5 scheme 1)
To a solution of N-phenyl-N-(piperidin-4-yl)propionamide (0.19 g, 0.77, 1.5 eq) in acetone at room temperature was added K2CO3 (0.1 g, 0.77mmol 1.5 eq) and followed by iodo compound 3 (0.2 g, 0.51 mmol 1.0 eq). The resultant reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was filtered through whatman filter paper, and solvent was removed from the filtrate. The residue obtained upon evaporation of solvent was chromatographed over silica gel and eluted with 60% ethyl acetate: hexane to give the title compound Boc-protected 4-anilidopiperdiine analogue 5 (0.2 g, 76% yield.) as a white colored solid. MS (ESI) m/z (M + H)+: 506, 1H NMR (499 MHz, Chloroform-d) δ 7.42 – 7.37 (m, 2H), 7.36 – 7.29 (m, 1H), 7.25 (d, J = 7.4 Hz, 1H), 7.16 – 7.12 (m, 2H), 7.09 – 7.06 (m, 1H), 7.00 (s, 1H), 4.82 (d, J = 7.7 Hz, 1H), 4.74 (d, J = 8.8 Hz, 1H), 3.62 (d, J = 7.1 Hz, 2H), 3.41 (s, 2H), 2.83 (d, J = 11.1 Hz, 2H), 2.78-2.66 (m, 2H), 2.03 (q, J = 7.2 Hz, 3H), 1.89 (m, 2H), 1.84 – 1.74 (m, 4H), 1.67 – 1.58 (m, 2H), 1.48 (s, 9H), 1.35 (t, J = 11.5 Hz, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.95, 155.40, 143.26, 137.16, 135.82, 129.60, 129.54, 128.41, 128.12, 127.62, 127.05, 79.23, 77.25, 77.20, 77.00, 76.95, 76.75, 62.83, 54.70, 53.17, 48.43, 34.40, 30.54, 29.90, 29.22, 28.43, 27.89, 19.91, 9.66.
(R)-tert-butyl (6-(2-(4-(N-phenylpropionamido)piperidin-1-yl)ethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (6 scheme 1)
To a solution of (R)-tert-butyl 6-(2-oxoethyl)-1,2,3,4-tetrahydronaphthalen-1-yl carabamate 4 (0.18 g, 0.62 mmol, 1eq) and 4-(N-phenylpropionamido)piperidin-1-ium chloride 2 (0.36 g, 1.55 mmol, 2.5 eq) in dichloroethane (10 mL) was added sodium triacetoxyborohydride (0.26 g, 1.24 mmol, 2 eq). After stirring overnight at room temperature, the reaction solution was diluted with ethyl acetate and washed with saturated sodium bicorbonate and brine. The organic phase was dried over sodium sulfate and evaporated to dryness in vacuo. The crude product was purified by flash chromatography 70% ethyl acetate:hexane to give the title compound 6 as a white powder (0.22 g, 70% yield) MS (ESI) m/z (M + H)+: 506, 1H NMR (499 MHz, Chloroform-d) δ 7.42 – 7.31 (m, 3H), 7.22 (d, J = 7.9 Hz, 1H), 7.10 – 7.04 (m, 2H), 6.95 (d, J = 8.0 Hz, 1H), 6.87 – 6.82 (m, 1H), 4.79 (m, 1H), 4.76 – 4.63 (m, 2H), 3.01 – 2.93 (m, 2H), 2.77 – 2.60 (m, 4H), 2.54 – 2.45 (m, 2H), 2.20 – 2.10 (m, 2H), 2.02 – 1.96 (m, 1H), 1.92 (q, J = 7.4 Hz, 2H), 1.79 (m, 4H), 1.46 (s, 9H), 1.41 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.46, 155.38, 139.07, 138.80, 137.35, 134.99, 130.40, 129.23, 129.08, 128.72, 128.20, 126.51, 79.20, 77.26, 77.21, 77.00, 76.75, 60.41, 53.05, 52.10, 48.36, 33.33, 30.59, 30.54, 29.19, 28.49, 28.42, 19.90, 9.59.
(R)-tert-butyl (6-((4-(N-phenylpropionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (7 scheme 1)
Prepared as described for 5 from (R)-tert-butyl (6-(iodomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate 3 (0.25 g, 0.64 mmol, 1eq) and 4-(N-phenylpropionamido)piperidin-1-ium chloride 2 (0.22 g, 0.96 mmol, 1eq) in 10 mL of acetone and 0.26 g of K2CO3 afforded the title compound 7 (0.25 g, 78% of yield) as a light brown colored solid. MS (ESI) m/z (M + H)+: 492. 1H NMR (499 MHz, Chloroform-d) δ 7.42 – 7.33 (m, 3H), 7.23 (d, J = 7.9 Hz, 1H), 7.04 (m, 3H), 6.93 (s, 1H), 4.79 (t, J = 6.5 Hz, 1H), 4.72 (d, J = 8.8 Hz, 1H), 4.65 (m, 1H), 3.37 (s, 2H), 2.86 (d, J = 11.8 Hz, 2H), 2.78 – 2.61 (m, 2H), 2.08 (m, 2H), 2.00 (m, 1H), 1.90 (q, J = 7.5 Hz, 2H), 1.83 – 1.69 (m, 4H), 1.46 (s, 9H), 1.43 – 1.32 (m, 2H), 1.00 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.4, 155.3, 138.9, 137.1, 136.9, 135.9, 130.3, 129.5, 129.1, 128.4, 128.1, 127.0, 79.1, 62.6, 53.0, 52.4, 48.4, 30.5, 29.1, 28.4, 28.4, 19.8, 9.5.
(R)-N-((1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)methyl)-N-phenylpropionamide (8 scheme 1)
To an ice-cold stirred solution of the 5 (0.26 g, 0.51 mmol, 1 eq) in dichloromethane (5 mL) was added trifluoroacetic acid (5 mL). The resulting reaction mixture was stirred for 2 h, and the solvent was stripped of under reduced pressure. The resultant residue was washed with diethyl ether a couple of times and dried afforded the amine trifluoroacetate derivative as a white solid (0.2 g, 74.9% of yield) ESI MS m/z 406 (MH)+. HRMS [M + H]+ 406.2855 (theoretical 406.2852); 1H NMR (499 MHz, DMSO-d6) δ 11.10 (d, J = 10.0 Hz, 1H), 8.79 – 8.51 (m, 3H), 7.67 (dd, J = 8.3, 4.6 Hz, 1H), 7.54 – 7.43 (m, 3H), 7.43 – 7.39 (m, 1H), 7.35 (m, J = 7.1 Hz, 3H), 4.42 (q, J = 5.6 Hz, 1H), 4.22 – 4.10 (m, 2H), 3.79 – 3.63 (m, 1H), 3.55 (d, J = 4.9 Hz, 1H), 3.52 – 3.45 (m, 1H), 3.44 – 3.33 (m, 2H), 3.23 (d, J = 11.8 Hz, 2H), 2.85 – 2.68 (m, 3H), 2.09 (m, 1H), 1.95 (m, 4H), 1.83 – 1.69 (m, 2H), 1.68 – 1.51 (m, 3H), 0.95 – 0.82 (t, 3H). 13C NMR (126 MHz, DMSO-d6) δ 173.05, 143.00, 138.41, 134.20, 132.78, 130.17, 130.13, 129.43, 128.73, 128.16, 72.62, 70.98, 66.81, 60.64, 58.85, 53.23, 51.47, 47.98, 44.08, 40.50, 40.43, 40.33, 40.26, 40.17, 40.09, 40.00, 39.83, 39.67, 39.55, 39.50, 32.83, 28.84, 27.77, 27.53, 26.75, 18.61, 10.03.
(R)-N-(1-(2-(5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)ethyl)piperidin-4-yl)-N-phenylpropionamide (9 scheme 1)
Prepared as described for 8 from 6 (R)-tert-butyl (6-((4-((N-phenylpropionamido)methyl)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (0.12 g, 0.23) afforded the title compound 9 as a white solid (0.085 g, 69% of yield). ESI MS m/z 406 (MH)+. HRMS [M + H]+ 406.2851 (theoretical 406.2852); 1H NMR (499 MHz, DMSO-d6) δ 9.63 – 9.46 (m, 1H), 8.32 (d, J = 5.7 Hz, 3H), 7.53 – 7.43 (m, 3H), 7.41 (d, J = 8.0 Hz, 1H), 7.28 – 7.23 (m, 2H), 7.11 (dd, J = 8.1, 1.8 Hz, 1H), 7.05 (s, 1H), 4.75 (m, 1H), 4.40 (q, J = 5.4 Hz, 1H), 3.61 – 3.51 (m, 2H), 3.22 – 3.07 (m, 4H), 2.93 – 2.83 (m, 2H), 2.80 – 2.63 (m, 2H), 2.08 – 1.94 (m, 3H), 1.90-1.80 (m, 4H), 1.73 (m, 1H), 1.60-1.49 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 172.69, 138.61, 138.39, 137.65, 131.71, 130.85, 130.02, 129.94, 129.76, 129.07, 126.93, 72.67, 71.04, 66.89, 60.66, 56.77, 51.31, 49.64, 47.99, 44.22, 40.50, 40.33, 40.24, 40.17, 40.00, 39.83, 39.67, 39.50, 29.52, 28.95, 28.28, 27.89, 27.81, 18.66, 9.97.
(R)-N-(1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-phenylpropionamide (10 scheme 1)
Prepared as described for 8 from 7 (R)-tert-butyl (6-((4-(N-phenylpropionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (0.15 g, 0.30) afforded the title compound 10 as a white solid (0.11 g, 71% of yield). ESI MS m/z 392 (MH)+. HRMS [M + H]+ 392.2696 (theoretical 392.2696); 1H NMR (499 MHz, DMSO-d6) δ 9.76 – 9.45 (m, 1H), 8.38 (d, J = 5.5 Hz, 3H), 7.51 (d, J = 8.0 Hz, 1H), 7.49 – 7.41 (m, 2H), 7.28 (d, J = 8.0 Hz, 1H), 7.24 (s, 1H), 7.21 (d, J = 6.8 Hz, 2H), 4.70 (m, 1H), 4.45 (q, J = 5.5 Hz, 1H), 4.17 (s, 2H), 3.30 (d, J = 10.8 Hz, 2H), 3.11 (q, J = 11.6 Hz, 2H), 2.84 – 2.63 (m, 2H), 2.05 (m, 1H), 1.98 – 1.77 (m, 6H), 1.77 – 1.69 (m, 1H), 1.56 – 1.42 (m, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ172.6, 159.3, 159.1.8, 158.8, 158.5, 138.6, 138.5, 134.3, 132.3, 130.7, 130.1, 129.8, 129.2, 129.0, 128.9, 120.4, 118.1, 115.7, 113.4, 51.3, 51.2, 49.2, 48.0, 28.7, 28.1, 27.7, 27.4, 18.5, 9.8.
(R)-N-((1-((5-acetamido-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)methyl)-N-phenylpropionamide (11 scheme 1)
To a solution of the amine 8 (R)-N-((1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)methyl)-N-phenylpropionamide (0.1 g, 0.24 mmol 1eq) in dry dichloromethane (6 mL) at 0°C under argon atmosphere was added triethylamine (0.085 mL, 0.72 mmol) followed by acetyl chloride (0.019 mL, 0.265 mmol) drop wise. The reaction mixture was stirred at room temperature for 1 h and was worked up by adding water followed by extraction with dichloromethane. The combined organic extracts were washed with water, brine and dried. The residue obtained upon evaporation of solvent was chromatographed over silica gel and eluted with 60% ethyl acetate:hexane to give the acetamide derivative 11 as a light yellow color solid (0.065 g, 59% yield). ESI MS m/z 448 (MH)+. HRMS [M + H]+ 448.2956 (theoretical 448.2958); 1H NMR (499 MHz, Chloroform-d) δ 7.40 (t, J = 7.7 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 7.18 – 7.10 (m, 2H), 7.03 (d, J = 6.9 Hz, 2H), 6.90 (d, J = 8.1 Hz, 1H), 5.77 (dd, J = 11.3, 6.7 Hz, 1H), 3.62 (d, J = 7.1 Hz, 2H), 3.39 (s, 2H), 2.92 – 2.69 (m, 4H), 2.34 – 2.19 (bs, 4H), 2.19 – 2.10 (m, 1H), 2.10 – 1.98 (m, 4H), 1.87 (m, 2H), 1.78 (m, 1H), 1.62 (d, J = 14.3 Hz, 2H), 1.33 (m, J = 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 174.11, 173.91, 143.28, 137.08, 129.87, 129.58, 128.10, 127.60, 127.39, 124.72, 77.25, 77.20, 77.00, 76.75, 62.78, 54.73, 54.14, 53.25, 34.47, 29.95, 29.41, 28.92, 27.87, 27.02, 22.96, 9.65.
(R)-N-(1-((5-acetamido-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-phenylpropionamide (12 scheme 1)
Prepared as described for 11 from 10 (R)-N-(1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-phenylpropionamide (0.17 g, 0.39 mmol) afforded the title compound 12 as a light yellow color solid (0.12 g, 69% of yield). ESI MS m/z 434 (MH)+. HRMS [M + H]+ 434.2801 (theoretical 434.2802); 1H NMR (499 MHz, Chloroform-d) δ 7.42 – 7.34 (m, 3H), 7.16 (d, J = 7.9 Hz, 1H), 7.08 – 7.04 (m, 2H), 7.03 (dd, J = 7.9 Hz, 1H), 6.96 (s, 1H), 5.65 (d, J = 8.4 Hz, 1H), 5.19 – 5.07 (m, 1H), 4.64 (m, 1H), 3.37 (s, 2H), 2.85 (d, J = 11.9 Hz, 2H), 2.80 – 2.64 (m, 2H), 2.08 (t, J = 12.3 Hz, 2H), 2.00 (s, 3H), 1.90 (q, J = 7.4 Hz, 2H), 1.81-1.76 (m, 3H), 1.76 – 1.69 (m, 3H), 1.44 – 1.32 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.44, 169.07, 138.93, 137.36, 137.24, 135.33, 130.38, 129.70, 129.22, 128.54, 128.20, 127.16, 77.25, 77.20, 77.00, 76.75, 62.65, 53.07, 53.04, 52.28, 47.28, 30.55, 30.13, 29.20, 28.51, 23.58, 19.87, 9.61.
(5S)-methyl 5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalene-2-carboxylate (15 scheme 2)
To a solution of (S)-methyl 5-hydroxy-5,6,7,8-tetrahydronaphthalene-2-carboxylate 14 (1.65 g, 8.0 mmol, 1 eq) in dry dichloromethane (20 mL) was added 3,4-Dihydro-2H-pyran (1.0 mL, 12 mmol) followed by pyridinium p-toluenesulfonate (0.2 g, 0.8 mmol, 0.1 eq) under argon atmosphere. The reaction mixture was stirred at room temperature for 4 h, and the reaction mixture was diluted with dichloromethane and washed with water twice and brine and dried. The residue obtained upon evaporation of solvent was chromatographed over silica gel and eluted with 15% ethyl acetate: hexane to afford the protected alcohol 15 as a viscous oil (2.1 g, 90% yield) ESI MS m/z 313 (MNa)+. 1H NMR (499 MHz, Chloroform-d; 1:1 diastereomeric ratio) δ 7.85 – 7.76 (m, 2H), 7.58 (d, J = 8.1 Hz, 0.5 H), 7.36 (d, J = 8.0 Hz, 0.5 H), 5.01 – 4.68 (m, 2H), 4.00 (m, 1H), 3.90 (s, 3H), 3.65 – 3.48 (m, 1H), 2.88 (m, 1H), 2.81 – 2.69 (m, 1H), 2.13 – 1.93 (m, 3H), 1.92 – 1.69 (m, 4H), 1.69 – 1.47 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 167.11, 167.05, 142.16, 141.93, 137.71, 137.54, 130.25, 130.04, 129.21, 128.88, 128.64, 128.61, 126.78, 126.49, 99.29, 95.63, 94.52, 77.25, 77.20, 77.00, 76.74, 73.79, 70.52, 62.90, 62.83, 62.67, 51.99, 51.96, 30.99, 30.91, 30.58, 30.28, 29.03, 28.70, 27.36, 25.39, 25.33, 19.69, 19.67, 18.94, 18.77.
((5S)-5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalen-2-yl)methanol (16 scheme 2)
To an oven dried 3-neck, 100 mL round-bottomed flask equipped with argon inlet/outlet, addition funnel, thermometer, was added THF (20 mL) and LAH (14.74 mL of a 1M solution in THF, 14.74 mmol). The reaction mixture was cooled in an ice-salt bath, and (5S)-methyl 5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalene-2-carboxylate 15 (2.1 g, 7.37 mmol) in THF (15 mL) was added over ca 30 min. The reaction mixture was warmed to room temperature overnight, and then cooled in an ice-salt bath the next morning. Water (1.4 mL) in THF (1.5 mL) was added to the reaction mixture over 3h. Vigorous gas evolution was occurred, 5N sodium hydroxide (1.4 mL) was added over 20 min followed by water (4.2 mL). After stirring for an additional 1 h, the reaction mixture was filtered, and the filtrate was concentrated in vacuo. The residue was reconstituted in methanol and acetonitrile, and concentrated in vacuo again to provide the title compound 16 as a color less liquid (1.7 g, 79% yield), the title compound used without further purification in the subsequent step.
2-(((S)-6-(iodomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)tetrahydro-2H-pyran (17 scheme 2)
To a solution of ((5S)-5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalen-2-yl)methanol 16 (1.3 g, 4.96 mmol) in dichloromethane/ether (1:1, 30 mL) at room temperature were added triphenylphosphine (1.9 g, 7.35 mmol) and imidazole (0.5 g, 7.35 mmol). To this stirred solution was then added iodine (1.86 g, 14.7 mmol). After stirring for 20 min, the reaction was quenched with 10% Na2S2O3 (20 mL) until it became a clear two-phase solution. The aqueous phase was extracted with ether, and combined organic phase was dried over sodium sulfate, filtered, and evaporated to dryness. Flash chromatography silica gel 10% ethyl acetate:hexane afforded the desired product 17 as a light yellow color liquid (1.1 g, 59% yield). ESI MS m/z 395 (MNa)+. 1H NMR (499 MHz, Chloroform-d; 1:1 diastereomeric ratio) δ 7.43 (d, J = 8.0 Hz, 0.5 H), 7.24 – 7.19 (m, 1.0 H), 7.18-7.15 (dd, J = 7.9, 2.0 Hz, 0.5 H), 7.13 – 7.09 (m, 1H), 4.89-4.83 (m, 1H), 4.80 – 4.65 (m, 1H), 4.42 (s, 2H), 4.05-3.95(m, 1H), 3.64 – 3.52 (m, 1H), 2.85 – 2.75 (m, 1H), 2.72-2.63 (m, 1H), 2.08 – 1.81 (m, 4H), 1.80 – 1.69 (m, 2H), 1.67 – 1.50 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 138.23, 138.16, 137.97, 137.95, 137.01, 136.75, 129.95, 129.41, 129.19, 128.90, 126.40, 125.99, 99.04, 95.56, 77.25, 77.00, 76.74, 73.63, 70.51, 62.72, 62.64, 31.10, 31.04, 30.49, 30.30, 29.11, 28.78, 27.56, 25.52, 19.77, 19.70, 18.89, 18.75, 5.95, 5.82.
N-phenyl-N-(1-(((5S)-5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)propionamide (18 scheme 2)
Prepared as described for 5 from 2-(((S)-6-(iodomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)tetrahydro-2H-pyran 17 (0.12 g, 0.33 mmol, 1eq) and 4-(N-phenylpropionamido)piperidin-1-ium chloride 2 (0.15 g, 0.67 mmol, 2eq) in 10 mL of acetone and 0.18 g of K2CO3 afforded the title compound 18 (0.11 g, 69% of yield) as a light yellow color viscous liquid. MS (ESI) m/z (M + H)+: 477. 1H NMR (499 MHz, Chloroform-d; 1:1 diastereomeric ratio) δ 7.42-7.32 (m, J = 7.0, 6.5, 3.4 Hz, 3.5 H), 7.18 (d, J = 7.8 Hz, 0.5H), 7.08-7.03 (m, 2.5 H), 7.01 (dd, J = 7.8, 1.8 Hz, 0.5H), 6.97 – 6.93 (m, 1H), 4.90 – 4.85 (m, 0.5H), 4.85 – 4.75 (m, 1H), 4.70-4.61 (m, 1.5H), 4.00 (m, 1H), 3.62 – 3.50 (m, 1H), 3.38 (s, 2H), 2.94 – 2.83 (m, 2H), 2.82 – 2.72 (m, 1H), 2.72 – 2.61 (m, 1H), 2.13 – 1.79 (m, 8H), 1.79 – 1.65 (m, 4H), 1.65 – 1.44 (m, 4H), 1.45 – 1.31 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.42, 138.96, 137.45, 137.35, 137.30, 137.08, 135.81, 135.55, 130.41, 129.66, 129.36, 129.25, 129.20, 128.78, 128.16, 126.83, 126.40, 98.95, 95.36, 77.25, 77.20, 77.00, 76.94, 76.75, 73.68, 70.56, 62.85, 62.67, 62.55, 53.12, 53.10, 52.33, 52.31, 31.10, 31.06, 30.62, 30.52, 29.23, 28.88, 28.50, 27.64, 25.56, 19.84, 19.61, 18.99, 18.87, 9.60.
N-phenyl-N-((1-(((5S)-5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)methyl)propionamide (19 scheme 2)
Prepared as described for 5 from 2-(((S)-6-(iodomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)oxy)tetrahydro-2H-pyran 17 (0.15 g, 0.40 mmol, 1eq) and 4-((N-phenylpropionamido)methyl)piperidin-1-ium chloride 2 (0.22 g, 0.80 mmol, 2eq) in 10 mL of acetone and 0.22 g of K2CO3 afforded the title compound 19 (0.10 g, 62% of yield) as a light brown color viscous liquid. MS (ESI) m/z (M + H)+: 491. 1H NMR (499 MHz, Chloroform-d; 1:1 diastereomeric ratio) δ 7.43 – 7.38 (m, 2.5H), 7.36 – 7.31 (m, 1H), 7.20 (d, J = 7.8 Hz, 0.5H), 7.16 – 7.12 (m, 2H), 7.11 – 7.05 (m, 1H), 7.03 – 6.99 (m, 1H), 4.90 (t, J = 3.7 Hz, 0.5H), 4.84 (dd, J = 4.9, 2.9 Hz, 0.5H), 4.81 (t, J = 4.8 Hz, 0.5H), 4.70 (t, J = 4.7 Hz, 0.5H), 4.05 (m, 0.5H), 3.98 (m, 0.5H), 3.66 – 3.52 (m, 3H), 3.47 – 3.37 (m, 2H), 2.90 – 2.75 (m, 3H), 2.68 (m, 1H), 2.03 (q, J = 7.4 Hz, 2H), 1.96 – 1.68 (m, 8H), 1.67 – 1.45 (m, 7H), 1.41 – 1.27 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.83, 173.82, 143.01, 137.40, 137.23, 137.13, 137.06, 135.42, 135.19, 129.54, 129.50, 129.26, 129.11, 128.63, 128.01, 127.53, 126.74, 126.29, 98.96, 95.14, 77.23, 77.18, 76.98, 76.72, 73.63, 70.33, 62.88, 62.64, 62.45, 54.54, 53.10, 53.06, 53.01, 34.28, 30.94, 30.55, 29.81, 29.14, 28.80, 27.78, 27.43, 25.40, 19.78, 19.49, 18.90, 18.72, 9.56.
(S)-N-(1-((5-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-phenylpropionamide (20 scheme 2)
To a solution of 18 (N-phenyl-N-(1-(((5S)-5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)propionamide) (0.13 g, 0.27 mmol) in dry methanol (10 mL) under argon atmosphere was added pyridinium p-toluenesulfonate (0.020 g, 0.08 mmol), and the resulting mixture was stirred at reflux temperature of methanol for 3 h. The reaction mixture was allowed to cool to room temperature and the solvent was evaporated under reduced pressure. The residue obtained upon evaporation of solvent was chromatographed over silica gel and eluted with 5% methanol: dichloromethane to give the title product 20 as a white color sticky solid (0.054 g, 54% yield.) ESI MS m/z 393 (MH)+. HRMS [M + H]+ 393.2534 (theoretical 393.2536); 1H NMR (499 MHz, Chloroform-d) δ 7.42 – 7.35 (m, 3H), 7.33 (d, J = 7.8 Hz, 1H), 7.07-7.05 (m, 3H), 6.97 (s, 1H), 4.75 (s, 1H), 4.65 (m, 1H), 3.38 (s, 2H), 2.92 – 2.82 (m, 2H), 2.80 -2.75 (m, 1H), 2.72 – 2.61 (m, 1H), 2.14 – 2.03 (m, 2H), 1.99 – 1.83 (m, 4H), 1.79 – 1.69 (m, 2H), 1.67-1.57 (m, 3H) 1.39 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ173.4, 138.7, 137.5, 136.9, 130.3, 129.6, 129.2, 128.4, 128.1, 127.1, 67.8, 62.7, 53.4, 53.0, 52.0, 32.2, 30.3, 29.1, 28.4, 18.7, 9.5.
(S)-N-((1-((5-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)methyl)-N-phenylpropionamide (21 scheme 2)
Prepared as described for 20 from 19 N-phenyl-N-((1-(((5S)-5-((tetrahydro-2H-pyran-2-yl)oxy)-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)methyl)propionamide (0.070 g, 0.14 mmol) afforded the title compound 21 as a light yellow colored solid (0.030 g, 52% of yield). ESI MS m/z 407(MH)+. HRMS [M+H]+ 407.2692 (theoretical 407.2693); 1H NMR (499 MHz, Chloroform-d) δ 7.43 – 7.38 (m, 2H), 7.34 (dd, J = 9.1, 7.5 Hz, 2H), 7.17 – 7.10 (m, 3H), 7.04 (s, 1H), 4.77 (t, J = 4.6 Hz, 1H), 3.62 (d, J = 7.1 Hz, 2H), 3.44 (s, 2H), 2.84 (dd, J = 12.2, 8.1 Hz, 2H), 2.80 – 2.76 (m, 1H), 2.74 – 2.65 (m, 1H), 2.08 – 1.85 (m, 6H), 1.83 – 1.73 (m, 1H), 1.68-1.59 (m, 4H), 1.35 (d, J = 12.8 Hz, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ173.9, 143.2, 136.8, 129.5, 128.3, 128.0, 127.5, 127.0, 67.9, 62.8, 54.5, 53.3, 53.1, 34.4, 34.2, 32.3, 29.9, 29.2, 27.8, 18.7, 9.6.
1-benzyl-N-(3,4-difluorophenyl)piperidin-4-amine (26 scheme 3)
N-Benzylpiperidone (1 g, 5.2 mmol, 1eq), 3, 4 difluoroaniline (0.52 mL, 5.2 mmol, 1eq) and sodium sulfate (3.7 g, 26.4 mmol, 5eq) were suspended in dichloromethane (20 mL). Sodium triacetoxyborohydride (1.3 g, 6.33 mmol, 1.2eq) was added and the reaction mixture was stirred at room temperature overnight. Aqueous sodium hydrogen carbonate was added followed by stirring for 30 min. The reaction mixture was extracted with dichloromethane and the combined organic phases were washed with brine, dried over sodium sulfate, filtrated and concentrated in vacuo. The residue obtained upon evaporation of solvent was chromatographed over silica gel and eluted with 35% ethyl acetate:hexane to give the 26 as a light yellow colored solid (1.2 g, 75% yield).
1-benzyl-N-(3,4-dichlorophenyl)piperidin-4-amine (27 scheme 3)
Prepared as described for 26 from N-Benzylpiperidone (1 g, 5.2 mmol, 1eq) and 3,4 dichloroaniline (0.85 g, 5.2 mmol, 1eq) afforded the title compound 27 as a yellow color solid (1.2 g, 68% of yield).
1-benzyl-N-(4-chloro-3-methoxyphenyl)piperidin-4-amine (28 scheme 3)
Prepared as described for 26 from N-Benzylpiperidone (1 g, 5.3 mmol, 1 eq) and 4-Chloro-3-methoxyaniline (0.81 g, 5.3 mmol, 1eq) afforded the title compound 28 as a white color solid (1.32 g, 75% of yield).
N-(1-benzylpiperidin-4-yl)-N-(3,4-difluorophenyl)propionamide (29 scheme 3)
1-benzyl-N-(3,4-difluorophenyl)piperidin-4-amine 26 (1.2 g, 3.64 mmol 1eq) was dissolved in toluene (15 mL). Propionic anhydride (0.93 mL, 7.28 mmol, 2eq) was added and the reaction mixture was heated to reflux temperature overnight. The reaction mixture was poured in to sodium hydroxide (1M, 25 mL) solution and stirred for 30 min. The organic layer was washed with water until pH 7, washed with brine and dried over sodium sulfate, filtrated and concentrated in vacuo to give product (0.95 g, 73% of yield) as a light yellow colored solid.
N-(1-benzylpiperidin-4-yl)-N-(3,4-dichlorophenyl)propionamide (30 scheme 3)
Prepared as described for 29 from 27 1-benzyl-N-(3,4-dichlorophenyl)piperidin-4-amine (1.2 g, 3.58 mmol, 1eq) afforded the title compound 30 as a yellow colored solid (0.85 g, 61% of yield).
N-(1-benzylpiperidin-4-yl)-N-(4-chloro-3-methoxyphenyl)propionamide (31 scheme 3)
Prepared as described for 29 from 28 1-benzyl-N-(4-chloro-3-methoxyphenyl)piperidin-4-amine (0.98 g, 3.0 mmol, 1 eq) afforded the title compound 31 as a white colored solid (0.70 g, 63% of yield).
N-(3,4-difluorophenyl)-N-(piperidin-4-yl)propionamide (32 scheme 3)
N-(3,4-difluorophenyl)-N-(piperidin-4-yl)propionamide 29 (0.95 g, 2.65 mmol, 1eq) was dissolved in dichloromethane (10 mL). 1-Chloroethyl chloroformate (1.43 mL, 13.25 mmol, 5eq) was added and the reaction mixture was heated to reflux temperature for 3 h and then concentrated in vacuo. The resulting residue was dissolved in methanol (10 mL) and heated to reflux temperature for 2 h, followed by concentration in vacuo. Diethyl ether was added and the resulting solid was filtrated and dried. The solid material was dissolved in water, washed with diethyl ether a couple of times, basified with sodium hydroxide (3 M). Extraction with diethyl ether, washing with brine, drying over sodium sulfate and concentration in vacuo gave N-(3,4-difluorophenyl)-N-(piperidin-4-yl)propionamide (0.5 g, 70% of yield) as a light yellow colored solid.
N-(3,4-dichlorophenyl)-N-(piperidin-4-yl)propionamide (33 scheme 3)
Prepared as described for 32 from N-(1-benzylpiperidin-4-yl)-N-(3,4-dichlorophenyl)propionamide (0.67 g, 0.23) afforded the title compound 33 as a light brown colored solid (0.4 g, 78% of yield).
N-(4-chloro-3-methoxyphenyl)-N-(piperidin-4-yl)propionamide (34 scheme 3)
Prepared as described for 32 from N-(1-benzylpiperidin-4-yl)-N-(4-chloro-3-methoxyphenyl)propionamide (0.63 g, 1.63 mmol, 1eq) afforded the title compound 34 as a light brown colored (0.35 g, 72% of yield).
(R)-tert-butyl (6-((4-(N-(3,4-difluorophenyl)propionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (35 scheme 3)
Prepared as described for 5 from (R)-tert-butyl (6-(iodomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate 3 (0.15 g, 0.38 mmol, 1eq) and N-(3,4-difluorophenyl)-N-(piperidin-4-yl)propionamide 32 (0.15 g, 0.58 mmol, 1.5eq) in 10 mL of acetone and 0.1 g of K2CO3 afforded the title compound 35 as a light brown colored solid (0.13 g, 64% of yield). MS (ESI) m/z (M + H)+: 528. 1H NMR (499 MHz, Chloroform-d) δ 7.24 (d, J = 7.9 Hz, 1H), 7.21 – 7.15 (m, 1H), 7.03 (d, J = 8.0 Hz, 1H), 6.96 – 6.90 (m, 2H), 6.83 (m, 1H), 4.80 (t, J = 6.2 Hz, 1H), 4.72 (d, J = 8.9 Hz, 1H), 4.67 – 4.56 (m, 1H), 3.37 (s, 2H), 2.87 (d, J = 11.1 Hz, 2H), 2.76-2.65 (m, 2H), 2.07 (m, 2H), 2.03 – 1.96 (m, 1H), 1.92 (q, J = 7.3 Hz, 2H), 1.84 – 1.59 (m, 5H), 1.47 (s, 9H), 1.36 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.0, 155.3, 151.1, 149.0, 148.9, 137.1, 136.9, 135.9, 135.2, 129.5, 128.4, 126.9, 126.8, 126.7, 126.3, 126.2, 119.7, 119.5, 117.6, 117.4, 79.2, 62.6, 53.0, 52.4, 48.4, 30.5, 29.2, 28.5, 28.4, 19.9, 9.5.
(R)-tert-butyl (6-((4-(N-(3,4-dichlorophenyl)propionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (36 scheme 3)
Prepared as described for 5 from (R)-tert-butyl (6-(iodomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate 3 (0.12 g, 0.32 mmol, 1eq) and N-(3,4-dichlorophenyl)-N-(piperidin-4-yl)propionamide 33 (0.14 g, 0.48 mmol, 1.5eq) in 10 mL of acetone and 0.08 g of K2CO3 afforded the title compound 36 (0.12 g, 67% of yield) as a light brown colored solid. MS (ESI) m/z (M + H)+: 560. 1H NMR (499 MHz, Chloroform-d) δ 7.48 (d, J = 8.4 Hz, 1H), 7.24 (d, J = 7.9 Hz, 1H), 7.19 (d, J = 2.4 Hz, 1H), 7.07 – 7.01 (m, 1H), 6.96 – 6.91 (m, 2H), 4.86 – 4.76 (m, 1H), 4.72 (d, J = 8.9 Hz, 1H), 4.68 – 4.57 (m, 1H), 3.37 (s, 2H), 2.93 – 2.82 (m, 2H), 2.79 – 2.63 (m, 2H), 2.08 (d, J = 11.8 Hz, 2H), 1.99 (m, 1H), 1.96 – 1.87 (m, 2H), 1.84 – 1.67 (m, 5H), 1.47 (s, 9H), 1.41 – 1.29 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ172.9, 155.4, 138.5, 137.2, 137.0, 136.0, 133.1, 132.9, 132.2, 130.9, 129.9, 129.5, 128.5, 127.0, 79.3, 62.6, 53.0, 52.9, 52.5, 48.5, 30.6, 29.2, 28.7, 28.5, 19.9, 9.5.
(R)-tert-butyl (6-((4-(N-(4-chloro-3-methoxyphenyl)propionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (37 scheme 3)
Prepared as described for 5 from (R)-tert-butyl (6-(iodomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate 3 (0.12 g, 0.32 mmol, 1eq) and N-(3-chloro-4-methoxyphenyl)-N-(piperidin-4-yl)propionamide 34 (0.14 g, 0.48 mmol, 1.5eq) in 10 mL of acetone and 0.09 g of K2CO3 afforded the title compound 37 (0.13 g, 75% of yield) as a light yellow colored solid. MS (ESI) m/z (M + H)+: 556. 1H NMR (499 MHz, Chloroform-d) δ 7.34 (d, J = 8.2 Hz, 1H), 7.21 (d, J = 7.8 Hz, 1H), 7.03 – 6.98 (m, 1H), 6.91 (s, 1H), 6.60 (dd, J = 8.2, 2.2 Hz, 1H), 6.56 (d, J = 2.2 Hz, 1H), 4.77 (s, 1H), 4.69 (d, J = 8.8 Hz, 1H), 4.63-4.56 (m, 1H), 3.85 (s, 3H), 3.34 (s, 2H), 2.84 (d, J = 11.4 Hz, 2H), 2.73-2.61 (m, 2H), 2.10 – 2.00 (m, 2H), 2.00 – 1.87 (m, 3H), 1.82 – 1.62 (m, 5H), 1.44 (s, 9H), 1.40 – 1.27 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.22, 155.41, 138.58, 137.21, 137.05, 136.01, 130.39, 129.55, 128.46, 127.03, 123.19, 122.69, 114.20, 79.27, 77.25, 77.20, 77.00, 76.94, 76.75, 62.65, 56.29, 53.00, 52.37, 48.47, 30.67, 30.57, 29.23, 28.45, 19.95, 9.64.
(R)-N-(1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-(3,4-difluorophenyl)propionamide (38 scheme 3)
Prepared as described for 8 from 35 (R)-tert-butyl (6-((4-(N-(3,4-difluorophenyl)propionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (0.12 g, 0.23) afforded the title compound 38 as a white solid (0.1 g, 78% of yield). ESI MS m/z 428 (MH)+. HRMS [M + H]+ 428.2509 (theoretical 428.2508); 1H NMR (499 MHz, DMSO-d6) δ 9.97 – 9.71 (m, 1H), 8.37 (d, J = 5.5 Hz, 3H), 7.59 – 7.44 (m, 3H), 7.29 (d, J = 8.0 Hz, 1H), 7.25 (s, 1H), 7.12 (d, J = 9.0 Hz, 1H), 4.74 – 4.61 (m, 1H), 4.45 (m, 1H), 4.18 (s, 2H), 3.37 – 3.24 (m, 2H), 3.17 – 3.04 (m, 2H), 2.80-2.67 (m, 2H), 2.12 – 2.00 (m, 1H), 2.00 – 1.79 (m, 6H), 1.74 (m, 1H), 1.50 (m, 2H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ172.5, 159.3, 159.0, 158.7, 158.5, 150.8, 150.5, 148.9, 148.5 138.6, 135.2, 134.2, 130.2, 129.2, 129.0, 128.37, 128.31, 120.5, 120.4, 118.3, 118.1, 115.9, 58.9, 51.2, 51.1, 49.2, 48.0, 28.7, 28.1, 27.7, 27.2, 18.5, 9.6.
(R)-N-(1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-(3,4-dichlorophenyl)propionamide (39 scheme 3)
Prepared as described for 8 from 36 (R)-tert-butyl (6-((4-(N-(3,4-dichlorophenyl)propionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (0.15 g, 0.26) afforded the title compound 39 as a white solid (0.1 g, 81% of yield). ESI MS m/z 460 (MH)+. HRMS [M + H]+ 460.1917 (theoretical 460.1916); 1H NMR (499 MHz, DMSO-d6) δ 9.69 (d, J = 11.1 Hz, 1H), 8.58 – 8.16 (m, 3H), 7.73 (d, J = 8.5 Hz, 1H), 7.66 – 7.61 (m, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.27 (m, 3H), 4.70 (d, J = 12.7 Hz, 1H), 4.50 – 4.39 (m, 1H), 3.30 (d, J = 12.0 Hz, 2H), 3.11 (m, 2H), 2.74 (m, 2H), 2.13 – 1.98 (m, 1H), 2.01 – 1.81 (m, 6H), 4.25 – 4.10 (m, 2H), 1.74 (m, 1H), 1.47 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 172.2, 159.2, 159.0, 158.7, 158.5, 138.6, 138.5, 134.2, 132.9, 132.3, 132.1, 131.56, 131.51, 130.2, 129.2, 129.0, 121.1, 118.7, 116.3, 113.9, 58.9, 51.2, 49.2, 48.0, 28.7, 28.3, 27.7, 27.2, 18.5, 9.6.
(R)-N-(1-((5-amino-5,6,7,8-tetrahydronaphthalen-2-yl)methyl)piperidin-4-yl)-N-(4-chloro-3-methoxyphenyl)propionamide (40 scheme 3)
Prepared as described for 8 from 37 (R)-tert-butyl (6-((4-(N-(4-chloro-3-methoxyphenyl)propionamido)piperidin-1-yl)methyl)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (0.13 g, 0.23) afforded the title compound 40 as a white solid (0.1 g, 75% of yield). ESI MS m/z 456 (MH)+. HRMS [M + H]+ 456.2416 (theoretical 456.2412); 1H NMR (499 MHz, DMSO-d6) δ 9.77 (d, J = 10.1 Hz, 1H), 8.40 (s, 3H), 7.50 (dd, J = 13.2, 8.1 Hz, 2H), 7.29 (d, J = 8.1 Hz, 1H), 7.24 (s, 1H), 7.01 (s, 1H), 6.86 – 6.74 (m, 1H), 4.66 (d, J = 12.5 Hz, 1H), 4.52 – 4.39 (m, 1H), 4.25 – 4.12 (m, 2H), 3.84 (s, 3H), 3.34 – 3.24 (m, 2H), 3.19 – 3.06 (m, 2H), 2.73 (m, 2H), 2.06 (m, 1H), 2.01 – 1.81 (m, 6H), 1.80 – 1.67 (m, 1H), 1.53 (m, 2H), 0.89 (t, J = 7.3 Hz, 3H), 13C NMR (125 MHz, DMSO-d6) δ172.5, 159.3, 159.0, 158.7, 158.5, 155.4, 138.6, 138.5, 134.3, 132.3, 130.5, 130.2, 129.2, 129.0, 123.6, 121.7, 121.0, 118.7, 116.3, 115.6, 113.9, 65.3, 58.9, 56.8, 51.3, 49.3, 48.0, 28.7, 28.1, 27.7, 27.4, 18.5, 15.6, 9.7.
Supplementary Material
Table 1.
Binding Affinities and functional activity of the amine, acetamide and hydroxyl substituted 4-anilidopiperidine analogues at DOR and MOR.
| Cpd | Binding Ki (nM) | MVD(δ) IC50c (nM) |
GPI (μ) IC50c (nM) |
Ki μ/δ | aLogP | |||
|---|---|---|---|---|---|---|---|---|
| log IC50 | MOR (μ) | log IC50 | DOR (δ) | |||||
| 8d | −5.88 ± 0.16 | 580 | −4.71 ± 0.12 | 6000 | 12.9% at 1 μM | 12.2% at 1 μM | 1/10 | 4.17 |
|
| ||||||||
| 9d | −5.76 ± 0.39 | 760 | nd | >10000 | 5.3 % at 1 uM | 8.0 % at 1 uM | 1/13 | 4.04 |
|
| ||||||||
| 10d | −7.02 ± 0.11 | 40 | nd | >10000 | 7.5% at 1 μM | 1020+/−360 | 1/250 | 3.53 |
|
| ||||||||
| 11 | −5.98 ± 0.16 | 460 | nd | >10000 | nd | nd | 1/21 | 4.61 |
|
| ||||||||
| 12 | −6.95 ± 0.12 | 50 | nd | >10000 | 24.4% at 1 uM | 25.5 % at 1 uM | 1/200 | 4.42 |
|
| ||||||||
| 20 | −8.58 ± 0.05 | 2 | nd | >10000 | 23% at 1 μM | 55.20+/−4.3 | 1/5000 | 3.93 |
|
| ||||||||
| 21 | −6.44 ± 0.13 | 190 | −4.59 ± 0.13 | 7800 | 16% at 1 μM | 600+/−92 | 1/41 | 4.29 |
|
| ||||||||
| 38d | −6.71 ± 0.05 | 110 | −4.91 ± 0.10 | 5700 | 12% at 1 μM | 1100+/−110 | 1/50 | 3.84 |
|
| ||||||||
| 39d | −5.92 ± 0.04 | 700 | −4.71 ± 0.08 | 5200 | nd | nd | 1/7 | 4.63 |
|
| ||||||||
| 40d | −5.24 ± 0.22 | 2600 | −5.00 ± 0.03 | 4500 | nd | nd | 1/2 | 3.91 |
Competition assays were carried out using rat brain membranes.
Competition against radiolabeled ligand, data collected from at least 2 independent experiments n.d.: not determined.
Concentration at 50% inhibition of muscle contraction at electrically stimulated isolated tissues; these values represent the mean of four tissues within 95% confidence limit.
Trifluoro acetate salts
Acknowledgments
The work was supported by grants from the U.S. Public Health Service NIDA (Grants 314450 NIDA 2P01 DA006284). We thank Christine Kasten for assistance with the manuscript.
Abbreviations
- ACN
Acetonitrile
- Boc
tert-butyloxycarbonyl
- BH3-SMe2
Borane dimethylsulfide
- CHO
Chinese hamster ovary
- DALEA
[D-Ala2, Leu5] enkephalin amide
- DCM
Dichloromethane
- DCE
Dichloroethane
- DOR
Delta opioid receptor
- DHP
Dihydropyran
- DIPEA
N,N-Diisopropylethylamine
- DAMGO
[D-Ala2,NMePhe4,Gly5-ol]enkephalin
- DPDPE
c[D-Pen2,DPen5]enkephalin
- Dmt
2,6-dimethyltyrosine
- GPI
guinea pig isolated ileum
- GPCR
G protein-coupled receptor
- HRMS
High resolution mass spectrometry
- HCl
Hydrochloric acid
- hDOR
human δ opioid receptor
- MOR
Mu opioid receptor
- MVD
mouse vas deferens
- RP-HPLC
reverse phase high performance liquid chromatography
- RT
Room temperature
- NaOtBu
Sodium tert-butoxide Pd2(dba)3
- SAR
structure-activity relationship
- Na(OAc)3BH
Sodium triacetoxyborohydride
- Pd2dba3
Tris(dibenzylideneacetone)dipalladium
- Et3N
Triethylamine
- Tyr
Tyrosine
- rMOR
rat μ opioid receptor
- TFA
Trifluoroacetic acid
- TLC
thin layer chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Kieffer BL. Trends Pharmacol Sci. 1999;20:19. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- 2.Eguchi M. Med Res Rev. 2004;24:182. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- 3.Mosberg HI, Yeomans L, Harland AA, Bender AM, Sobczyk-Kojiro K, Anand JP, Clark JM, Jutkiewicz EM, Traynor JR. J Med Chem. 2013;56:2139. doi: 10.1021/jm400050y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anand JP, Purington LC, Pogozheva ID, Traynor JR, Mosberg HI. Chem Biol Drug Des. 2012;80:763. doi: 10.1111/cbdd.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C, McFadyen IJ, Traynor JR, Mosberg HI. Bioorg Med Chem Lett. 1998;8:2685. doi: 10.1016/s0960-894x(98)00472-7. [DOI] [PubMed] [Google Scholar]
- 6.Janssen PAJ. Brit J Anaesthesia. 1962;34:260. doi: 10.1093/bja/34.4.260. [DOI] [PubMed] [Google Scholar]
- 7.Gentilucci L, Tolomelli A, De Marco R, Artali R. Curr Med Chem. 2012;19:1587. doi: 10.2174/092986712799945030. [DOI] [PubMed] [Google Scholar]; Kaczor A, Matosiuk D. Curr Med Chem. 2002;9:1567. doi: 10.2174/0929867023369394. [DOI] [PubMed] [Google Scholar]
- 8.Weltrowska G, Chung NN, Lemieux C, Guo J, Lu Y, Wilkes BC, Schiller PW. J Med Chem. 2010;53:2875. doi: 10.1021/jm9019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vučković S, Prostran M, Ivanović M, Došen-Mićović L, Todorović Z, Nešić Z, Stojanović R, Divac N, Miković Z. Curr Med Chem. 2009;16:2468. doi: 10.2174/092986709788682074. [DOI] [PubMed] [Google Scholar]
- 10.Jagerovic N, Cano C, Elguero J, Goya P, Callado LF, Meana JJ, Girón R, Abalo R, Ruiz D, Goicoechea C, Martin MA. Bioorg Med Chem. 2002;10:817. doi: 10.1016/s0968-0896(01)00345-5. [DOI] [PubMed] [Google Scholar]
- 11.Vardanyan RS, Hruby VJ. Future Med Chem. 2014;6:385. doi: 10.4155/fmc.13.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Podolsky AT, Sandweiss A, Hua J, Bilskyc EJ, Cain JP, Kumirov VK, Lee YS, Hruby VJ, Vardanyan RS, Vanderaha TW. Life Sci. 2013;93:1010–1016. doi: 10.1016/j.lfs.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramanian G, Paterlini MG, Portoghese PS, Ferguson DM. J Med Chem. 2000;43:381. doi: 10.1021/jm9903702. [DOI] [PubMed] [Google Scholar]
- 14.Bagley JR, Kudzma LV, Lalinde NL, Colapret JA, Huang BS, Lin BS, Jerussi TP, Benvenga MJ, Doorley BM. Med Res Rev. 1991;11:403. doi: 10.1002/med.2610110404. [DOI] [PubMed] [Google Scholar]
- 15.Essawi MYH, Portoghese PS. J Med Chem. 1983;26:348. doi: 10.1021/jm00357a007. [DOI] [PubMed] [Google Scholar]
- 16.Lee YS, Nyberg J, Moye S, Agnes RS, Davis P, Ma S-W, Lai J, Porreca F, Vardanyan R, Hruby VJ. Bioorg Med Chem Lett. 2007;17:2161. doi: 10.1016/j.bmcl.2007.01.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrov RR, Vardanyan RS, Lee YS, Ma SW, Davis P, Begay LJ, Lai J, Porreca F, Hruby VJ. Bioorg Med Chem Lett. 2006;16:4946. doi: 10.1016/j.bmcl.2006.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee YS, Petrov RR, Park C, Ma SW, Davis P, Lai J, Porreca F, Hruby VJ. J Med Chem. 2007;50:5528. doi: 10.1021/jm061465o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee YS, Kulkarani V, Cowell SM, Ma SW, Davis P, Lai J, Porreca F, Vardanyan R, Hruby VJ. J Med Chem. 2011;54:382. doi: 10.1021/jm100982d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrov RR, Lee YS, Vardanyan RS, Liu L, Ma S-W, Davis P, Lai J, Porreca F, Vanderah TW, Hruby VJ. Bioorg Med Chem Lett. 2013;23:3434. doi: 10.1016/j.bmcl.2013.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dan P, Birgitte EL, Gordon M, Ostergaard NE, Paul RJ. WO 2007093603. 2007
- 22.Ben AC, Toshihiro A, Kaustav B, Jian CJ, Brooks HJ, Wenyuan Q. WO 2006041888. 2006
- 23.Corey EJ, Bakshi RK, Shibata S. J Am Chem Soc. 1987;109:5551. [Google Scholar]
- 24.Misicka A, Lipkowski AW, Horvath R, Davis P, Kramer TH, Yamamura HI, Hruby VJ. Life Sci. 1992;51:1025. doi: 10.1016/0024-3205(92)90501-f. [DOI] [PubMed] [Google Scholar]
- 25.Kramer TH, Davis P, Hruby VJ, Burks TF, Porreca F. J Pharmacol Exp Ther. 1993;266:577. [PubMed] [Google Scholar]
- 26.Yaksh TL, Rudy TA. Physiol Behav. 1976;17:1031. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 27.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. Pain. 1988:3277. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.